Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
Considering the limited benefit of current therapies for NF2-associated tumors, immunotherapy prevails as a promising treatment strategy with the potential to selectively eliminate tumor cells. This chapter focuses on the concepts of cancer immunotherapy, specific challenges unique to nervous system tumors, and current preclinical studies and clinical trials. We highlight several promising advances which may help to overcome the unmet therapeutic need in NF2-associated tumors.
Departments of Neurological Surgery and Neurology, University of Southern California, Los Angeles, CA, USA
Thomas C. Chen
Department of Neurological Surgery, University of Southern California, Los Angeles, CA, USA
Frances E. Chow*
Departments of Neurological Surgery and Neurology, University of Southern California, Los Angeles, CA, USA
*Address all correspondence to: frances.chow@med.usc.edu
1. Introduction
Neurofibromatosis type 2 (NF2) is characterized by the development of nervous system tumors including vestibular schwannomas, meningiomas, and ependymomas [1, 2, 3]. Although these tumors are typically histologically benign, they may exhibit aggressive growth, cause devastating neurological disability, and lead to complications that result in early mortality [4, 5]. Current treatments for NF2 associated tumors involve multidisciplinary collaborations and include surgical resection, possible radiation, and chemotherapy—all of which are non-curative and are associated with the risk of permanent hearing loss or future development of malignancy [6, 7]. The identification of two-hit alterations to the NF2 gene on chromosome 22q12 [8, 9] and subsequent aberrations in the merlin and PI3k/Akt/mTOR pathways have led to extensive investigations into targeted therapies, yet none have demonstrated significant benefit to garner approval [10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20]. Therefore, a significant unmet need exists in the management of NF2 associated tumors.
Immunotherapy prevails as an appealing treatment strategy due to its potential to generate tumor-specific responses to selectively eliminate tumor cells. Since 2014, growing success in liquid and solid tumors has resulted in over 50 separate United States Food and Drug Administration approvals for immunotherapies [21]. Notable exceptions to the list of indications are central and peripheral nervous system tumors. This chapter focuses on the underpinning principles of immunotherapy, specific challenges unique to nervous system tumors, and promising emerging advances which may overcome the unmet therapeutic need in NF2 associated tumors.
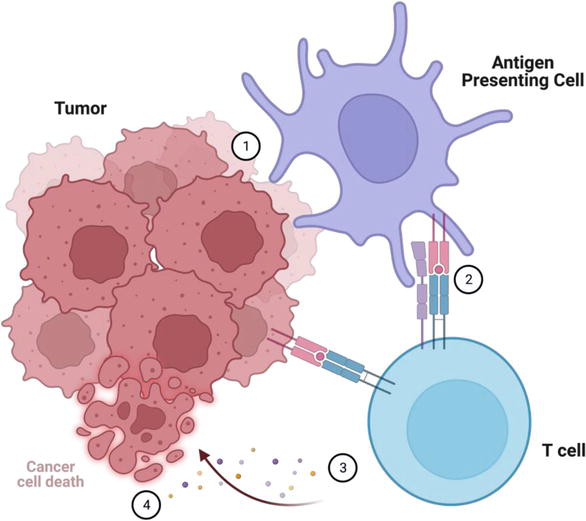
The immune system recognizes deviations from natural homeostasis as dangerous and employs the innate and adaptive immune systems to protect the body from “non-self” such as bacteria, viruses, and cancer. While innate immunity drives rapid responses through evolutionarily conserved mechanisms, adaptive immunity develops over days and is honed against specific antigens. In applying this concept to cancer immunology, it is possible to leverage each of the key executors—the cancer cell, antigen presenting cell, and effector T cell—as an immunotherapeutic strategy (Figure 1).
Figure 1.
Cancer immunology and principles of immunotherapy. (1) Tumor cells undergo phagocytosis by an antigen presenting cell (APC). (2) The APC presents tumor associated antigens on an MHC molecule to a naïve T cell. Dendritic cells have the most effective antigen presentation and T cell activation. Macrophages are subtyped into M1 anti-tumor and M2 pro-tumor/anti-inflammatory. Natural killer cells are part of the innate immune system and are capable of both antigen presentation and T cell activation. (3) T cell activation occurs when the appropriate T cell receptor (TCR) pairs with a matching tumor-associated antigen. Additional costimulatory signals such as B7-CD28 and cytokines are necessary to guide the differentiation and expansion of effector cells into cytotoxic T cells, helper T cells, regulatory T cells, and B cells. Each of these effector cells have unique markers and functions (Table 1). (4) The cytotoxic T cell eliminates the cancer cell in a process involving perforins, granzymes, and lysozymes.
Cell type
Markers
Function
Cytotoxic T cell
TCR, CD8+
Kill cells
Helper T cell
TCR, CD4+
Enhance immune function
Regulatory T cell
TCR, CD4+
Dampen immune function
B cell
BCR
Antibody production
Table 1.
Effector cell types.
2.1 Leveraging tumor: cancer vaccines and oncolytic viruses
Direct targeting of the tumor cell has been attempted through methods such as cancer vaccines and oncolytic viruses.
Vaccines are designed to prime the immune system to induce a tumor-directed response. Crucial to this mechanism is the identification of a pre-identified tumor specific antigen. Cautionary tales in tumor antigen escape were identified through experience in glioblastoma with rindopepimut (CDX-110), an epidermal growth factor receptor variant III (EGFRvIII) “vaccine” composed of EGFRvIII conjugated to the potent immunogenic substance KLH. Rindopepimut effectively activated humoral immunity through the endogenous formation of EGFRvIII antibodies. In a series of early phase clinical trials in recurrent glioblastoma, rindopepimut demonstrated improvement in overall survival [22, 23, 24, 25]. However, the phase 3 study was terminated early for futility, as survival was similar in the experimental and control arms [26]. Although 84% of tumors that originally expressed EGFRvIII lost expression at recurrence, this phenomenon of antigen escape was observed in both the control and treatment arms—thereby challenging the notion that EGFRvIII targeting therapies were solely responsible for the outgrowth of EGFRvIII-deficient glioblastoma cells.
The challenge with cancer vaccines remains difficulty in identifying a tumor-associated antigen and the intrinsic evolution of antigens over time through antigen escape. However, a potential alternative to vaccines based on pre-identified high quality endogenous neoantigens is through in-situ introduction of tumor-associated antigens via oncolytic viruses.
Oncolytic viruses are engineered to selectively infect cancer cells, leading to cell death and endogenous release of tumor antigens to promote a secondary immune response. An example of an oncolytic virus is the recombinant nonpathogenic polio-rhinovirus chimera, which infects malignant cells via CD155 receptor and causes tumor cell death. This releases tumor antigens into the microenvironment, attracting immune cells which then are sub-lethally infected by the virus to create a sustained pro-inflammatory cytokine response [27]. Additional viruses modified for oncolytic purposes include herpes, adeno, reo, vaccinia, measles, newcastle, and parvovirus [28].
Dendritic cells represent the most effective antigen presenting cell in phagocytosing cancer cells and training T cells. Dendritic cell vaccines have been a mainstay of immunotherapy through the leukapheresis, ex vivo loading or pulsing with various antigen sources (including peptides, DNA, RNA, liposomes), and then delivery of dendritic cells intradermally as a “vaccine” administered to the patient. The concept of dendritic cell vaccines has been explored in several groups, with recent promise demonstrated in newly diagnosed glioblastoma [29].
Macrophages make up the predominant immune infiltrate within intracranial tumors. For example, in vestibular schwannomas, macrophages—rather than Schwann cells—constitute up to 70% of proliferating cells [30]. Macrophages confer both anti-tumor (M1) and pro-tumor (M2) properties. Tumor-associated macrophage markers remain under investigation, but M1 macrophages (iNOS+, CD86+, CD80+, HLA-DR+) are classically tumor-resistant in the setting of phagocytotic and antitumor inflammatory signatures. Alternatively, M2 macrophages (CD206+, CD204+, CD163+) promote immunosuppression, angiogenesis, and neovascularization [31]. The unique balance of immune-stimulating and immunosuppressive properties marks macrophages as an ideal candidate for immune modulation and directed polarization.
Natural killer (NK) cells represent 5–15% of circulating lymphocytes [32]. As members of the innate immune system, they are not limited by MHC restriction and do not require priming. To date, NK cells have encountered minimal issues with cytokine release syndrome (as with CAR T cells) and maintain antigen specificity such as for antibody-mediated cell engagers. NK cells therefore serve as appealing targets of immunotherapy.
2.3 Leveraging effector cytotoxic T cells: adoptive transfer, CAR T cells, checkpoint inhibitors, BITE therapies
T cells serve as the end effector to trigger tumor cytotoxicity through lysozymes, granzymes, and perforins. Potential mechanisms of T cell manipulation include adoptive transfer of genetically engineered CAR T cells, reversal of T cell exhaustion through checkpoint inhibitors, and localization of T cells to targets through bispecific T cell engagers (BITEs).
Adoptive transfer precipitates T cell activation by enlisting T cells through harvesting of autologous T cells, which are trained and expanded ex vivo against tumor, and transferred back to patients. However, there is significant difficulty in generating large numbers of functional tumor-specific T cells.
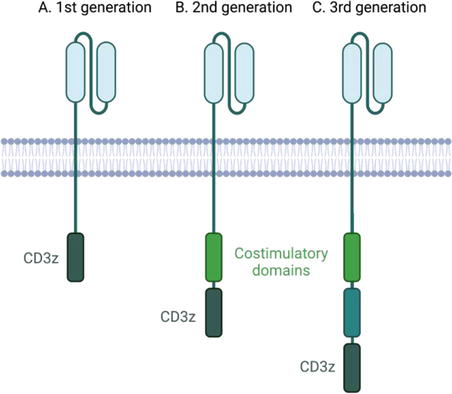
Out of this need arose the concept of CAR T cells, which are genetically modified T cells expressing chimeric antigen receptors (CARs). Such engineered CARs are chimeric because they combine both antigen-binding and T-cell activating functions into a single receptor (Figure 2). CARs are thereby programmed to recognize antigen without the need for MHC presentation or costimulatory signals to activate proliferation or clonal expansion in situ. Additional CAR modifications include bispecific CARs that target multiple tumor-associated antigens to minimize off-tumor effects or mitigate antigen escape; masked CARs with tumor-expressed proteases capable of cleaving linkers to scFvs of CARs for selective activation specifically by tumor cells; and switchable CARs to target peptide neo-epitopes present on a tumor-associated antigen-binding antibody (rather than the tumor cell) [33].
Figure 2.
CAR T cells of 1st generation, 2nd generation, 3rd generation. (A) 1st generation CARs exhibit single chain variable fragment to target a tumor antigen moiety and are limited by poor persistence. (B) 2nd generation CARs are supplemented by the presence of a co-stimulatory domain and are associated with increased toxicity. (C) 3rd generation CARs have both co-stimulatory domains.
In a case report from a phase 1 clinical trial for IL13Ra2 CAR T, a patient with recurrent multifocal intracranial glioblastoma underwent resection and direct intratumoral infusion with CAR T cells with local control. However, he subsequently developed continued progression of non-resected areas and developed leptomeningeal spread with spinal metastases. Additional intraventricular infusions of CAR T cells via a rickham reservoir led to dramatic clinical and radiographic response that lasted 7.5 months; however his disease ultimately recurred [34]. CAR T cells have thus far had limited efficacy in brain tumors due to antigen escape, limited T cell persistence, exhaustion, and adequate delivery to the tumor.
An effective mechanism of crosslinking T cells to their target is through bispecific T cell engagers (BITEs). BITEs are antibody constructs comprised of a CD3 antigen binding segment and a tumor-antigen binding segment.
All T cells, including engineered CAR T cells, demonstrate limited persistence due to exhaustion over time. T cell expression of CTLA-4 and PD-1, as well as engagement with PD-L1, deactivates T cells and triggers apoptosis. Checkpoint blockade with ipilimumab, nivolumab, atezolizumab, or pembrolizumab inhibits these inhibitory signals, thereby re-activating exhausted T cells. Checkpoint inhibitors are of interest because they are off the shelf; however, they have had limited benefit as a monotherapy in nervous system tumors. There remains significant promise in the use of checkpoint inhibitors (and other checkpoints such as IDO1 and TIM3) neoadjuvant to surgery [35, 36], with radiation [37], or in combination with other immune modulating therapies.
Despite the elegant simplicity of the immune system, many intrinsic and extrinsic obstacles pose therapeutic challenges to immunotherapy via immune escape, immune privilege, low tumor antigenicity, and a cold tumor microenvironment. Immunotherapy aims to counteract each of these intrinsic hurdles and tumor evasion techniques.
3.1 Cancer immune escape
Cancer immune escape is based upon the theory of immune surveillance, in which the immune system recognizes and destroys transformed cells before they give rise to detectable tumors. However, the building burden of genetic instability and immune pressure leads to immune escape and ultimately tumor progression.
3.2 Immune privilege and the blood-brain barrier
Historically, the central nervous system (CNS) has been considered an immune privileged organ due to an intact blood brain barrier and absence of a typical lymphatic system. Early studies in rabbits demonstrated that allogeneic skin grafts placed on different organs of the body were rapidly rejected, but skin grafts placed on the brain escaped rejection [38]. However, this assumption of immune privilege has been challenged considering recent evidence for CNS lymphatics, or glymphatics [39, 40, 41]. Evidence for CNS lymphatics was based on observations that radiolabeled polyethylene glycol and albumin injected in the brain could be detected in the cervical lymphatics and along the olfactory nerve [42, 43, 44, 45, 46]. Further work described an additional route for the egress of soluble antigen via perivenous and periarterial structures [39, 40].
Therefore, the brain is not as immunologically privileged as once thought. But it continues to hold true that the blood-brain barrier limits the traversing of molecules, recombinant proteins, or gene-based medicines that are larger than 50 Da. Poor penetration across the blood-brain barrier occurs with approximately 98% of small molecule drugs and nearly all large molecules. Most chemotherapies, targeted therapies, and drug-antibody conjugates unfortunately do not cross the blood-brain barrier [47, 48].
3.3 Low tumor antigenicity
A second mechanism of immune escape is inherent to the tumor itself. Nervous system tumors have low antigenicity, making it difficult for the immune system to recognize the tumor as non-self to generate an immune response. Tumor-associated antigens may be identified through genetic, biochemical, and predictive computational approaches [49].
3.3.1 Low tumor mutational burden
The success of immunotherapy in melanoma and NSCLC is in part due to the high tumor mutational burden from UV- and smoking-related DNA damage. A high tumor mutational load provides a high availability of neoantigens, which are easily recognized by the patrolling immune system to induce an antitumor immune response [50, 51, 52, 53]. However in glioblastoma, fewer than 4% of glioblastomas have an inherent high tumor mutational burden, as the median tumor mutational burden is 4 [54].
3.3.2 Rarity of common tumor-specific antigens
As an alternative to overall mutational load, antigens of high tumoral specificity may serve as an optimal target for the immune system. Across patients there unfortunately exists a low number of shared common mutations in NF2 associated tumors. Additionally, in gliomas there is significant heterogeneity within a single tumor, as a tumor-associated antigen is not necessarily expressed on every cell across a tumor. Furthermore, as observed in the example of the tumor-specific antigen EGFRvIII, expression of tumor antigens varies not only with location, but also with time. Serial evaluation of tumors demonstrates transformation over time, such as with the loss of the tumor-associated antigen EGFRvIII in glioblastoma [26], thereby contributing to immune escape.
3.4 Cold tumor microenvironment
Gliomas notoriously create a highly immunosuppressive microenvironment characterized by few functional antigen presenting cells, in addition to abundant immunosuppressive APCs and regulatory T cells [55]. We continue to learn more about the abundant monocytic and dendritic cell populations which predominate the bulk of tumor infiltrating immune cells [56, 57, 58]. Brain tumors such as glioblastoma are therefore considered immunologically “cold.”
Over the past several decades, significant research has been conducted to understand the underlying mechanisms of vestibular schwannoma development and to identify effective treatments for these tumors. It has been long recognized that regions of inflammatory cells penetrate vestibular schwannomas (Antoni B regions) [59]. Serum and tumor extracts from patients with vestibular schwannomas overexpress immunogenic mediators including IL-1B, IL-6, TNF-a, ICAM-1, and CXCR4 [60, 61, 62, 63, 64, 65, 66, 67]. Fast-growing vestibular schwannomas express elevated IL-34 and M-CSF, which may be responsible for chemotaxis of tumor associated macrophages [68] and higher levels of inflammation correlating with longer duration of symptoms [69].
More recent work has explored the role of inflammation [30] and identification of precise tumor infiltrating immune populations in vestibular schwannomas [70, 71]. CD163+ tumor-associated macrophages (M2) are associated with the volumetric growth of vestibular schwannomas [72] and shorter progression-free survival [73, 74, 75, 76]. Regulatory T cells (CD4+, CD25+, FOXP3+) suppress tumor-specific immunity [77] and are more prominent in progressive vestibular schwannomas [78, 79]. Progressive vestibular schwannomas in patients demonstrate increased expression of PD-L1 and other checkpoints such as TIM-3 [80], potentially implicating a mechanism of PD-L1 mediated immune evasion [81, 82].
4.2 Immunotherapy strategies
4.2.1 Checkpoint inhibitor
Preclinical and clinical investigations have explored the potential role of checkpoint inhibitors in the management of vestibular schwannomas. Mouse models treated with PD-1 blockade successfully demonstrated reduction in schwannoma size, underscoring the potential therapeutic effect of checkpoint inhibitors in vestibular schwannomas [83].
4.2.2 Oncolytic bacteria
A recent preclinical study employed the bacterial cancer theory to demonstrate the effectiveness of intratumoral injections of bacteria within hypoxic areas of angiogenic tumors, thereby triggering lysis of tumor cells and antitumor responses [84] such as shift of macrophages from M2 (pro-tumor) to M1 (anti-tumor) subtypes [85, 86]. In human xenograft and mouse syngeneic models, a highly attenuated strain of Salmonella typhimurium (VNP00002) was capable of inducing cytokine and effector cell profiles toward enhanced innate and adaptive immune responses to control the growth of intrasciatic benign schwannomas [87]. The mechanism is consistent with a vaccination-based immunotherapy and supports further evaluation in NF2 vestibular schwannomas.
4.3 Future directions
These preclinical results provide important evidence to support the potential role of immunotherapy in the treatment of NF2-associated vestibular schwannomas. Clinical trials are needed to fully understand the safety and efficacy of this approach and to determine the optimal treatment regimen for patients with this condition.
An understanding of the intricate interaction between meningiomas and the immune system offers opportunities for immunotherapy-based treatments. Within meningiomas, up to one quarter of all cells are macrophages [88, 89, 90, 91]. Macrophages make up the largest fraction of immune infiltrates (up to 80%) [89] and are predominantly the immunosuppressive M2 phenotype [92]. High grade meningiomas and recurrent meningiomas harbor a higher proportion of M2 macrophages [88, 93], and M2 macrophages are independently associated with worse prognosis [94]. Other immune infiltrates include T cells and B cells [95]. Although the degree of Treg infiltration has not yet been demonstrated as an independent prognostic factor, grade 3 meningiomas have increased penetration of Tregs, supporting the potential role of immunosuppression in developing resistance and immune escape [96]. Further contribution to an immunosuppressive response in meningiomas is the high expression of several checkpoint molecules including NY-ESO-1, PC-L1, PD-L2, B7-H3, and CTLA-4 [96, 97, 98, 99, 100], which have been associated with tumor recurrence, progression, and worse prognosis [99].
5.2 Immunotherapy strategies
5.2.1 Interferon-alpha
The initial use of immune modulating therapies in meningiomas employed the cytokine interferon-alpha (IFN-a). A pilot study in both low and high grade meningiomas demonstrated that treatment with IFN-a demonstrated a slight regression or stable disease lasting from 6 to 14 months (n = 6) [101]. An additional phase 2 trial in grade 1 meningiomas demonstrated a 6-month progression free survival (PFS-6) of 54% and a median progression free survival (mPFS) of 7 months [102]. However, a retrospective cohort study in high grade meningiomas demonstrated a limited PFS-6 of only 17% and mPFS of 3 months [103]. These studies served as proof-of-concept in the potential role of immune modulation for meningiomas.
5.2.2 Checkpoint inhibitors
In a recent phase 2 trial evaluating the efficacy of pembrolizumab in sporadic recurrent grade 2 and 3 meningiomas (n = 24), PFS-6 was 0.48 (90% CI: 0.31–0.66) and mPFS reached 7.6 months (90% CI: 3.4–12.9 months). The adverse event profile matched that of other PD-1 inhibitor studies, including fatigue, pruritis, and 20% experienced CTCAE grade 3 or higher adverse events [104]. Another recent phase 2 trial evaluating the efficacy of nivolumab in recurrent grade 2 and 3 meningiomas (n = 25) demonstrated PFS-6 of 42.4% (95% CI: 22.8–60.7) and median overall survival of 30.9 months (95% CI: 17.6-NA). The investigators concluded that a subset of patients benefitted from therapy but overall, the study did not meet its predefined endpoint [105]. Although these studies were not strictly for NF2 associated meningiomas, the results warrant further investigation, particularly in a dedicated NF2 population.
5.2.3 Adoptive transfer
We currently await results from a recently-completed phase 2 trial which evaluated anti-NY-ESO1 T cell receptor-gene engineered lymphocytes [106]. In this study, 11 patients with NY-ESO1-expressing tumors (melanoma, meningioma, breast cancer, non-small cell lung cancer, and hepatocellular cancer) were treated with T cells that had undergone TCR-engineering via retroviral transfection. The patients completed lymphodepletion with cyclophosphamide and fludarabine prior to infusion of the modified T cells with enhancement by aldesleukin. If beneficial, this study may support prior preclinical data in targeting NY-ESO-1 as an immunotherapeutic strategy for the treatment of meningiomas [107].
5.3 Future directions
These clinical trials provide evidence of the potential of immunotherapy as a treatment option for meningiomas. However, it is important to note that these studies are preliminary and that more research is needed to determine the best approach for incorporating immunotherapy into the treatment of NF2-associated meningiomas.
Genomic analysis has demonstrated that ependymomas are molecularly distinct from gliomas such as glioblastoma. However, as a subtype of gliomas, significant interest and investigation is ongoing in the ependymoma tumor microenvironment and potential immunotherapy targets. Qualitative analysis of tumor-infiltrating immune populations reveals that increased cytotoxic T cells (CD8+), a high ratio of CD4+/CD8+ cells, and increased number of IDO+ cells (dendritic cells) are associated with favorable prognosis. Alternatively, elevated FOXP3+ Tregs, CD68+ TAMs, and M2 polarization (high ratio of CD163/AIF1+ cells) are associated with poor prognosis [108].
Ependymomas express a subset of unique tumor-associated antigens which may serve as potential targets for vaccine or oncolytic therapies due to their selective overexpression in tumor compared to normal brain tissue, including EphA2, IL-13Ra2, Survivin, and WT1 [109, 110]. Response to bevacizumab in NF2 associated ependymomas [111, 112] suggests that VEGF may serve as another target antigen for further immunotherapy development. This builds upon prior work demonstrating that more significant immune responses may correlate with improved survival [113].
6.2 Immunotherapy strategies
6.2.1 Peptide vaccine
There is an ongoing phase 1 trial of the SurVaxM vaccine in children with progressive or relapsed ependymoma, medulloblastoma, high grade glioma or newly diagnosed diffuse intrinsic pontine glioma (NCT04978727) [114]. The SurVaxM vaccine targets Survivin, a protein that stabilizes microtubules during spindle formation and whose high expression in several malignancies including ependymomas is associated with unfavorable prognosis [115]. The vaccine employs several strategies to create an antitumor effect, including through the incorporation and enhanced binding of multiple MHC class I epitopes, cytokine support, and antibody-mediated cell killing. SurVaxM has completed a phase 2 trial in newly diagnosed glioblastoma, with no serious adverse events [116].
Under investigation for children with recurrent ependymoma is an additional tumor antigen peptide-mediated vaccine. In this phase 1 trial, HLA-A2 restricted synthetic tumor antigens will be administered in combination with the immunoadjuvant imiquimod to stimulate an immune response (NCT01795313) [117]. We eagerly await results from these trials.
6.2.2 Oncolytic virus
The ongoing phase 1 trial of oncolytic HSV-1 G207 in combination with single dose radiation for children with recurrent or refractory cerebellar brain tumors seeks to use an engineered herpes simplex virus-1 to selectively replicate in and kill tumor cells (NCT03911388) [118]. While several past and ongoing oncolytic HSV brain tumor trials have been completed, therapeutic resistance has remained a challenge in relation to intratumoral delivery of the viral vector, the tumor microenvironment, and failure of the host’s immune response [119].
6.2.3 CAR T cell therapy
Adoptive transfer with CAR T cells is an active area of investigation for ependymomas. Microarray analysis of pediatric ependymomas identify the human epidermal growth factor receptor 2 (HER2) as a potential tumor-associated antigen target, with preclinical experiments confirming antigen recognition [120]. There is an ongoing phase 1 trial of HER2 specific CAR T cells in children with recurrent or refractory ependymomas (NCT04903080) [121]. Additionally, there is an ongoing phase 1 trial of IL-13Ra2 CAR T cells in adults with ependymoma, leptomeningeal glioblastoma, or medulloblastoma (NCT04903080) [122]. Up to 67% of ependymoma cells express IL-13Ra2, which is associated with poor prognosis [123]. Early benefit with IL-13Ra2 CAR T cells in refractory glioblastoma [34] supports its use as a feasible therapy in ependymomas.
However, considering lessons learned from glioblastoma, concerns for an immunosuppressive microenvironment and tumor heterogeneity may limit the effectiveness of single-target CAR T therapies in ependymomas [124]. Tandem CAR T cells targeting more than 1 tumor-associated antigen may help to mitigate tumor antigen escape [125].
6.2.4 Checkpoint inhibitor
An ongoing phase 1 trial is evaluating the safety and preliminary efficacy of pembrolizumab (anti-PD1) in young patients with recurrent, progressive, or refractory brain tumors including ependymoma (NCT02359565) [126]. Additional novel checkpoint inhibitors such as the humanized IgG4 anti-PD-1 monoclonal antibody tiselizumab (BGB-A317) are under investigation (NCT02407990), with promising preliminary efficacy in stabilizing a case of extensive ependymoma with spinal and lung metastases for a duration of 18 months [127].
6.2.5 Macrophage modulation
Magrolimab is a first-in-class humanized IgG4 monoclonal antibody that blocks the macrophage checkpoint CD47, thereby inhibiting exhaustion of phagocytic macrophages. In a first-in-human basket trial of solid tumors (n = 84), magrolimab (Hu5F9-G4) was well tolerated [128]. No maximum tolerated dose was reached, and toxicities were mild to moderate, including transient anemia (57%), fatigue (64%), headache (50%), fever (45%), or chills (45%). An ongoing phase 1 trial of magrolimab in recurrent or progressive brain tumors is currently enrolling (NCT05169944) [129].
6.3 Future directions
Although extensive preclinical and clinical investigations have explored immunotherapy strategies in ependymoma, none are exclusively in the setting of NF2 mutations. Further investigation into the differences between sporadic and NF2-mediated ependymomas are necessary.
Immunotherapy has established itself as the fourth pillar of cancer therapy, following surgery, radiation, and chemotherapy. Thus far, there has been limited exploration into the potential of immunotherapy for NF2-associated tumors. No immunotherapy clinical trials have been conducted in vestibular schwannomas, a few studies are ongoing in meningiomas, and increasing clinical investigation is underway in ependymomas, borrowed predominantly from prior studies in glioblastoma. Potential strategies to harness the immune system include cancer vaccines, oncolytic viruses, dendritic cell vaccines, macrophage modulators, NK cell therapies, adoptive transfer, CAR T cells, checkpoint inhibitors and bispecific T cell engagers. Although current trials are not specifically restricted to patients with underlying NF2, future investigations may offer more insight in this unique population. Additional challenges include effective delivery of immunotherapy to the nervous system, reliable assessment of response versus progression through imaging and biomarkers, and rapid clinical trial accrual in this small and rare population. We hope that insights and limitations from our past experiences [130] will inform and pave the way for future advances toward a more effective treatment for NF2 associated tumors.
This work was supported by grant KL2TR001854 from the National Center for Advancing Translational Science (NCATS) of the U.S. National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
1.Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Archives of Neurology. 1988;45(5):575-578
2.Evans DG, King AT, Bowers NL, Tobi S, Wallace AJ, Perry M, et al. Identifying the deficiencies of current diagnostic criteria for neurofibromatosis 2 using databases of 2777 individuals with molecular testing. Genetics in Medicine. 2019;21(7):1525-1533
3.Plotkin SR, Messiaen L, Legius E, Pancza P, Avery RA, Blakeley JO, et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Genetics in Medicine. 2022;24(9):1967-1977
4.Wilding A, Ingham SL, Lalloo F, Clancy T, Huson SM, Moran A, et al. Life expectancy in hereditary cancer predisposing diseases: An observational study. Journal of Medical Genetics. 2012;49(4):264-269
5.Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V, et al. A clinical study of type 2 neurofibromatosis. The Quarterly Journal of Medicine. 1992;84(304):603-618
6.Baser ME, Evans DG, Jackler RK, Sujansky E, Rubenstein A. Neurofibromatosis 2, radiosurgery and malignant nervous system tumours. British Journal of Cancer. 2000;82(4):998
7.Balasubramaniam A, Shannon P, Hodaie M, Laperriere N, Michaels H, Guha A. Glioblastoma multiforme after stereotactic radiotherapy for acoustic neuroma: Case report and review of the literature. Neuro-Oncology. 2007;9(4):447-453
8.Rouleau GA, Wertelecki W, Haines JL, Hobbs WJ, Trofatter JA, Seizinger BR, et al. Genetic linkage of bilateral acoustic neurofibromatosis to a DNA marker on chromosome 22. Nature. 1987;329(6136):246-248
9.Narod SA, Parry DM, Parboosingh J, Lenoir GM, Ruttledge M, Fischer G, et al. Neurofibromatosis type 2 appears to be a genetically homogeneous disease. American Journal of Human Genetics. 1992;51(3):486-496
10.Phadnis S, Hagiwara M, Yaffe A, Mitchell C, Nicolaides T, Akshintala S, et al. Phase II study of axitinib in patients with neurofibromatosis type 2 and progressive vestibular schwannomas. Neuro-Oncology. 2020;22(Suppl 3):iii419
11.Karajannis MA, Legault G, Hagiwara M, Ballas MS, Brown K, Nusbaum AO, et al. Phase II trial of lapatinib in adult and pediatric patients with neurofibromatosis type 2 and progressive vestibular schwannomas. Neuro-Oncology. 2012;14(9):1163-1170
12.Goutagny S, Raymond E, Esposito-Farese M, Trunet S, Mawrin C, Bernardeschi D, et al. Phase II study of mTORC1 inhibition by everolimus in neurofibromatosis type 2 patients with growing vestibular schwannomas. Journal of Neuro-Oncology. 2015;122(2):313-320
13.ClinicalTrials.gov. A Study of Nilotinib in Growing Vestibular Schwannomas [Internet]. 2023. Available from: https://clinicaltrials.gov/ct2/show/NCT01201538 [Accessed: February 20, 2023].
14.Welling DB, Collier KA, Burns SS, Oblinger JL, Shu E, Miles-Markley BA, et al. Early phase clinical studies of AR-42, a histone deacetylase inhibitor, for neurofibromatosis type 2-associated vestibular schwannomas and meningiomas. Laryngoscope Investigative Otolaryngology. 2021;6(5):1008-1019
15.Collier KA, Valencia H, Newton H, Made EM, Sborov DW, Cavliere R, et al. A phase 1 trial of the histone deacetylase inhibitor AR-42 in patients with neurofibromatosis type 2-associated tumors and advanced solid malignancies. Cancer Chemotherapy and Pharmacology. 2021;87(5):599-611
17.Zhao Y, Liu P, Zhang N, Chen J, Landegger LD, Wu L, et al. Targeting the cMET pathway augments radiation response without adverse effect on hearing in NF2 schwannoma models. Proceedings of the National Academy of Sciences of the United States of America. 2018;9:E2077-E2084
18.Plotkin S, Allen J, Babovic-Vuksanovic D, Dinh C, Nghiemphu L, Trippa L, et al. INTUITT-NF2, an adaptive platform-basket trial for neurofibromatosis 2 patients with progressive tumors: Interim results of the brigatinib treatment arm. Neuro-Oncology. 2022;24(s7):vii88
19.Plotkin S, Jordan J, Beauchamp R, Muzikansky A, Stemmer-Rachamimov A, Ramesh V. A single arm phase 2 study of the dual mTORC1/mTORC2 inhibitor vistusertib provided on an intermittent schedule for neurofibromatosis 2 patients with progressive or symptomatic meningiomas. Neuro-Oncology. 2018;20(Suppl 6):vi19
20.ClinicalTrials.gov. Trial of Selumetinib in Patients With Neurofibromatosis Type II Related Tumors (SEL-TH-1601) [Internet]. 2023. Available from: https://clinicaltrials.gov/ct2/show/NCT03095248 [Accessed: February 20, 2023]
21.The Cancer Research Institute. FDA Approval Timeline [Internet]. 2023. Available from: https://www.cancerresearch.org/fda-approval-timeline-of-active-immunotherapies [Accessed: February 20, 2023]
22.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. Journal of Clinical Oncology. 2010;28(31):4722-4729
23.Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-Oncology. 2011;13(3):324-333
24.Lai RK, Recht LD, Reardon DA, Paleologos N, Groves M, Rosenfeld MR, et al. Long-term follow-up of ACT III: A phase II trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma. Neuro-Oncology. 2011;13(Suppl 3):iii34-iii40
25.Reardon DA, Desjardins A, Vredenburgh JJ, O'Rourke DM, Tran DD, Fink KL, et al. Rindopepimut with bevacizumab for patients with relapsed EGFRvIII-expressing glioblastoma (ReACT): Results of a double-blind randomized phase II trial. Clinical Cancer Research. 2020;26(7):1586-1594
26.Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. The Lancet Oncology. 2017;18(10):1373-1385
27.Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. The New England Journal of Medicine. 2018;379(2):150-161
28.Rius-Rocabert S, García-Romero N, García A, Ayuso-Sacido A, Nistal-Villan E. Oncolytic virotherapy in glioma tumors. International Journal of Molecular Sciences. 2020;21(20):7604
29.Liau LM, Ashkan K, Brem S, Campian JL, Trusheim JE, Iwamoto FM, et al. Association of autologous tumor lysate-loaded dendritic cell vaccination with extension of survival among patients with newly diagnosed and recurrent glioblastoma: A phase 3 prospective externally controlled cohort trial. JAMA Oncology. 2023;9(1):112-121
30.Lewis D, Roncaroli F, Agushi E, Mosses D, Williams R, Li KL, et al. Inflammation and vascular permeability correlate with growth in sporadic vestibular schwannoma. Neuro-Oncology. 2019;21(3):314-325
31.Liu J, Geng X, Hou J, Wu G. New insights into M1/M2 macrophages: Key modulators in cancer progression. Cancer Cell International. 2021;21(1):389
32.Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. 2017;47(5):820-833
33.Siegler EL, Wang P. Preclinical models in chimeric antigen receptor-engineered T-cell therapy. Human Gene Therapy. 2018;29(5):534-546
34.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. The New England Journal of Medicine. 2016;375(26):2561-2569
35.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nature Medicine. 2019;25(3):477-486
36.Chow F, Mochizuki A, Lee A, Galvez M, Orpilla J, Everson R, et al. Validation of response to neoadjuvant anti-PD-1 immunotherapy in recurrent glioblastoma. Neuro-Oncology. 2020;21(6):vi4-vi5
37.Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clinical Cancer Research. 2017;23(1):124-136
38.Medawar PB. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. British Journal of Experimental Pathology. 1948;29(1):58-69
39.Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Science Translational Medicine. 2012;4(147):147ra111
40.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337-341
41.Iliff JJ, Nedergaard M. Is there a cerebral lymphatic system? Stroke. 2013;44(6 Suppl 1):S93-S95
42.Cserr HF, Cooper DN, Suri PK, Patlak CS. Efflux of radiolabeled polyethylene glycols and albumin from rat brain. The American Journal of Physiology. 1981;240(4):F319-F328
43.Johnston M, Zakharov A, Papaiconomou C, Salmasi G, Armstrong D. Evidence of connections between cerebrospinal fluid and nasal lymphatic vessels in humans, non-human primates and other mammalian species. Cerebrospinal Fluid Research. 2004;1(1):2
44.Murtha LA, Yang Q , Parsons MW, Levi CR, Beard DJ, Spratt NJ, et al. Cerebrospinal fluid is drained primarily via the spinal canal and olfactory route in young and aged spontaneously hypertensive rats. Fluids and Barriers of the CNS. 2014;11:12
45.Szentistvanyi I, Patlak CS, Ellis RA, Cserr HF. Drainage of interstitial fluid from different regions of rat brain. The American Journal of Physiology. 1984;246:F835-F844
46.Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathologica. 2009;117:1-14
47.Pardridge WM. CNS drug design based on principles of blood–brain barrier transport. Journal of Neurochemistry. 1998;70:1781-1792
48.Pardridge WM. Blood–brain barrier delivery. Drug Discovery Today. 2007;12:54-61
49.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nature Reviews. Cancer. 2014;14(2):135-146
50.Meléndez B, Van Campenhout C, Rorive S, Remmelink M, Salmon I, D'Haene N. Methods of measurement for tumor mutational burden in tumor tissue. Translational Lung Cancer Research. 2018;7(6):661-667
51.McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Kumar Saini S, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463-1469
52.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415-421
53.Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. The New England Journal of Medicine. 2017;377:2500-2501
54.Fusco M, Macaulay RJ, Forsyth PAJ, Walko CM. Detection of targetable somatic alterations in glioblastoma (GBM) and clinical impact. Journal of Clinical Oncology. 2019;37:2058
55.Li B et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biology. 2016;17:174
56.Lee AH, Sun L, Mochizuki AY, Reynoso JG, Orpilla J, Chow F, et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nature Communications. 2021;12(1):6938
57.Lu Y, Ng AHC, Chow F, Everson RG, Helmink BA, Tetzlaff MT, et al. Resolution of tissue signatures of therapy response in patients with recurrent GBM treated with neoadjuvant anti-PD1. Nature Communications. 2021;12(1):4031
58.Mochizuki A, Lee A, Orpilla J, Kienzler J, Galvez M, Chow F, et al. Myeloid populations and the effect of neoadjuvant PD-1 inhibition in the glioblastoma microenvironment: A surfaceomic and transcriptomic dissection at the single-cell level. Neuro-Oncology. 2019;21(Suppl 6):vi248
59.Antoni NRE. Über Rückenmarkstumoren und Neurofibrome. Munich: JF Bergmann; 1920
60.Rasmussen N, Bendtzen K, Thomsen J, Tos M. Specific cellular immunity in acoustic neuroma patients. Otolaryngology and Head and Neck Surgery. 1983;91(05):532-536
61.Harker LA, Nysather J, Katz A. Immunologic detection of acoustic neuroma: Preliminary report. The Laryngoscope. 1978;88(5):802-807
62.Rossi ML, Jones NR, Esiri MM, Havas L, Nakamura N, Coakham HB. Mononuclear cell infiltrate, HLA-Dr expression and proliferation in 37 acoustic schwannomas. Histology and Histopathology. 1990;5(4):427-432
63.Archibald DJ, Neff BA, Voss SG, Splinter PL, Driscoll CL, Link MJ, et al. B7-H1 expression in vestibular schwannomas. Otology & Neurotology. 2010;31(6):991-997
64.Dilwali S, Briët MC, Kao SY, Fujita T, Landegger LD, Platt MP, et al. Preclinical validation of anti-nuclear factor-kappa B therapy to inhibit human vestibular schwannoma growth. Molecular Oncology. 2015;9(7):1359-1370
65.Taurone S, Bianchi E, Attanasio G, Di Gioia C, Ierinó R, Carubbi C, et al. Immunohistochemical profile of cytokines and growth factors expressed in vestibular schwannoma and in normal vestibular nerve tissue. Molecular Medicine Reports. 2015;12(1):737-745
66.Constantin G, Piccio L, Bussini S, Pizzuti A, Scarpini E, Baron P, et al. Induction of adhesion molecules on human schwann cells by proinflammatory cytokines, an immunofluorescence study. Journal of the Neurological Sciences. 1999;170(2):124-130
67.Breun M, Schwerdtfeger A, Martellotta DD. CXCR4: A new player in vestibular schwannoma pathogenesis. Oncotarget. 2018;9(11):9940-9950
68.de Vries WM, Briaire-de Bruijn IH, van Benthem PPG, van der Mey AGL, Hogendoorn PCW. M-CSF and IL-34 expression as indicators for growth in sporadic vestibular schwannoma. Virchows Archiv. 2019;474(3):375-381
69.Labit-Bouvier C, Crebassa B, Bouvier C, Andrac-Meyer L, Magnan J, Charpin C. Clinicopathologic growth factors in vestibular schwannomas: A morphological and immunohistochemical study of 69 tumours. Acta Oto-Laryngologica. 2000;120(08):950-954
70.Schulz A, Büttner R, Hagel C, Baader SL, Kluwe L, Salamon J, et al. The importance of nerve microenvironment for schwannoma development. Acta Neuropathologica. 2016;132(2):289-307
71.de Vries M, Hogendoorn PCW, De Bruyn IB, Malessy MJA, Van Der Mey AGL. Intratumoral hemorrhage, vessel density, and the inflammatory reaction contribute to volume increase of sporadic vestibular schwannomas. Virchows Archiv. 2012;460(6):629-636
72.de Vries M, Briaire-de Bruijn I, Malessy MJ, de Bruïne SF, van der Mey AG, Hogendoorn PC. Tumor-associated macrophages are related to volumetric growth of vestibular schwannomas. Otology & Neurotology. 2013;34(2):347-352
73.Tamura R, Morimoto Y, Sato M. Difference in the hypoxic immunosuppressive microenvironment of patients with neurofibromatosis type 2 schwannomas and sporadic schwannomas. Journal of Neuro-Oncology. 2020;146(02):265-273
74.Hannan CJ, Lewis D, O’Leary C, Donofrio CA, Evans DG, Roncaroli F, et al. The inflammatory microenvironment in vestibular schwannoma. Neuro-Oncology Advances. 2020;2:vdaa023
75.Tamura R, Morimoto Y, Sato M, Kuranari Y, Oishi Y, Kosugi K, et al. Difference in the hypoxic immunosuppressive microenvironment of patients with neurofibromatosis type 2 schwannomas and sporadic schwannomas. Neuro-Oncology. 2020;146:265-273
76.Nisenbaum E, Misztalm C, Szczupak M, Thielhelm T, Peña S, Mei C, et al. Tumor-associated macrophages in vestibular schwannoma and relationship to hearing. OTO Open. 2021;5:2473974X211059111
77.Tamura R, Tanaka T, Akasaki Y, Murayama Y, Yoshida K, Sasaki H. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: Perspectives for therapeutic implications. Medical Oncology. 2019;37:2
78.Tamura R, Fujioka M, Morimoto Y, Ohara K, Kosugi K, Oishi Y, et al. A VEGF receptor vaccine demonstrates preliminary efficacy in neurofibromatosis type 2. Nature Communications. 2019;10:5758
79.Jacobs JF, Idema AJ, Bol KF, Nierkens S, Grauer OM, Wesseling P, et al. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro-Oncology. 2009;11:394-402
80.Li Z, Liu X, Guo R, Wang P. TIM-3 plays a more important role than PD-1 in the functional impairments of cytotoxic T cells of malignant schwannomas. Tumor Biology. 2017;39:1010428317698352
81.Perry A, Graffeo CS, Carlstrom LP, Raghunathan A, Driscoll CLW, Neff BA, et al. Predominance of M1 subtype among tumor-associated macrophages in phenotypically aggressive sporadic vestibular schwannoma. Neurosurgery. 2020;133(6):1637-1645
82.Wang S, Liechty B, Patel S, Weber JS, Hollmann TJ, Snuderl M, et al. Programmed death ligand 1 expression and tumor infiltrating lymphocytes in neurofibromatosis type 1 and 2 associated tumors. Neuro-Oncology. 2018;138(1):183-190
83.Chang JH, Tseng CY, Lee JH, Hsueh C. Anti-PD-1 immune checkpoint inhibition for vestibular schwannoma therapy. OncoImmunology. 2018;7(12):e1469564
84.Guo Y, Chen Y, Liu X, Min JJ, Tan W, Zheng JH. Targeted cancer immunotherapy with genetically engineered oncolytic Salmonella typhimurium. Cancer Letters. 2020;469:102-110
85.Zheng JH, Nguyen VH, Jiang SN, Park SH, Tan W, Hong SH, et al. Two-step enhanced cancer immunotherapy with engineered Salmonella typhimurium secreting heterologous flagellin. Science Translational Medicine. 2017;9(376):eaak9537
86.Yang M, Xu J, Wang Q , Zhang AQ , Wang K. An obligatory anaerobic Salmonella typhimurium strain redirects M2 macrophages to the M1 phenotype. Oncology Letters. 2018;15(3):3918-3922
87.Ahmed SG, Oliva G, Shao M, Wang X, Mekalanos JJ, Brenner GJ. Intratumoral injection of schwannoma with attenuated Salmonella typhimurium induces antitumor immunity and controls tumor growth. Proceedings of the National Academy of Sciences of the United States of America. 2022;119(24):e2202719119
88.Proctor DT, Huang J, Lama S, Albakr A, Van Marle G, Sutherland GR. Tumor-associated macrophage infiltration in meningioma. Neuro-Oncology Advances. 2019;1(01):1-10
89.Rossi ML, Cruz Sanchez F, Hughes JT, Esiri MM, Coakham HB. Immunocytochemical study of the cellular immune response in meningiomas. Journal of Clinical Pathology. 1988;41:314-319
90.Bo L, Mork SJ, Nyland H. An immunohistochemical study of mononuclear cells in meningiomas. Neuropathology and Applied Neurobiology. 1992;18:548-558
91.Asai J, Suzuki R, Fujimoto T, Suzuki T, Nakagawa N, Nagashima G, et al. Fluorescence automatic cell sorter and immunohistochemical investigation of CD68-positive cells in meningioma. Clinical Neurology and Neurosurgery. 1999;101:229-234
92.Pinton L, Solito S, Masetto E, Vettore M, Cane S, Puppa AD, et al. Immunosuppressive activity of tumor-infiltrating myeloid cells in patients with meningioma. Oncoimmunology. 2018;7:e1440931
93.Domingues PH, Teodósio C, Ortiz J. Immunophenotypic identification and characterization of tumor cells and infiltrating cell populations in meningiomas. The American Journal of Pathology. 2012;181(05):1749-1761
94.Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: A marker of alternative immunologic macrophage activation. The Journal of Experimental Medicine. 1992;176(01):287-292
95.Fang L, Lowther DE, Meizlish ML, Anderson RC, Bruce JN, Devine L, et al. The immune cell infiltrate populating meningiomas is composed of mature, antigen-experienced T and B cells. Neuro-Oncology. 2013;15:1479-1490
96.Du Z, Abedalthagafi M, Aizer AA, Mchenry AR, Sun HH, Bray MA, et al. Increased expression of the immune modulatory molecule PD-L1 (CD274) in anaplastic meningioma. Oncotarget. 2015;6:4704-4716
97.Proctor DT, Patel Z, Lama S, Resch L, Van Marle G, Sutherland GR. Identification of PD-L2, B7-H3 and CTLA-4 immune checkpoint proteins in genetic subtypes of meningioma. Oncoimmunology. 2019;8:e1512943
98.Baia GS, Caballero OL, Ho JS, Zhao Q , Cohen T, Binder ZA, et al. NY-ESO-1 expression in meningioma suggests a rationale for new immunotherapeutic approaches. Cancer Immunology Research. 2013;1:296-302
99.Han SJ, Reis G, Kohanbash G, Shrivastav S, Magill ST, Molinaro AM, et al. Expression and prognostic impact of immune modulatory molecule PD-L1 in meningioma. Neuro-Oncology. 2016;130:543-552
100.Deng J, Ma M, Wang D, Zhu H, Hua L, Sun S, et al. Expression and clinical significance of immune checkpoint regulator B7-H3 (CD276) in human meningioma. World Neurosurgery. 2019;135:e12-e18
101.Kaba SE, DeMonte F, Bruner JM, Kyritsis AP, Jaeckle KA, Levin V, et al. The treatment of recurrent unresectable and malignant meningiomas with interferon alpha-2B. Neurosurgery. 1997;40:271-275
102.Chamberlain MC, Glantz MJ. Interferon-alpha for recurrent World Health Organization grade 1 intracranial meningiomas. Cancer. 2008;113:2146-2151
103.Chamberlain MC. IFN-a for recurrent surgery- and radiation-refractory high-grade meningioma: A retrospective case series. CNS Oncology. 2013;2:227-235
104.Brastianos PK, Kim AE, Giobbie-Hurder A, Lee EQ , Wang N, Eichler AF, et al. Phase 2 study of pembrolizumab in patients with recurrent and residual high-grade meningiomas. Nature Communications. 2022;13(1):1325
105.Bi WL, Nayak L, Meredith DM, Driver J, Du Z, Hoffman S, et al. Activity of PD-1 blockade with nivolumab among patients with recurrent atypical/anaplastic meningioma: Phase II trial results. Neuro-Oncology. 2022;24(1):101-113
106.ClinicalTrials.gov. T Cell Receptor Immunotherapy Targeting NY-ESO-1 for Patients With NY-ESO-1 Expressing Cancer [Internet]. 2023. Available from: https://clinicaltrials.gov/ct2/show/study/NCT01967823 [Accessed: February 20, 2023]
107.Sun M, Orpilla J, Contreras E, Treger J, Molaie D, Tsang J, et al. Systemic adoptive transfer immunotherapy with TCR-transduced T-cells targeting NY-ESO-1 for meningioma. Neuro-Oncology. 2019;21(Suppl 6):vi129-vi130
108.Nam SJ, Kim YH, Park JE, Ra YS, Khang SK, Cho YH, et al. Tumor-infiltrating immune cell subpopulations and programmed death ligand 1 (PD-L1) expression associated with clinicopathological and prognostic parameters in ependymoma. Cancer Immunology, Immunotherapy. 2019;68(2):305-318
109.Yeung JT, Hamilton RL, Okada H, Jakacki RI, Pollack IF. Increased expression of tumor-associated antigens in pediatric and adult ependymomas: Implication for vaccine therapy. Neuro-Oncology. 2013;111(2):103-111
110.Hatano M, Eguchi J, Tatsumi T, Kuwashima N, Dusak JE, Kinch MS, et al. EphA2 as a glioma-associated antigen: A novel target for glioma vaccines. Neoplasia. 2005;7(8):717-722
111.Morris KA, Afridi SK, Evans G, Hensiek AE, McCabe MG, Kellett M, et al. The response of spinal cord ependymomas to bevacizumab in patients with neurofibromatosis type 2. Journal of Neurosurgery. Spine. 2017;26(4):474-482
112.Farschtschi S, Merker VL, Wolf D, Schuhmann M, Blakeley J, Plotkin SR, et al. Bevacizumab treatment for symptomatic spinal ependymomas in neurofibromatosis type 2. Acta Neurologica Scandinavica. 2016;133(6):475-480
113.Donson AM, Birks DK, Barton VN, Wei Q , Kleinschmidt-Demasters BK, Handler MH, et al. Immune gene and cell enrichment is associated with a good prognosis in ependymoma. Journal of Immunology. 2009;183(11):7428-7440
114.ClinicalTrials.gov. A Pilot Study of SurVaxM in Children Progressive or Relapsed Medulloblastoma, High Grade Glioma, Ependymoma and Newly Diagnosed Diffuse Intrinsic Pontine Glioma [Internet]. 2023. Available from: https://clinicaltrials.gov/ct2/show/NCT04978727 [Accessed: February 20, 2023]
115.Preusser M, Wolfsberger S, Czech T, Slavc I, Budka H, Hainfellner JA. Survivin expression in intracranial ependymomas and its correlation with tumor cell proliferation and patient outcome. American Journal of Clinical Pathology. 2005;124(4):543-549
116.Ahluwalia MS, Reardon DA, Abad AP, Curry WT, Wong ET, Figel SA, et al. Phase IIa study of SurVaxM plus adjuvant temozolomide for newly diagnosed glioblastoma. Journal of Clinical Oncology. 2023;41(7):1453-1465
117.ClinicalTrials.gov. Immunotherapy for Recurrent Ependymomas in Children Using Tumor Antigen Peptides With Imiquimod [Internet]. 2023. Available from: https://clinicaltrials.gov/ct2/show/NCT01795313 [Accessed: February 20, 2023]
118.ClinicalTrials.gov. HSV G207 in Children With Recurrent or Refractory Cerebellar Brain Tumors [Internet]. 2023. Available from: https://clinicaltrials.gov/ct2/show/NCT03911388 [Accessed: February 20, 2023]
119.Totsch SK, Schlappi C, Kang KD, Ishizuka AS, Lynn GM, Fox B, et al. Oncolytic herpes simplex virus immunotherapy for brain tumors: Current pitfalls and emerging strategies to overcome therapeutic resistance. Oncogene. 2019;38(34):6159-6171
120.Nellan A, Griesinger A, Witt D, Donson A, Amani V, Foreman NK. Study of anti-HER2 CAR T cells in the immunosuppressive ependymoma tumor microenvironment. Neuro-Oncology. 2018;20(Suppl 2):i80
121.ClinicalTrials.gov. HER2-specific Chimeric Antigen Receptor (CAR) T Cells for Children With Ependymoma [Internet]. 2023. Available from: https://clinicaltrials.gov/ct2/show/NCT04903080 [Accessed: February 20, 2023]
122.ClinicalTrials.gov. Brain Tumor-Specific Immune Cells (IL13Ralpha2-CAR T Cells) for the Treatment of Leptomeningeal Glioblastoma, Ependymoma, or Medulloblastoma [Internet]. 2023. Available from: https://clinicaltrials.gov/ct2/show/NCT04903080 [Accessed: February 20, 2023]
123.Kawakami M, Kawakami K, Takahashi S, Abe M, Puri RK. Analysis of interleukin-13 receptor alpha2 expression in human pediatric brain tumors. Cancer. 2004;101(5):1036-1042
124.Nellan A, Donson A, Calhoun J, Griesinger A, Fry T, Foreman N. Evaluation of CAR T cells in epenydmoma. Neuro-Oncology. 2020;22(Suppl 3):iii364
125.Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. The Journal of Clinical Investigation. 2016;126(8):3036-3052
126.ClinicalTrials.gov. Pembrolizumab in Treating Younger Patients With Recurrent, Progressive, or Refractory High-Grade Gliomas, Diffuse Intrinsic Pontine Gliomas, Hypermutated Brain Tumors, Ependymoma or Medulloblastoma [Internet]. 2023. Available from: https://clinicaltrials.gov/ct2/show/NCT02359565 [Accessed: February 20, 2023]
127.Tapia Rico G, Townsend A, Price T, Patterson K. Metastatic myxopapillary ependymoma treated with immunotherapy achieving durable response. BML Case Reports. 2020;13(12):e236242
128.Sikic BI, Lakhani N, Patnaik A, Shah SA, Chandana SR, Rasco D, et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. Journal of Clinical Oncology. 2019;37(12):946-953
129.ClinicalTrials.gov. Magrolimab in Children and Adults with Recurrent or Progressive Malignant Brain Tumors (PNOC025) [Internet]. 2023. Available from: https://clinicaltrials.gov/ct2/show/NCT05169944 [Accessed: February 20, 2023]
130.Chuntova P, Chow F, Galvez M, Watchmaker P, Prins R, Okada H, et al. Unique challenges for glioblastoma immunotherapy: Discussions across neuro-oncology and non-neuro-oncology experts in cancer immunology. Neuro-Oncology. 2021;23(3):356-375
Written By
Shyam Patel, Thomas C. Chen and Frances E. Chow
Submitted: 24 February 2023Reviewed: 02 March 2023Published: 28 March 2023
 Open access peer-reviewed chapter
Open access peer-reviewed chapter