 Open access peer-reviewed chapter
Open access peer-reviewed chapter
Abstract
Neurofibromatosis (NF1) is a rare genetic disease that predisposes to tumors in the peripheral and central nervous systems and other neoplasms. Neurofibromas are complex benign tumors involving various cell types. Among them, plexiform neurofibromas are locally invasive peripheral nerve sheath tumors (PNs) that can cause disfigurement and functional limitations. Based on histopathological and magnetic resonance imaging (MRI) data, PNs display variable morphology and behavior. The appearance of distinct nodular lesions (DNLs) in PNs raises concerns about an increased risk of transformation into malignant peripheral nerve sheath tumors (MPNSTs). Surgery represents the primary treatment option for NF1-related PN, although recently, specific targeted agents, that is, MEK inhibitors, have been shown to be partially effective. Several surgical techniques have been proposed for PNs to decrease intraoperative bleeding and facilitate tumor excision of either diffuse PNs or rapidly growing nodular PN. However, despite improving surgical methods, complete tumor excision can be achieved in only a few cases. In this frame, it emerges evident that searching for appropriate surgical and pharmacological treatments for neurofibromas is a priority challenge. In this chapter, we will review current treatments approved by the scientific/clinical community, emphasizing the most recent progress in this field.
Keywords
- neurofibromatosis type 1
- neurofibroma
- treatment
- neurofibroma surgery
- tumour progression
- MPNST
- MEK inhibitors
- Selumetinib
1. Introduction
Neurofibromatosis type 1 (NF1; OMIM 613113) is an autosomal dominant disease affecting approximately 1 in 2500 individuals worldwide. NF1 is associated with cutaneous, neurologic, and orthopedic symptoms, some of which are progressive and lead to significant morbidity or mortality. The latest consensus has been reached for diagnosis and resumed in the guidelines for neurofibromatosis type 1 recently published [1, 2]. Among the clinical features considered was the development of characteristic tumors such as neurofibromas or gliomas; the presence of cutaneous findings including cafè au lait macules (CALMs), skinfold freckling, juvenile xanthogranulomas, and nevus anemicus, ophthalmologic markers such as Lisch nodules and choroidal abnormalities and the occurrence of characteristic osseous lesions such as sphenoid wing dysplasia, scoliosis, anterolateral bowing of the lower leg, pseudarthrosis.
This syndrome is caused by pathogenic variants in the NF1 gene encoding neurofibromin enzyme (Nf1). Nf1 operates as a GTPase-activating protein (GAP) that negatively regulates the RAS/MAPK pathway activity by accelerating the hydrolysis of RAS-bound GTP [3]. All alterations in the NF1 gene result in loss-of-function of the Nf1, causing an uncontrolled cell proliferation and growth. The neurofibromin haploinsufficiency, and the consequent Ras proto-oncogene hyperactivation, make NF1 a tumor predisposing disease [4]. Nf1 affected individuals, display increased risk of certain cancers, including female breast cancer <50 years, gastrointestinal, neuroendocrine brain and hematopoietic tumors [5, 6, 7].
The most relevant manifestation of NF1 is the appearance of tumors in both the Central (CNS) and Peripheral Nervous Systems (PNS) as gliomas and neurofibromas, respectively. Of the tumors involving the nervous system, peripheral nerve sheath tumors (cutaneous and plexiform neurofibromas) are frequently found in between 30 and 60% of subjects with NF1, depending on diagnostic modalities (symptomatic Vs screening) and can present at a various age range from congenital to young adulthood (median age 5.5 years). In addition, 8–13% of individuals harboring a PN can develop a malignant peripheral nerve sheath tumor (MPNSTs) [8, 9]. The appearance of distinct nodular lesions (DNLs) in PNs raises clinical concerns, as these changes are correlated to increased risk of transformation into Atypical Neurofibroma (AN) or MPNSTs [10], the prognosis of which is ominous due to delayed diagnosis, early metastasis, and poor response to systemic therapy [11]. Atypical neurofibromatous neoplasms of uncertain biological potential (ANNUBPs), are potentially premalignant lesions with unique biology still under investigation but are often characterized by specific histological features including cytological atypia, hypercellularity, loss of neurofibroma architecture, and an increased mitotic index [12]. Also ANNUBP can be associated with additional somatic genetic events such as heterozygous or homozygous loss of CDKN2A/B in addition to NF1 alteration [13] and often display a more aggressive growth behavior. Furthermore, individuals with AN/ANNUBP are at higher risk for the development of malignant peripheral nerve sheath tumors (MPNSTs), with a 33% incidence in one study versus the cumulative MPNST risk of 15.8% in the general NF1 population [6, 14].
Even though a large number of histological changes between PNs and MPNSTs has been described, a clear molecular definition of the stepwise characteristics of PN evolution towards MPNSTs is currently under investigation [15, 16]. The effort to fully elucidate the development and progression of these tumors, finalized to identify genes suitable for diagnostic markers and pharmacological targets, is made even more difficult by their structural heterogeneity and a complex tumor-host interaction.
Neurofibromas arise within nerves and comprise multiple cell types, including Schwann cells, fibroblasts, perineural cells, mast cells, and macrophages [17]. The PNST development requires both biallelic loss of NF1 in Schwann cells, and a haplo-insufficient microenvironment. Whereas the knowledge on the molecular basis of Schwann cell transformation is increasing, a deeper understanding of the exact contribution of the microenvironment in the Schwann cells neoplastic transformation [18] it is still elusive. The current concept in the NF1 field states that the tumorigenic proliferation of Nf1 haplo-insufficient Schwann cells depends on an inflammatory milieu triggering a powerful burst of mixed mitogenic signals from both collagen-secreting fibroblasts and pericytes stimulated by mast cells [19]. A considerable amount of data speak in favor of this pro-inflammatory model, in primis the discovery that mast cell have a central role in sustaining chronic inflammation, cell proliferation, and potent extracellular matrix deposition (ECM) leading to fibrotic tissue deposition; however, this model does not entirely fulfill the molecular signature proper of “tumorigenic” cells able to grow in the absence of basal membrane binding, possessing higher proliferative potential and own metabolism [17] and further investigation are required to fully understand how microenvironmental cells contribute to Schwann cell transformation.
In general, PNs are characterized by a slow-growing behavior tending to exhaust after a definite period of time. In addition, many PN can manifest prolonged quiescent phases, or sometimes even undergo a spontaneous partial regression, rendering its natural history often unpredictable [20].
From a prospective NF1-PN study (“NCI natural history”) [21] the median PN volume change was +21.3%/year. Other studies suggested that infants or young children at PN diagnosis tend to present with more rapidly growing lesions compared to young adults [11, 20, 22].
While PNs may be asymptomatic, a proportion of PNs can lead to various morbidities, ranging from disfigurement, pain, functional motor-sensitive deficits, which can also be associated with life-threatening complications such as bleeding and vital structures (e.g., airways) obstruction.
In most cases, stable PN not causing morbidity should be observed, as they may never progress or cause symptoms and an active intervention should be offered only to those patients who manifest progressive growth over time causing significant symptoms which cannot be treated otherwise (e.g. pain medications ineffective) [23].
Furthermore, as morbidity is often secondary to the PN anatomical distribution, some locations (e.g., paraspinal, orbital, neck) may cause more symptoms than others (limbs, pelvis) and more often require an active treatment, irrespectively from their actual size.
PNs rapidly growing (>20% increase in volume in the prior year) are painful and with high risk of malignant transformation, thus should be considered for treatment [23]. In addition, PNs can exert a mass effect, compressing nearby structures (such as the trachea or blood vessels), and either stimulate bone growth or lead to bone erosion. PNs can also cause spinal cord compression, weakness, cranial neuropathy, disfigurement, and pain.
Although surgical treatment is the only curative treatment option, and it is indicated for symptomatic PNs, the tumor’s complete removal is frequently not possible or associated with severe morbidity, especially in deep-seated tumors involving multiple nerves [24]. For instance, diffuse PNs spread extensively along connective tissue and surrounding normal tissue structures with indistinct borders, whereas nodular lesions are well-demarcated. Also, PNs have a rich vascular network and can bleed profusely during surgery.
Several surgical techniques have been proposed for PNs to decrease intraoperative bleeding and facilitate tumor excision of either diffuse PNs or rapidly growing nodular PN (DNL) [25, 26]; however, despite the improvement in surgical methods, reports on post-surgical outcomes suggest that complete tumor excision can be achieved in only low rate of cases (about 10–15%). Sometimes PN re-growth occurs in those who underwent either partial or subtotal resection [27, 28].
In this chapter, we will provide an overview of the most recent advances of tumor biology knowledge and treatment of neurofibromatosis derived peripheral tumors.
1.1 Non-surgical management of NF1-associated plexiform neurofibroma (PNs)
About 85–95% of PN are considered inoperable. Most of the times a complete surgery is not feasible due to the PN intimate anatomy and growth within the nerve fascicles as well as a significant vascularity leading to an increased risk of bleeding. Moreover, PN regrowth is frequent after incomplete surgery, particularly in young children, therefore any benefit of debulking surgery should be carefully balanced against risks.
Consequently, non-surgical treatment has been historically used to treated inoperable and symptomatic PN.
Unfortunately, until recent times, multiple trials have so far failed to identify any active agent against inoperable and symptomatic PN representing thus a clearly unmet medical need for a significant proportion of NF1-PNs.
First Roberston in 2012 published a phase II trial with series of 23 children or adults with NF1-PN in which 6/23 (26%; 95% CI: 10–48%) experienced ≥20% decrease in volume of one or more plexiform tumors and 30% had symptomatic improvement. Main predictive factors for Imatinib response were a smaller tumor (<20 cm3 volume), head and neck location, while age was not predictive of response [29].
Few years later Weiss et al, published their experience (n = 46) on a phase II trial with the mTOR inhibitor Sirolimus, which showed Median TTP was 15.4 months compared to the historical placebo cohort (tipifarnib trial) of 11.9 months. No subjects achieved a partial response, and the largest volume decrease was 17%. Overall, Sirulimus was well tolerated (stomatitis, increased liver and cholesterol level) and it appeared as a reasonable alternative to imatinib with the advantage of a liquid formulation available [30].
Another trial with peg-interferon-alfa-2b compared to placebo also showed minimal response rate [31].
A tremendous advance in understanding of NF1 PN pathophysiology was only possible with the use of pre-clinical mouse model of NF1 mimicking patients’ tumors, particularly those from N. Ratner’s group and NCI (Bethesda) and allowing to test biological-based pre-clinical drug screening. These advances have rapidly identified the MAPK pathway signaling as the main driver of NF1-PN, focusing on MEK as an actionable target of MAPK downstream pathway, which subsequently led to the pre-clinical testing of MEK inhibitors in mouse models showing a remarkable and unprecedented response rate with significant tumor shrinkage in PN models [32, 33].
Following such preclinical evidence provided, a phase I/II trial (SPRINT) was launched at NCI testing a selective orally available MEK1–2 inhibitor currently under development for other indications (uveal melanoma), named Selumetinib [22]. Volumetric MRI assessments and functional outcomes such as quality of life were included in the study methods as primary and secondary outcomes respectively.
The exceptional response rate for a phase I trial, with a volumetric Partial Response (PR) of 20% volume reduction in 71% of the 50 enrolled children which was confirmed by the phase II data (PR = 68%) with relatively favorable toxicity profile [34]. A reduction of PN volume by a median change of 27.9% (−55.1% – +2.2%) was documented in the SPRINT trial. Median time to initial response, cycles (range) was 8 monthly cycles (range 4–20), and time to best response, was 16 cycles (range 4–36) suggesting that many patients tended to respond many months or even years after Selumetinib initiation. Progression-free survival (PFS) at 3 years after therapy start was of 84%. Mean duration of treatment was 24 months. However, at the time of analysis 23/50 (46%) children remained on treatment for a median of 4 years, experiencing a durable and/or ongoing response.
From the cohort of 50 children with NF1-PN suffering from a median of 3 target morbidities at baseline, before treatment. A functional effect was also demonstrated, including a significant improvement/amelioration of patient’s symptoms, including pain, disfigurement, and quality of life, as well as an objective improvement in strength, range of motion and pulmonary function based on REiNS standardized response criteria.
Based on the SPRINT data on safety, radiological and clinical efficacy, Selumetinib (Koselugo) at the doses of 25 mg/m2 BID has been now granted approval from FDA and EMA as the first licensed drug in children aged 2 or more, with NF1 and inoperable and symptomatic PN. Despite at present Koselugo can be only offered to those children able to swallow capsules, a currently ongoing phase I/II trial is evaluating an alternative sprinkle formulation in young children (>1 year) and will likely extend its indication to younger children not able to swallow capsules.
An additional stratum from the SPRINT trial evaluated the role of preventive use of Selumetinib in children with NF1-PN at risk of developing severe symptoms and showed similar efficacy (PR = 72%) and absence of symptoms during treatment and rate of volumetric shrinkage (−32,7, range − 16,8 to – 39,7%). From this specific subgroup of asymptomatic PN, younger patients were more likely to respond in terms of radiological tumor shrinkage when compared to older children suggesting a potential extended indication to start Selumetinib early in selected young children at risk, and before they develop PN-related symptoms [34].
1.2 Selumetinib real world data and ongoing studies
After its approval in US, UK ad EU, few retrospective cases series based on Early Access Program and compassionate use have contributed with real world data, including large institutional series from Espirito Santo et al. [35] in which all but one among the 19 patients who were treated with Selumetinib had a sustained clinical and/or radiological response mainly within the first 2–3 months from start.
Also, Cacchione et al. [36] from 13 patients (7 males, 6 females) -median age 3 yr-treated with either selumetinib [12] or Tranetinib (1) documented a PR rate of 23%, with a median time to onset of response to treatment of 12 cycles (6–30 range). All other patients were stable (SD = 69%) except in one case who had a rapid progression, and who was subsequently diagnosed with a malignant nerve sheath tumor (MPNST). A smaller series from Baldo et al. showed PN reduction of >20% in 16 out of 17 PN (94%). One PN remained stable and no PN progressed while on active treatment. Mean follow-up was relatively short (12 months) at the time of publication [37].
In addition to the SPRINT study, other Selumetinib trials are currently ongoing in US, UK or Europe. In adults, KOMET randomized trial of Selumetinib against placebo is open and enrolling patients with symptomatic and inoperable PN (NCT04924608). Moreover, a PASS (post authorization safety)- study will evaluate long-term Koselugo safety and other possible side effects including endocrine function in children. Two trials are evaluating alternative dosing, either intermittent (5 days on/2 days off, UK-GOSH (NCT 03326388) or a lower dose maintenance phase (US only, MJ Fisher personal communication) both aiming at optimizing treatment compliance and long-term tolerability.
1.3 Other MEKi efficacy data
Other MEKi have been tested against NF1-PN in other ongoing or more recently completed trials (cancel). One of the most relevant is the phase II trial (TRAM-01) including 46 children treated with a median of 16 months of Trametinib, a capsules and liquid formulation available MEKi, and showing that 17 out of 28 evaluable children with NF1-PN had a confirmed PR (60%) and a median decrease of - 30% as documented from a semi-automated volumetric calculation. In the meanwhile, Ronsley et al. [38] published their experience with Trametinib compassionate use showing response in 3/6 pediatric patients with NF1-PN.
Caution in direct comparison of various MEKi studies as study design, outcome measure, and enrolment criteria were slightly different by age, progressive/non progressive prior to start. Therefore, given the lack of direct comparison between various MEKi used to treat PN, it is not possible to assign a clear superiority of any MEKi at current stage.
With this in mind, other agents have shown promising data against NF1-PN both in children, such as the MEK inhibitors Binimetinib (19 children 2–16 yr., and a PR rate of 74%) or Mirdametinib (19 young adults 16–25 yr. and PR 42%, SD 53%, PD 5%) or Cabozantinib, a multiTKI targeting c-Kit, VEGFR2, MET, RET, FLT3 which was associated with a PR of 42% in 19 adults (>16 yr) with NF1-PN [39, 40, 41].
1.4 Selumetinib and other MEKi toxicities
Side effects from selumetinib were overall mild, with 10% who interrupted treatment due to intolerable or recurrent toxicities, but up to 30% of patients required in a dose reduction.
Notably, there were no AEs resulting in patient death and side effects were always reversible with drug interruption. Most frequently encountered and clinically detectable adverse effects (AE) were predominantly skin toxicity such as follicular or acneiform rash, paronychia in 52–58% (grade I/II) and 4–10% (Gr 3) often presenting within the first 2 weeks of initiation of therapy and gastrointestinal side effects such as nausea, abdominal pain, diarrhea and, less frequently vomiting. In addition, elevation of creatinine kinase was frequently seen during monitoring of blood tests, but most often asymptomatic and not requiring any active intervention or drug interruption [42].
The most serious dose-limiting side effects of MEK inhibitors may involve cardiac- with decrease of ejection fraction, which was reported in 6–10% of children from SPRINT trial. Differently from the adult series, ophthalmic- with retinal fluid and detachment-toxicity was not reported within the SPRINT dataset [32, 42, 43].
Main side effects from Trametinib were similar to other MEKi, and including skin, gastrointestinal toxicities and asymptomatic CPK elevation, generally mild and manageable with an adequate supportive care or dose interruption and further adjustments.
While various MEKi seem to share a class-effect of similar toxicities, potential differences in frequency and severity of side effects may perhaps be due to the specific dose needed to achieve response for each MEKi.
A preventive strategy as well as regular monitoring of AE are of paramount importance as AEs are frequent and might require dose adjustments and dose reductions, due to AEs or other reasons, can lead to PN regrowth. Prevention of side-effects since the very first start of MEKi, particularly in high-risk subjects such as post-pubertal and young adolescent patients is a key factor to potentially help maintaining a durable response [23].
Other relevant aspects of MEKi treatment: Optimal duration, events after interruption, prevention of malignant degeneration.
While in the SPRINT trial treatment with Selumetinib was planned for an undetermined period of duration, limited to 24 months in case of stable disease only, long-term data available from a subset of patients showed that 23/50 (46%) patients remained on treatment beyond the initial 24 months period of scheduled treatment phase (Median TTP: 36 cycles, range 16–48), thus adding useful information regarding effect durability and long-term safety and tolerability. ORR and safety profile were confirmed. Furthermore, it was shown that among those patients who interrupted treatment, a proportion of cases with PN regrowth was seen, after a median of 3–5 months from interruption [32].
In addition, from the Trametinib TRAM-01 trial out of the 36 patients (80%) who discontinued treatment after the planned 18 months initial duration, 7 (19,4%) suffered from PN progression after a median of 6.1 months (range 5.2–17,9) from end of Trametinib, the majority of whom were successfully rechallenged with Trametinib [44].
While these data do not provide a clear nor evidence-based indication on the optimal duration of treatment with various MEKi it appears clear that interrupting it too early may lead to PN symptomatic regrowth, particularly in young children and that the majority of children with NF1-PN who need treatment, will need to stay on drug for a relatively long period of time, posing significant challenges in terms of long-term tolerability, safety and, last but not least sustainability for the health system and/or other reimbursement mechanisms.
Early data on response after re-challenge at the same time, suggest that it is also possible, after a planned period of time (2–3 years), to consider a “drug holiday” based on clinician, or parent’s shared decision, with the back-up option to restart MEKi in case of rebound growth after treatment interruption [23].
The role of MEKi against atypical Neurofibroma (AN/ANNUBP) is still unclear but some cases may still respond while on treatment. However, it seems also clear from SPRINT and other dataset that MEKi cannot prevent the malignant degeneration of PN to a MPNST. Therefore, any PN which progress while on MEKi should raise the suspicion of an ongoing degeneration whose additional somatic mutations (CDKN2A, PRC, TP53) render the treatment ineffective [45].
1.5 Conclusions
The treatment paradigm of PN has dramatically changed since the use of MEKi, which proved able to alter the natural growth trajectory of PN in children and, in many cases, a significant short-term clinical benefit.
Long-term side effects, overall benefit as well as optimization of treatment indication, duration and tolerability represent key unanswered questions which need to be addressed.
Upcoming collaborative projects (Eu-Pearl) are in the pipelines paving the way towards a more patient-centered approach to identify and prospectively validate PRO/ObsRO measures that specifically assess PN-related morbidities focusing on QOL as a first important step to significantly ameliorate and mitigate PN morbidities.
2. Surgical management of neurofibromatosis type
2.1 The plastic surgery contributes to therapeutic neurofibroma treatment
The distinctive tumor of NF1 is the neurofibroma. Histologically, neurofibromas are tumors that develop from peripheral nerves and are made up of a variety of cell types, including Schwann cells, myelinated and unmyelinated axons, fibroblasts, endothelial cells, and mast cells [46]. Discrete cutaneous neurofibromas develop at dermal nerve terminals. They present as soft, well-defined, sessile or pedunculated nodules with clinical findings that range in size from a few millimeters to several centimeters. Although stinging and itching sensations can be annoying, they rarely cause pain or discomfort. Subcutaneous neurofibromas are firm rubbery nodules that arise along the course of the peripheral nerve beneath the dermis and they can be painful and tender.
Numerous lesions, even thousands, appear primarily on the face, trunk, and proximal limbs.
Malignant transformation in these tumors is rare although the emotional and social impact can affect a patient’s quality of life [47, 48]. Plexiform neurofibromas represent a significant cause of patient morbidity. Plexiform neurofibromas occur in approximately 30–50% of patients who have NF1.
They are composed of the same cell types as cutaneous neurofibromas [49]. Massive tumors known as diffuse plexiform neurofibromas spread to nearby organs, bones, and soft tissues. A deep tumor may be indicated by thicker and hyperpigmented skin that is overlaying the area. Plexiform neurofibromas can develop within the first year of life or be present at birth in patients with neurofibromatosis, with a frequency of 24,1–32%. These tumors’ size and location affect how clinically significant they are. They may show up as painful lesions under the skin or asymptomatic bumps. They have the ability to fill space and produce a mass impact when close to significant structures because of their growing potential.
They are also related to a 10% risk of malignant degeneration. The progression tendency of these neurofibromas cannot be established a priori, however, associated risk factors have been identified. Head and neck localization, age < 10 years, and incomplete surgical excision are in fact associated with an increased progression rate [27, 47].
Malignant Peripheral Nerve Sheath Tumors are quite rare in the general population, with an incidence of 0.001%. However, as many as 60% occur in patients with N1, with an approximately 10% lifetime risk of developing it. These are highly aggressive tumors that usually evolve from plexiform neurofibromas. In NF1-affected patients, they generally have an earlier onset, around the second and third decades of life, and a worse prognosis [47, 50]. Unfortunately, surgery is still the only available therapeutic option for this type of neurofibroma because it is impossible to forecast how it will develop over time.
The time and type of surgery are the two most challenging considerations about surgical treatment. Large neurofibromas can require subtotal surgical resection, which may require a staged procedure (Figures 1 and 2).
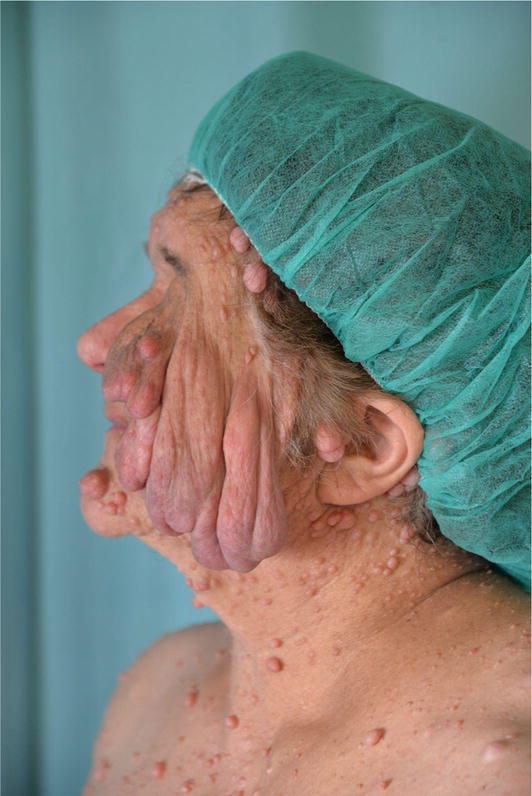
Figure 1.
Voluminous plexiform neurofibroma of the upper eyelid in patient with NF1.
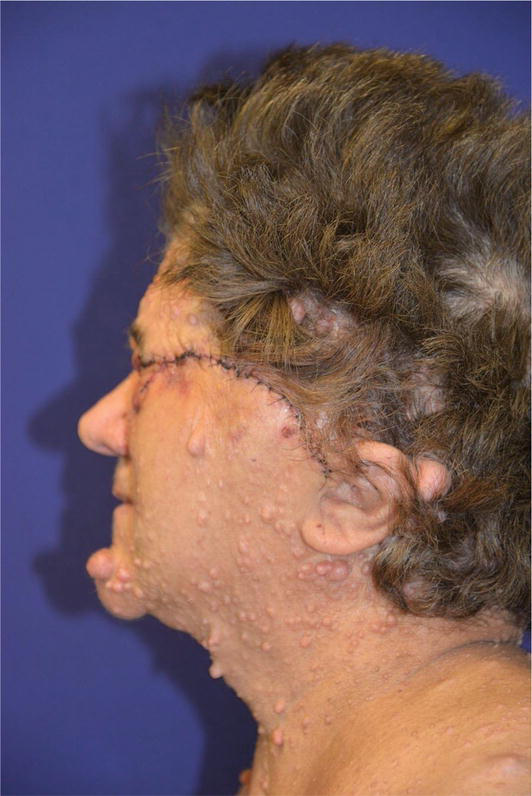
Figure 2.
One-week follow-up after neurofibroma removal and eyelid reconstruction surgery.
According to our experience, early radical surgery during the initial procedure will result in the greatest long-term outcomes. Each patient must receive individualized care, taking into account the needs that are dictated by their particular diseases. A multidisciplinary approach is the best form of care for people with neurofibromatosis, both in children and adults. Each patient is different and presents particular difficulties for the patient’s healthcare professionals. Making timely and suitable judgments requires knowledge of the disease, how it progresses, and any potential morbidities. Timing of the operation will depend on symptoms, deformities, disability, and disease development.
Surgery as the last choice is frequently not the most effective option and can produce more severe morbidity. A well-timed operation can significantly reduce long-term deformity and impairment.
The surgical approach of cutaneous and subcutaneous neurofibromas is generally simpler. Since the extension is limited to the epidermal-dermal level, pre-operative imaging investigations are generally not required. The surgery goal is not only to radically remove the lesion but to ensure an aesthetical outcome. The type of reconstruction depends both on the size and the excision area. Minor excisions without skin tension can be addressed with direct sutures. Following the surgical ladder, major lesions on the other hand may require graft usage, collecting the donor skin from a neurofibromas-free area, and local, pedunculated or free flaps.
The NF1-affected patient will often present with multiple neurofibromas to be removed. In cases of multiple modest-size cutaneous neurofibromas, a considerable number of lesions can be removed within the same surgical session. The limit of removable lesions under local anesthesia depends on the amount of anesthetic that can be safely used, and on the patient’s tolerance. In our experience, it is not rare to reach a dozen removed neurofibromas per single surgical session (Figure 3). To avoid misunderstandings and exaggerated expectations, however, a priority of the lesions being removed should be agreed upon with the patient before the surgery, leaving any others for future sessions.
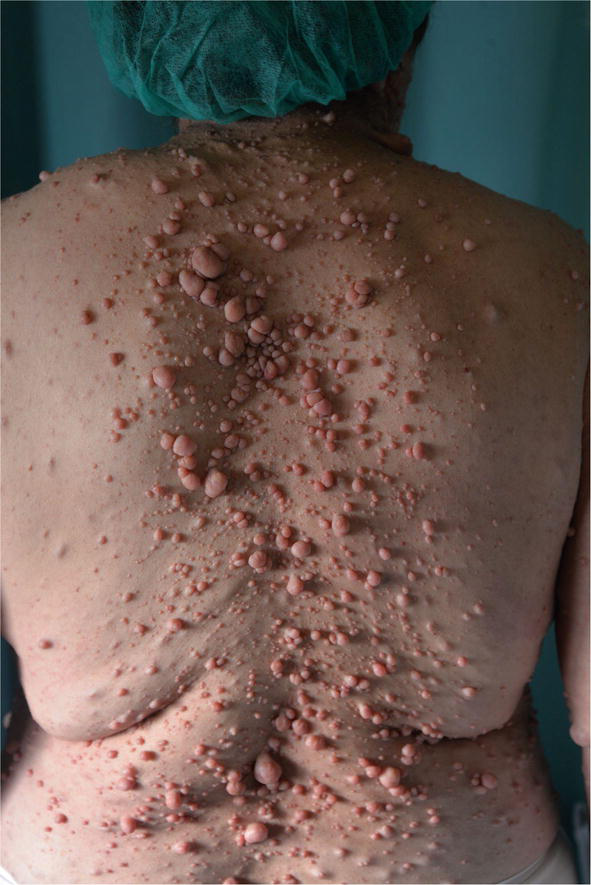
Figure 3.
Numerous cutaneous neurofibromas in patient with NF1.
Likewise, especially for neurofibromas involving high esthetic interest areas, such as the face, it is advisable to make sure that the patient understands that he will have a scar in place of the neoformation. Scar massages with silicon-based gels should be recommended for the 2–3 months after surgery, to promote proper scar maturation.
In case of unsatisfactory results, the patient should be invited to wait at least six months or one year before a possible touch-up. Both scars and blemishes can improve over time, making surgery superfluous. The risks relating to these surgeries are rather limited. They mainly concern possible recurrence and surgical wound complications, such as delayed healing, dehiscence, or local infection. No absolute indications for antibiotic therapy are recommended, but if comorbidities that can favor infection onsets are present, the operator may recur to pre-operative antibiotic prophylaxis [51].
The histological examination should always be requested for each excision, even once the diagnosis has already been formulated. Plexiform neurofibromas, on the other hand, represent a tougher surgical challenge. For surgical excision indications, in addition to the esthetic and pain, which they share with cutaneous neurofibromas, they present functional impairment. Following significant volumetric growth, they can compress nerve structures and lead to functional deficits.
A periodic follow-up is the only valid method to monitor their evolution and evaluate surgery indications. Preoperative imaging is required and MRI is considered the gold standard. Compared to other methods such as ultrasound or CT, MRI allows a better margins definition, which is often unclear, a better tissue distinction, and the evaluation of nerve/vascular infiltration and local invasion. MRI can also be used to classify plexiform neurofibromas into three classes: superficial, displacing, and invasive [52].
The excision should be as radical as possible to avoid tumor recurrences.
Early interventions seem to improve both the outcome, as the exeresis operation remains more confined without extending to other districts, and the recurrence rate.
Ideally, the tumor should be removed at its superficial level, as this appears to effectively prevent a recurrence. Once the tumor is in the invasive phase, the recurrence rate is in fact around 20%, rising to 46% for incompletely excised cases [53].
For advanced neoformations, when the tumor grows to invade nerve fascicles, it could be unfeasible for the surgeon to remove it without having to sacrifice the nerve, thus causing a possible functional deficit of which the patient must be informed during the collection of informed consent for surgery.
Among the potential risks, the vascularization status of these neoformations should be investigated during pre-operative routines.
In the case of particularly vascularized neurofibromas, known as hemangioneurofibromas, angiography with pre-operative embolization can be planned, to significantly reduce bleeding risk during and after surgery [54].
Acting as subcutaneous expanders, a skin laxity could remain after these tumors’ enucleation. If this happens, a skin lozenge could be removed around the incision to re-tension the tissues.
Especially for large volumes tumors, it is possible, after performing accurate hemostasis, to place a drain to prevent hematomas and seromas formation.
In the case of voluminous neurofibromas involving lower limbs, the possibility of prescribing anticoagulant therapy or the use of elastic stockings until normal walking is resumed is advisable to prevent deep vein thrombosis. Antibiotic prophylaxis may be indicated if there are concurrent risk factors for infection or a drain is in place.
MRI is the reference diagnostic to evaluate the tumor extent and its relationship with adjacent structures. However, to confirm the lesion malignancy, it is necessary to resort to other methods like F-2-deoxy-D-glucose positron emission tomography (FDG-PET), which can also detect the presence of distant metastases. Nevertheless, a biopsy is required for histological confirmation. Since at least 2 centimeters of tumor-free margins are recommended for these lesions, the biopsy can be excisional for lesions smaller than 3–4 cm. For larger lesions, multiple core-needle biopsies, possibly ultrasound or CT-guided, are recommended. Incisional biopsies are less used since they are burdened by a greater complications risk, and are often limited to a single sample collection, albeit of larger dimensions.
Adjuvant radiotherapy may be used to address medium and high-grade lesions. Although it does not improve overall survival, it seems to improve the local management of the tumor. Adjuvant chemotherapy is reserved for systemic metastatic diseases instead [47, 55].
The requirement to perform a wide resection to ensure disease-free margins often results in major nerve sacrifice and consequent functional damage. Although regarded as the last resort, limb amputation may sometimes be necessary to ensure adequate cancer radicalization [56].
Complications are similar to those resulting from the enucleation of large plexiform neurofibroma. Nonetheless, the patient should be informed about the bad prognosis and the high recurrence rate of these tumors. Tumors bigger than 5 centimeters, local invasiveness, distant metastases, and non-radical surgery are all negative prognostic factors [57].
The 5-year local recurrence rate ranges from 27–49%. Distant metastases are instead reported between 26–65%, and mainly involve the lungs, brain, skeletal system, and liver [58].
Because of this high recurrence rate, careful follow-up is mandatory. Clinical evaluations should be performed every three months for the first two years, and an imaging investigation is recommended at least twice a year.
2.2 Role of neurosurgery in the treatment of nerve tumors in patients with neurofibromatosis type 1
The treatment of nerve tumors in patients with NF1 is multidisciplinary; it involves geneticists, oncologists, neurosurgeons and plastic surgeons mainly.
Surgery is in all cases reserved for symptomatic lesions or malignant evolution; the neurosurgeon is involved in lesions of nerve plexuses and trunks or in case of extension of the pathology within the spinal canal with resultant myelopathy.
Special attention and a dedicated experience in the treatment of these lesions is essential as the functioning nerve fascicles in these lesions are incorporated in the tumor and may not always be dissected from it. This surgery is potentially at risk of increasing additional neurological deficits and the extent of the removal must be limited to what is necessary.
2.2.1 Neurofibroma
Neurofibromas can be located in any peripheral nerve or root; they originate from Schwann cells and grow in the nerve by dividing the nerve fibers without infiltrating them; due to this characteristic, it is generally possible to remove them without determining a reduction in neurological function as they only require the sacrifice of the usually non-functioning original fiber. On the contrary, neurofibromas and plexiform neurofibromas (PNs) are complex conglomerates of intraneural neurofibromas that create tortuous and huge masses arising in nerve trunks (Figures 4 and 5). PNs are pathognomonic and typical manifestation of NF1 (30% patients with NF1 have clinical evident PNs; with RM study incidence grows to 50–60%) [23], potentially involving almost every part of the body outside of the brain and spinal cord. They can also occur on the face (including around the eyes), neck, arms, legs, back, chest, abdomen, and internal organs. Large tumors cause the nerves (also superficial sensory nerves) to become thick and misshapen, which can affect the structure of nearby bone, skin, and muscle. Especially large and deforming PNs can cause severe pain, mobility problems, vision and hearing loss, high blood pressure and other medical problems. They commonly occur in children and most of them are not cancer, but some may later become cancer. When present on major nerve trunks they involve multiple fascicles. Therefore, the removal of such lesions can lead to sensorimotor deficits, including neuropathic pain, in patients that can develop in the future other lesions. This kind of surgery to be safely performed requires experience and expertise in this field. For all the above-mentioned reasons surgery in cases of NPs is limited to lesions with suspected malignant degeneration or resulting in neurological deficits although, not infrequently some patients ask for the removal of large PNs also for esthetic and functional problems.
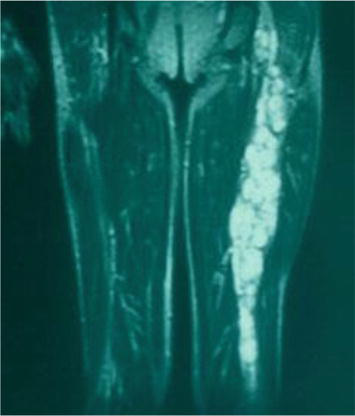
Figure 4.
MRI imaging of plexiform neurofibroma of left sciatic nerve.
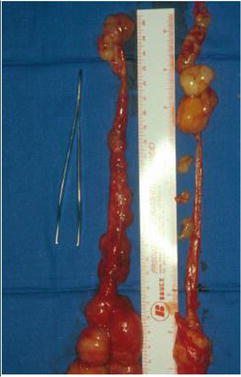
Figure 5.
Left sciatic plexiform neurofibroma after surgical removal.
MPNSTs are the malignant variant of peripheral nerve tumors; although they also originate from Schwann cells, due to their aggressive behavior, they are mostly considered as sarcomas. These lesions, that are very rare in the general population, have a high incidence among patients with NF1 and are one of the most frequent causes of death in patients with this disease [59]. The chance of a patient with NF1 developing an MPNST is estimated to be between 8 and 15% [2]. Among NF1 patients with a microdeletion, this percentage rises to 16–26% [59]. The 5-year overall survival between patients with MPNST is about 20–50% [12] with a worst outcome in metastatic or unresectable lesions; actually, the survival in the population with NF1 is like similar to the general one. This progress in related to a better follow up and an early diagnosis in patients with NF1.
2.2.2 Indications for surgery
The indication for surgery for nerve tumors in patients with NF1 is limited to cases symptomatic of neurological deficits or suspected malignancy of the mass. Since there is growing evidence that MEK inhibitors may play a significant role in their treatment (of PNs). The risks of surgery needs to be balanced against the possibility to offer a conservative management based on pharmacological treatments now available for selected cases [59, 60]. This is especially true for those large disfiguring multi-embryonic derivation PNs (affecting nerves, cartilage, bone and dermis) where the surgical results leave much to be desired. However, considering the period of several weeks required for the medical treatment to show the first results, it is straight forward that for the following conditions, a drug treatment approach cannot be the solution.
2.3 Malignancy
The lifetime chance of developing a malignancy (MPNST) in a NF1 patient is reportedly 8–15%; at least in 1/3 of cases, they arise from PNs and bulky masses (≥6 cm) seem predisposed to turn malignant. The factors that may favor malignancy are not known except a previous radiation treatment [23]. Clinical signs of a malignant evolution are rapid growth, spontaneous sharp pain and neurological deficit of the affected nerve. The growth of a neurofibroma is a common event especially in children and adolescents in which the masses often grow consensually with physical development; on the other hand, it is unusual to have a significant growth in adults, in whom a volumetric increase greater than 20% per year represents an important index of suspicion [23]. Currently, patients with NF1 undergo periodic clinical and instrumental checks in specialized centers to identify early clinical or radiological manifestations of possible malignant evolution. This is to ensure prompt diagnosis and treatment. An early diagnosis, in fact, can significantly improve the life prognosis. Sometimes the MPNST arises de novo but, in most cases, it is a progressive degeneration of a nodule within plexiform neurofibromas that accumulate genetic alterations becoming atypical neurofibromatous neoplasm of uncertain biological potential (ANNUBP), lesions with potential malignant evolution. From the anatomopathological point of view, this entity, also defined as atypical neurofibroma, is gaining importance. This lesion, which from the radiological and clinical point of view seems to be indistinguishable from a benign one, presents histological features (atypia, loss of neurofibroma architecture, high cellularity and/or mitotic activity between 1:50 and 3:10 high power fields) that are similar to MPNST and seems to be a precursor of malignant nerve sheath tumors [12, 61]. The use of PET CT with fludeoxyglucose (FDG) and volumetric MRI [62] can precociously identify lesions harboring these characteristics, so allowing their complete removal if the mass is easily reachable, or a diagnostic biopsy if the tumor is placed in less accessible sites. In detail, PET CT is capable of early identification of masses undergoing malignant transformation, as they have a more active metabolism; however, this type of investigation has a better sensitivity but rather low specificity since there is a non-negligible overlapping of uptake values between lesions undergoing a malignant evolution and lesions that are still benign [12, 23, 62, 63]. The removal in the case of MPNST must be aggressive, sacrificing the nerve of origin regardless of its function. Radicality with free surgical margins must be obtained and this is of paramount importance; if the lesion, on the other hand, is an ANNUBP, the removal can be confined to the tumor without the extension beyond its margins. Therefore, an early diagnosis not only improves the prognosis, but also allows avoidance of destructive interventions if it is not the case.
2.4 Neurological deficits
Neurinomas, neurofibromas and PNs generally are benign lesions, and they do not cause neurological deficits; neurological deficits appear either because they cause compression on the spinal cord having an intracanal growth or because the nerve itself becomes deficient as the tumor grows within an anatomical bottleneck.
2.4.1 Spinal cord compression
Spinal cord compression from root neurinomas or neurofibromas represents an important clinical problem in patients with NF1; it generally occurs at the cervical or at lumbosacral level due to an intracanal growth of huge NFS of brachial and lumbar plexus. Patients develop progressive quadriparesis or paraparesis, incontinence and spinal pain. The objective of the surgery is the decompression of the spinal cord which is obtained not only through the removal of the mass (in the case of a neurinoma) but also through a decompressive hemilaminectomy and a large dural plastic. Performing a widening of the spinal canal during the surgical procedure ensures that even a possible regrowth of the neurofibroma is better tolerated by the patient and does not interfere with neurological integrity. These surgical procedures generally lead to a regression of spinal compression symptoms [64, 65, 66, 67, 68], but the removal of root lesions can cause a major sensory damage to the nerves involved. The recurrence of pyramidal symptoms is rare since normally the growth of even these intracanal lesions tends to slow down with increasing age.
2.4.2 Nerve deficits
Diffuse enlargement of all major peripheral nerve trunks occurs in some individuals with particularly aggressive forms of NF1 due to the development of multiple plexiform neurofibromas, isolated neurofibromas, and schwannoma [68, 69]. When examined, these subjects often present partial deficits of peripheral nerves which have arisen slowly and progressively and of which often the patients are unaware. We are dealing with deficits affecting nerves that grow into anatomical bottlenecks such as the olecranon canal, Froehse’s arcade or the peroneal arch at the head of the fibula; the growth of any neoplasm within an anatomical narrowing may cause deficit of the nerve trunk itself and pain. The treatment of this type of problem consists of removing all or part of the tumor mass to reduce the volume of the nerve along with decompression at the physiological anatomical narrowing. While widening the anatomical narrowing appears to be a safe maneuver, the removal of the NPs in an already deficient nerve is not risk-free and must be carefully evaluated. This type of procedure mostly serves to prevent a further worsening of the deficit and must be performed only at the request of the patient. The worsening of these deficits is slow and progressive, sometimes accompanied by modest pain; from this point of view it clearly differs from the growth of malignancies which, on the contrary, appear sudden and extremely painful.
2.5 Aesthetical reasons
At light of the new therapeutic proposals, an attempt with the so-called RAS therapy (selumetinib is in a phase II trial) is in our opinion always worth to be firstly attempted.
We have witnessed some astonishing results especially in those cases where the aesthetical problem seems to be prevalent. Surgery, when a complex malformation of the superficial layers is involved, where the dermis and the convoluted superficial nerves often infiltrate and deform the underlying muscles, very rarely attains a satisfactory result.
3. Surgery
3.1 General consideration
According to these guidelines, surgery is performed. The patients are usually under general anesthesia in the absence of curare or in spinal anesthesia; magnified vision with microscope or loupes is required; the exposure of the tumor must be adequate with the possibility of identifying both the nerve entering and exiting the mass. This is easily obtained in limb lesions, while in large masses in deep sites, this type of exposure may be difficult to achieve [59]. Once exposed, the mass is inspected under a microscope and mapped to identify the eloquent nerve bundles that need to be spared. The tumor capsule should be opened in the direction of the fibers in areas silent to stimulation and then the main tumor nodules and their fibers of origin should be identified. The possibility of radical removal is linked to the specific histology of the lesion.
3.2 Neurinomas
Neurinomas of peripheral nerves practically always appear to be radically removable as the tumor grows by dividing the nerve fibers which are distributed on the surface of its capsule. An intracapsular removal of the mass allows a complete and safe en bloc removal, leaving the nerve fibers intact on the capsule left in place. An intracapsular removal is currently accepted and recognized as safe; moreover, it is absolutely not associated with a risk of recurrence (Figures 6 and 7).
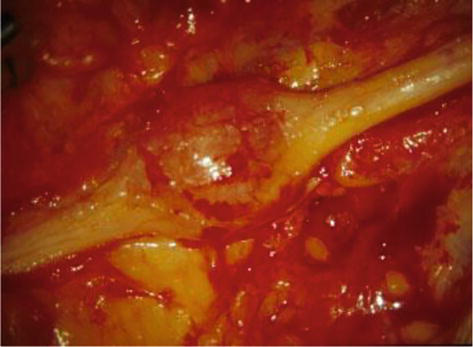
Figure 6.
Surgical appearance of a neurinoma.
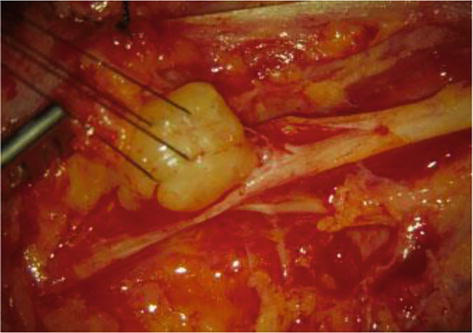
Figure 7.
Removal of a neurinoma: Surgical technique.
An exception is represented by cystic schwannomas, a very insidious subgroup as a surgical outcome. In fact, in neurinomas with these characteristics, the thin wall of the tumor mass does not always seem recognizable concerning the capsule. In an attempt to remove it altogether, sometimes, part of the capsular wall is removed, damaging the fibers contained therein, thus creating a neurological deficit. For this reason, in cystic neurinomas, it is sometimes advisable to subtotal excision and emptying of the cyst to avoid nerve damage.
3.3 Neurofibromas and plexiform Neurofibromas
Neurofibroma and plexiform neurofibroma appears as fusiform and multiple enlargements of the nerve and the fibers appear dispersed within the neoplasm (Figures 8–10).
Figure 8.
Clinical appearance of plexiform neurofibroma of the neck involving brachial plexus.
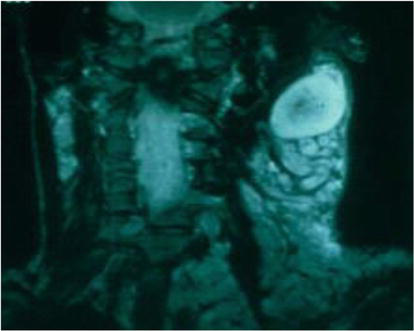
Figure 9.
Its radiological appearance.
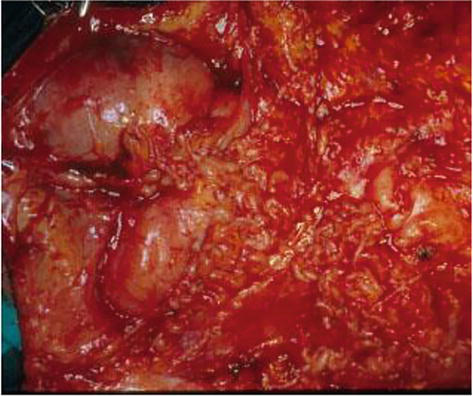
Figure 10.
Intraoperative imagine of plexiform neurofibroma of the neck involving brachial plexus.
Sometimes the removal of the nodules in plurinodular neoplasms entails the sacrifice of some fascicles conducting when stimulated. In this case it is necessary to carefully evaluate the contribution of each fascicle to the general function of the nerve and limit the removal to the most voluminous or suspicious nodules, at times preparing for a subtotal debulking to avoid neurological deficits [70]. In fact, the functioning fascicles can be sometimes the origin of the neoplasm and appear inseparable from the rest of the tumor: therefore, an accurate balance with direct stimulation or with the use of NAPs (nerve action potentials) is mandatory [59]. The surgical treatment of these lesions challenges the surgeon’s ability to approach localized masses in every part of the body and sometimes in deep thoracic, abdominal or pelvic areas for which an anatomical knowledge of these regions may not be straightforward. A basic expertise of general or urologic surgery is often required coupled with experience in the field of nerve surgery. It is important to be able to remove the lesion in the desired way already at the first surgical procedure; having to re-operate on nerve tumors in which there has been previous manipulation with consequent development of scar tissue, makes it much more difficult and significantly increases the risk of post-surgical neurological deficits. The first surgery is the best possibility we have to obtain a total removal: this must be the mantra. Therefore, the best possible removal must be accurately planned since the very first observation of the case. In addition to the risk of causing neurological deficits, the removal of large plexiform neurofibromas involving large parts of the body and skin can cause massive blood loss with acute hypovolemia and significant post-operative hematomas in the surgical cavity.
3.4 MPNST, ANNUBP and atypical neurofibromas
Particular attention is required for the surgical technique to be used in case of suspected or proven malignancy of the mass. Even more, in this situation, wide exposure of the lesion is necessary.
In the case of atypical neurofibromas or ANNUBP, the mass is often asymptomatic and morphologically identical to a benign neurofibroma. Surgery only requires the complete removal of the mass without the need to sacrifice the nerve in its entirety. The removal may be restricted to the fascicle of origin as the malignant component is inside the nodule [12, 59]. While orthodoxy, in the case of MPNST, dictates a complete removal with margins free from disease (Figure 11), the possibility of saving part of the nerve of origin, that is not involved in the neoplasm, should be considered only for small lesions and to safeguard very delicate functions (es. ulnar of median intrinsic function of the hand) where the nerve cannot be easily repaired or replaced with a nerve transfer technique.
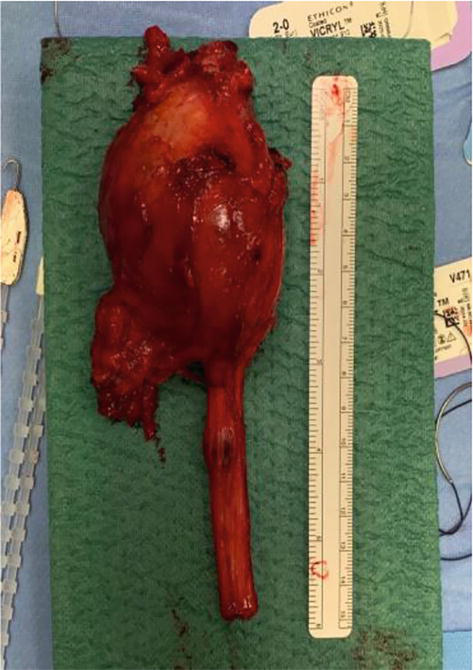
Figure 11.
Sciatic MPNST removed with sacrifice of the nerve.
The patient should be informed in advance and eventually accept the risk of a local recurrence, with the possibility of reintervention in the short term and, theoretically, the occurrence of distant metastases. Obviously, the patient will undergo close follow-up to detect an early recurrence of the illness. In the experience of our department, the use of this surgical technique has not been affected by an increase in mortality and morbidity; on the contrary, it was possible to preserve the nervous function without worsening the prognosis and with only one out of eight patient who needed reoperation. Complete healing was obtained after the second surgery. The attitude towards those cases of MPNST coming to attention in an advanced stage and originating from the lumbar or lumbosacral plexus is completely different; often the lesions are large (>10 cm) and have engulfed arteries and veins.
Accurate staging of the disease is necessary before proceeding with surgery. In the surgical approach it is important to consider the relationship and the tumor infiltration of the vessels which must be investigated previously with angiography or angioCT. If there is arterial or venous encasement, the main vessels must be prepared upstream and downstream of the lesion both to control bleeding and to proceed with a vascular bypass if the vessel cannot be sacrificed. Once more, every effort is made to obtain a complete removal of the mass and radicality as much as possible. Unfortunately, experience has demonstrated that when a malignant lesion presents these characteristics, despite excellent surgical treatment, the prognosis is in any case poor; the only result is a prolongation of survival. On the other hand, surgery currently appears to be the only valid treatment available in the case of MPNST; conventional radiotherapy, hadron therapy (proton beam seems to be more effective) and chemotherapy treatments have very poor results in slowing the progression of the disease and have no curative chance. This underlines the importance of early diagnosis of MPNST, to detect the disease as long as it appears surgically removable in a radical way [61, 70, 71]. Individuals with NF1 appear to be the population at greatest risk for this pathology. The attending physicians and specialists who treat such patients with NF1 must be vigilant and pay attention to signs and symptoms that may herald the onset of this pathology for a prompt diagnosis.
3.5 Reconstructive technique
In the event of a destructive surgery such as required by MPNSTs or in the unfortunate exceptions of neurological deficits following the removal of neurofibromas and neurinomas, the surgeon must be able to schedule reconstructive or secondary surgery to guarantee the patient the best possible quality of life. Patients with NF1 present specific problems as it is not always easy to respect one of the basic concepts of nerve surgery, that the sutured nerves must be completely healthy; an anatomical classical repair would involve suturing nerve stumps that are not completely tumor-free.
Therefore, the first choice in these injuries, if technically possible, is palliative surgery. This consists in surgical muscle transfer techniques which act by modifying the insertions and levers of the muscles spared from injury. The new tendon insertions change their effect and restore lost functions. Examples of such techniques are the posterior tibialis muscle transfer in case of foot drop due to common peroneal nerve palsy or the triple transfer according to Merle d’Aubignè to restore wrist and finger dorsiflexion in the event of a radial nerve injury [72]. These techniques also have the advantage of giving an almost immediate result as they do not need to wait for the natural regrowth of the nerve. In very extensive lesions such as in cases of MPNST of the brachial or lumbosacral plexus, it may be necessary to apply reconstructive techniques following the same principles as in traumatic nerve or plexus injuries [72]. In fact, when possible, nerve transfers are an even better alternative to muscle transfers, since they ensure a natural reinnervation of the lost muscles. First-choice techniques are the direct reinnervations of distally sectioned nerves; They do not require grafts nor nerve sutures in surgical site where radiotherapy will or could be performed. For example, in complete injuries of the brachial plexus, multiple extraplexual reinnervation can be used, such as an accessory-suprascapular nerve for the shoulder and intercostal to muscolocutaneous nerve to recover the bicipital function [73]. The site of repair is so completely outside the region to be irradiated and a good chance of nerve repair is assured.
4. Illustrative cases
4.1 Case 1
An 18-year-old woman with NF1 due to a de novo variant was referred to our department after a 6 month of sharp right arm pain, right brachial plexus palsy and a rapidly growing bulky supraclavicular mass. On physical examination, the patient presented an initial tetraparesis and the MRI of the right brachial plexus showed a voluminous supraclavicular and infraclavicular mass lesion involving the entire brachial plexus, with encasement of the subclavian artery and vein and intracanal extension at the level of C7 with radiological signs of cervical myelopathy (Figures 12 and 13). The investigations clearly suggested a diagnosis of MPNST. Staging performed showed no further localization of disease. After an angio-CT study of the vessels, the patient underwent surgery to remove the mass in two stages. In the first stage, the patient underwent right C4-T1 hemilaminectomy with the aid of sensory and motor evoked potential monitoring and removal of the intracanal component of the lesion. The posterior roots of C7 from which the tumor originated were highlighted (Figure 14); they were isolated and detached from the spinal cord and the intracanal component was completely removed (Figure 15). Dural plasty and closure of the dura of the C7 foramen was performed. The second approach consisted of a large anterior exposure with removal of the right half of the sternal manubrium and 2/3 of the clavicle without disarticulating the sternoclavicular joint; the subclavian vessels have been exposed up to their origin from the anonymous trunks (Figure 16). The tumor was isolated by sectioning the roots at the entrance and the nerves at the exit; the vessels were freed from the neoplasm and their walls did not appear to be infiltrated, therefore bypass was not performed. At the end of the procedure, the removal appeared macroscopically complete. In the same session, an accessory suprascapular reinnervation was performed with a view to a future reconstruction as the suture would have been outside the radiotherapy treatment field. Subsequently, in case of a good survival, reinnervation of the musculocutaneous would have been planned with intercostals and a gracilis free flap to recover some function of the hand. The patient was referred for radiotherapy treatment on the surgical focus. Unfortunately, 8 months after surgery, a dissemination of disease was observed in the intradural spinal area: no further surgical options were possible, and the patient was referred for oncological treatment.
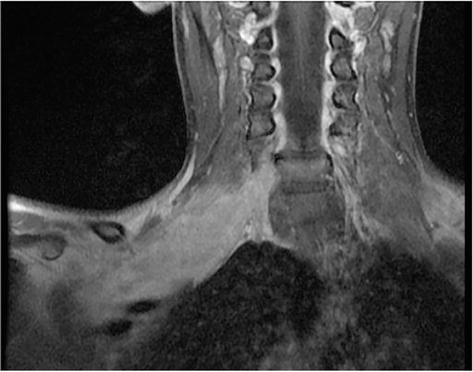
Figure 12.
MR imaging of a right brachial plexus MPNST.
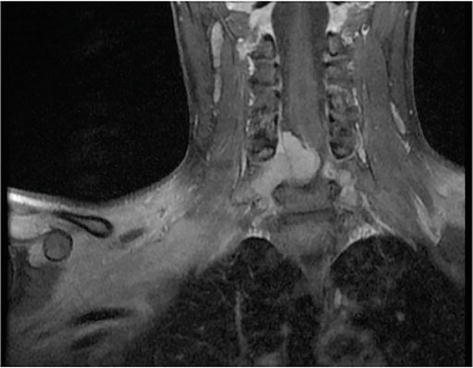
Figure 13.
MR imaging of a right brachial plexus MPNST.
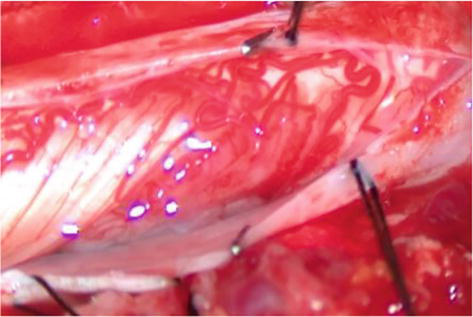
Figure 14.
C7 root with MPNST infiltration.
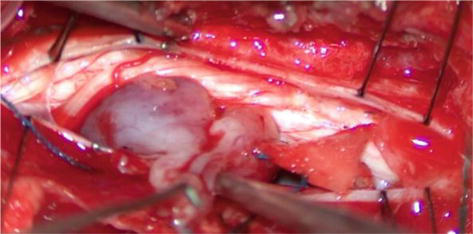
Figure 15.
Removal of intradural extension of MPNST.
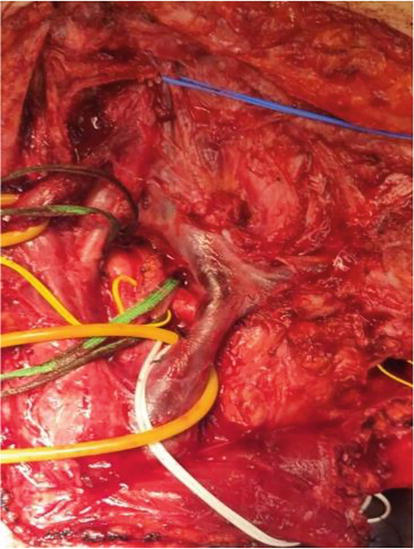
Figure 16.
Anterior approach with exposition of subclavian vessels encased in the MPNST.
4.2 Case 2
A 14-year-old girl with NF1 was referred to our department for evaluation of a rapidly growing mass in a previously known plexiform neurofibroma of the femoral region of the right leg. The patient did not present any pain or neurological deficit but the rapid evolution of the lesion and its MR appearance gave evidence for a possible malignant evolution (Figures 17 and 18). Surgical removal was planned. The removal of the mass was radical with the possibility of preserving most of the fibers of the femoral nerve (Figure 19); the patient did not present any neurological deficit. Histological examination indicated an atypical neurofibroma with foci of malignancy. After the surgical procedure, the patient with a follow-up of more than 5 years did not present any disease recurrence.
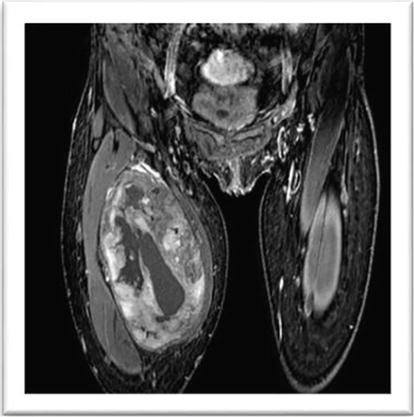
Figure 17.
MR appearance of a low grade MPNST of right femoral nerve.
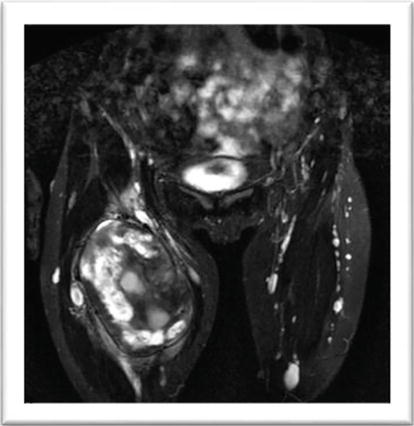
Figure 18.
MR appearance of a low grade MPNST of right femoral nerve.
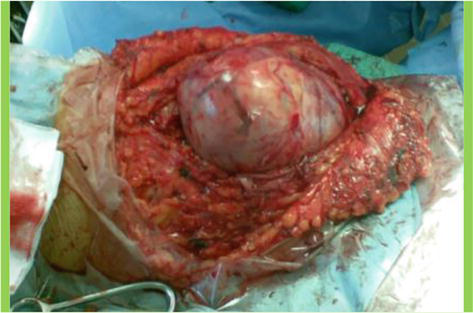
Figure 19.
Low grade MPNST of the femoral nerve; intraoperative image.
5. Conclusion
The treatment of peripheral nerve tumors in patients with NF1 appears complex when these lesions develop in the main nerve trunks and the plexuses; nevertheless, there are some general principles to respect:
Surgery is not definitively curative; therefore, it must be limited to severely symptomatic lesions or when a possible malignant evolution is dreaded.
Neurinomas are generally removable without damage and radically; however, it is advisable to pay attention to the predominantly cystic forms, whose total removal can often be achieved at the expense of a nerve lesion.
In the case of PNs, despite the present indication that medical therapy is only when a mass is declared inoperable, we think that apart from cases where malignancy is suspected or an immediate result is expected (such as spinal cord compression), surgery must stay in a second plane.
It is necessary to evaluate and limit the removal of the PNs not to worsen the patient’s condition with post-surgical neurological deficits.
Any lesion rapidly growing, severely painful, and that causes neurological deficits should be considered suspicious for MPNST until proven otherwise.
All patients with NF1 should undergo periodic check-ups to detect lesions with potential malignant evolution in a very early phase and treat them promptly.
For all the above considerations, the patient affected by NF1 needs to be followed and treated by a highly specialized multidisciplinary team that can offer all the treatments and therapies available at the state-of-the-art.
6. Future directions and developments in NF1-PN research
6.1 Extracellular matrix and tumor development: a new therapeutic scenario
NF1 is caused by a pathogenic variant in the NF1 tumor suppressor gene, which is located at chromosome 17q11.2.5 In 90% of the cases, the mutation leads to a loss of function of the NF1 gene product neurofibromin type I (Nf1, comprising 2818 residues, UniProt: P21359–2) [74]. This giant protein (320 kDa) is a GTPase-activating protein promoting the conversion of active guanosine triphosphate (GTP)-bound RAS to its inactive guanosine diphosphate (GDP)-bound conformation [8]. Unexpected layers of regulation for biological regulation Nf1 have been opened by recent studies on cryo-Electron microscopy (cryo-EM). Nf1 works as a dimeric architecture resting in an equilibrium between open and close conformational states (including an auto-inhibited closed conformation [75]. The structure derives from a dynamic interaction of known Nf1 domains as the GAP-related domain (GRD), the phosphatidylinositol-transfer protein Sec14p-homologous domain, and the Pleckstrin-homology domain module, together referred to as Sec14-PH domain. The GRD carries out Ras GAP activity and interacts via its GAP-extra domain (GAPex) with SPRED proteins [76]. Remarkably, genetic variants within these regions have been linked to NF1 or Legius syndromes, proposing the importance of all domains for neurofibromin function [77].
The GRD is locked down into a crevice formed by the alpha-helical repeat scaffold by an interprotomer interaction. The GAPex domain is formed by residues flanking the GAP core. Strikingly, in this position, the GRD-binding site for Ras is occluded by the surrounding CDM, suggesting that the observed neurofibromin conformation does not represent its active state.
Moreover, Nf1 can directly bind glycerophospholipids and phosphoinositide lipids on the plasma membrane through the Sec14-PH domain and the C-terminal Sprouty domain of SPRED proteins [78, 79]. These data account for functional interaction with the signaling platforms active in cell homeostasis composed of multiple molecules such as tyrosine kinase receptors and their downstream effectors; likewise, they suggest new potential Nf1 plasma membrane targets on the membrane as Src, integrin/Fak complexes.
Haploinsufficiency for Nf1 causes growth factor-dependent hyperactivation of Ras (11), which further increases when loss of heterozygosis (LOH) of NF1 occurs [3]. LOH is considered the primary process behind neurofibromas onset due to the activation of several signaling axes downstream Ras as Raf /ERK, PI3K/AKT, and mTOR in Schwann cells. In addition to its role in the negative regulation of Ras, neurofibromin is a positive regulator of adenylyl cyclase, the enzyme responsible for the generation of intracellular cyclic AMP (cAMP) [4, 80, 81]. This knowledge leads to pharmacologically targeting these upstream and downstream effectors such as growth factor receptors [82, 83] mTOR [30, 84], and MAP Kinase MEK [10]. MEK inhibitors-based treatment is the most successful treatment. Among MEK inhibitors, selumetinib has been recently approved for PNST treatment by the Food and Drug Administration [10, 33]. The partial therapeutic response of MEK inhibitors has been ascribed to the multifaced nature of tumor biology.
As learned from other tumor models, Ras hyperactivation is insufficient per se to promote neoplastic growth and progression: it requires a second hit to promote neoplastic growth. According to the advances in other tumors, cooperation of Ras signaling with biochemical and biophysical signals provided by a heterozygous niche (NF1+/-) [4] induces neoplastic transformation of a cell [85]. Accordingly, growing literature states that increasing matrix stiffness generated in abnormal tissues as fibrotic tissues functionally contribute to tumor progression and dissemination through profound modification of the tumor cell genome landscape [85, 86]. Indeed, the main feature of neurofibroma is a potent extracellular matrix (ECM) structure due to the massive deposition of insoluble and soluble proteins by abnormally activated fibroblasts; nevertheless, its role in tumor progression is still poorly defined. Recently, Errico et al. explored whether Ras signaling could be potentiated by ECM-mediated inward signaling. They demonstrated that Focal adhesion Kinase is a crucial enzyme able to integrate and potentiate both ECM-dependent of inward signaling and the Ras/Raf/Erk and PI3K downstream signaling, thus creating a potent tumorigenic axis. Remarkably, combinatorial treatment with the FAK inhibitor defactinib (VS-6063) and selumetinib (AZD6244) selectively suppressed the colony growth of Schwann cells. These data pave the way for a new therapeutic route based on disrupting the ECM-sensing machinery [18]. PNs appear with a serpentine pattern with multiple patchy nodules with thick collagen fibers [87]. In pancreatic ductal adenocarcinoma (PDAC), these fibers, typical of fibrotic tissues, have been shown to potently induce both FAK and RAS family GTP hydrolases (RHO GTPases) activity influencing the genome landscape of the cancer cells.
In general, an abnormal, stiff matrix is generated by active fibroblasts or Fibrosis Associated Fibroblasts (FAF) or myofibroblasts, robustly secreting many of the constituents of the ECM and basement membranes — such as type I, type III, and type IV and type V collagens, laminins and fibronectin and growth factors. Indeed, activated fibroblasts were first described in the setting of wound healing and were recognized predominantly by their ability to form a scar and later to be critical cells in the chronic tissue wound healing response, also known as tissue fibrosis.
In neurofibromas, many myofibroblasts in the stroma implicate their essential role in fibrosis development. However, both the cellular type from which they originate and the local biological process sustaining their transition are still under debate [88]. Parrinello et al. in 2008 have launched the idea that neurofibroma begins from a wound healing process initiated by Nf1 loss sufficient
Several pieces of evidence corroborate the hypothesis that neurofibroma resembles a wounded nerve that never heals, although they do not explain the neoplastic progression of Schwann cells: a common feature possessed by Schwann cells and many other cells is the injury-induced activation of genes associated with epithelial–mesenchymal transitions and stemness (see below), differentiation states that are linked to cellular plasticity and that help injury-induced tissue remodeling. PN’s Schwann cells coexist in distinct subpopulations of Schwann cells at various differentiation states and remarkable plasticity [90]; Nerve repair depends on myelin dedifferentiation but also on the activation of transcriptional mechanisms that were not necessarily involved in Schwann cell development; two transcriptional controls, c-Jun and chromatin modifications involving H3K27 demethylation, H3K27 deacetylation, and H3K4 methylation, were found to be essential for the normal execution of the Schwann cell injury response. The H3K27 demethylation correlates to tumor progression towards malignancy, since is considered a suitable marker in Atypical Neurofibromatous Neoplasm with Uncertain Biological Potential (ANNUBP) [14]; Choi et al. found that the gene expression profiles of neurofibroma from genetically engineered mouse (Nf1flox/flox; DhhCre, [91]) and rat sciatic nerve, following crush injury were similar [92]; this mouse model, generated neurofibromas with wild type niche, enriched by macrophages; macrophages are critical determinants in Schwann cell reprogramming towards mesenchymal state during peripheral nerve regeneration. However, the macrophage population is poorly represented in human plexiform neurofibromas and scarcely investigated in other mouse models of PN (Nf1flox/flox; Krox20-Cre, [93] Nf1flox/flox; PlpCre [94]; these inflammatory cells, in malignant evolution of PN, the MPNSTs, are significantly characterized suggesting that they do not appear to be necessary for tumorigenesis, but likely contribute to aspects of plexiform neurofibroma progression [95]. By contrast, Nf1-haploinsufficient mast cells are abundantly recruited by stem cell factors (SCFs) secreted by Schwann cells that lack neurofibromin (Nf1−/−). Moreover, compared with normal mast cells, SCF-stimulated heterozygous mast cells (Nf1+/−) secreted 2.5-fold more transforming growth factor beta (TGF-β) [96]. Thus, the direct cooperation of Schwann cells and myofibroblasts generates a continuous mast cell recruitment in the neurofibroma that may explain the onset and maintenance of a chronic inflammation disrupting homeostatic regulation of the wound healing (Figure 20).
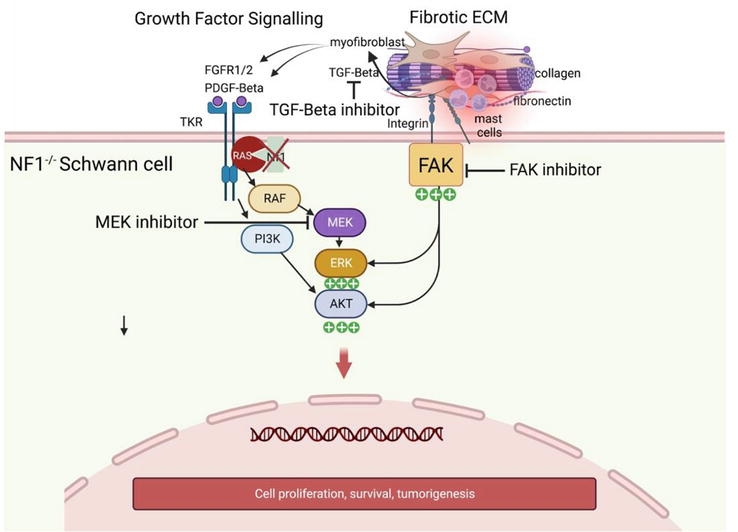
Figure 20.
Ras hyperactivation- and fibrotic extracellular matrix-dependent signaling cooperate to promote tumorigenesis. (Figure made with biorender).
A self-renewing loop is triggered, since mast cell-derived TGFβ promotes epithelial-mesenchymal transition (EMT)-like changes in fibroblasts and other cell type as pericytes or endothelial cells, that trans-differentiates into highly secreting TGFβ myofibroblasts. TGFβ is a master EMT regulator and wound healing regulator and has been previously linked to peripheral nervous system (PNS) regeneration and wound healing and fibrosis [97]. Schwann cells from PNs consistently harbor phosphorylated Smad3 (p-Smad3), active PI3K, and protein kinases (MAPK), the canonical downstream effectors of the TGFβ pathway. In this view, the paracrine feedback loop of Schwann cell-myofibroblasts-mast cell leads to tumor formation due to the combination of Ras hyperactivity and potent secretion of growth factors in a fibrotic milieu. Similarly, in PDAC, TGFβ is a direct modulator of cancer-Schwann cell interactions. Indeed, Schwann cells, chemoattracted by myofibroblasts, are found to directly enhance the aggressiveness of pancreatic cancer cells by secreting TGFβ through SMAD activation; high levels of TGFβ signaling activation positively correlated with perineural invasion by cancer cells suggesting that TGFβ is an essential supporter of both fibrosis and transforming potential [98]. Imatinib, designed to block tyrosine phosphorylation of Smad4 and restores TGF-β growth-suppressive signaling in BCR-ABL1-positive leukemia, has been tested in a phase 2 clinical trial in a patient with inoperable PNs, given its ability to inhibit also tyrosine activity of Kit and PDGF- β receptors. The main trial outcome was tumor shrinkage. The authors observed in a significant subgroup of patients around 20% of tumor reduction, in a subset of pre-existing PNs. These results suggest that TGF-β inhibitors against SMAD- and non-SMAD downstream signaling must represent an alternative or combinatorial therapeutic solution to MEK inhibitors to achieve therapeutic success [99].
Acknowledgments
We thank all patients for their advice, humanity, and patience, represented by patient Associations such as Linfa OdV https://www.linfaneurofibromatosi.com/.
References
- 1.
Legius E, Messiaen L, Wolkenstein P, Pancza P, Avery RA, Berman Y, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genetics in Medicine. 2021; 23 :1506-1513. DOI: 10.1038/s41436-021-01170-5 - 2.
Bergqvist C, Servy A, Valeyrie-Allanore L, Ferkal S, Combemale P, Wolkenstein P, et al. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet Journal of Rare Diseases. 2020; 15 :37. DOI: 10.1186/s13023-020-1310-3 - 3.
Harrisingh MC, Lloyd AC. Ras/Raf/ERK signalling and NF1. Cell Cycle. 2004; 3 :1255-1258. DOI: 10.4161/cc.3.10.1182 - 4.
Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nature Reviews. Disease Primers. 2017; 3 :17004. DOI: 10.1038/nrdp.2017.4 - 5.
Ruggeri RM, Benevento E, De Cicco F, Fazzalari B, Guadagno E, Hasballa I, et al. Neuroendocrine neoplasms in the context of inherited tumor syndromes: A reappraisal focused on targeted therapies. Journal of Endocrinological Investigation. 2023; 46 :213-234. DOI: 10.1007/s40618-022-01905-4 - 6.
Uusitalo E, Rantanen M, Kallionpaa RA, Poyhonen M, Leppavirta J, Yla-Outinen H, et al. Distinctive cancer associations in patients with Neurofibromatosis type 1. Journal of Clinical Oncology. 2016; 34 :1978-1986. DOI: 10.1200/JCO.2015.65.3576 - 7.
Cimino PJ, Ketchum C, Turakulov R, Singh O, Abdullaev Z, Giannini C, et al. Expanded analysis of high-grade astrocytoma with piloid features identifies an epigenetically and clinically distinct subtype associated with neurofibromatosis type 1. Acta Neuropathologica. 2023; 145 :71-82. DOI: 10.1007/s00401-022-02513-5 - 8.
Cichowski K, Jacks T. NF1 tumor suppressor gene function: Narrowing the GAP. Cell. 2001; 104 :593-604.1 - 9.
Stucky CC, Johnson KN, Gray RJ, Pockaj BA, Ocal IT, Rose PS, et al. Malignant peripheral nerve sheath tumors (MPNST): The Mayo Clinic experience. Annals of Surgical Oncology. 2012; 19 :878-885. DOI: 10.1245/s10434-011-1978-7 - 10.
Gross AM, Dombi E, Widemann BC. Current status of MEK inhibitors in the treatment of plexiform neurofibromas. Child's Nervous System. 2020; 36 :2443-2452. DOI: 10.1007/s00381-020-04731-2 - 11.
Dombi E, Solomon J, Gillespie AJ, Fox E, Balis FM, Patronas N, et al. NF1 plexiform neurofibroma growth rate by volumetric MRI: Relationship to age and body weight. Neurology. 2007; 68 :643-647. DOI: 10.1212/01.wnl.0000250332.89420.e6 - 12.
Miettinen MM, Antonescu CR, Fletcher CDM, Kim A, Lazar AJ, Quezado MM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Human Pathology. 2017; 67 :1-10. DOI: 10.1016/j.humpath.2017.05.010 - 13.
Pemov A, Hansen NF, Sindiri S, Patidar R, Higham CS, Dombi E, et al. Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define premalignant neurofibromatosis type 1-associated atypical neurofibromas. Neuro-Oncology. 2019; 21 :981-992. DOI: 10.1093/neuonc/noz028 - 14.
Higham CS, Dombi E, Rogiers A, Bhaumik S, Pans S, Connor SEJ, et al. The characteristics of 76 atypical neurofibromas as precursors to neurofibromatosis 1 associated malignant peripheral nerve sheath tumors. Neuro-Oncology. 2018; 20 :818-825. DOI: 10.1093/neuonc/noy013 - 15.
Prudner BC, Ball T, Rathore R, Hirbe AC. Diagnosis and management of malignant peripheral nerve sheath tumors: Current practice and future perspectives. Neuro-oncology Advances. 2020; 2 :i40-i49. DOI: 10.1093/noajnl/vdz047 - 16.
Brohl AS, Kahen E, Yoder SJ, Teer JK, Reed DR. The genomic landscape of malignant peripheral nerve sheath tumors: Diverse drivers of Ras pathway activation. Scientific Reports. 2017; 7 :14992. DOI: 10.1038/s41598-017-15183-1 - 17.
Staser K, Yang FC, Clapp DW. Pathogenesis of plexiform neurofibroma: Tumor-stromal/hematopoietic interactions in tumor progression. Annual Review of Pathology. 2012; 7 :469-495. DOI: 10.1146/annurev-pathol-011811-132441 - 18.
Errico A, Stocco A, Riccardi VM, Gambalunga A, Bassetto F, Grigatti M, et al. Neurofibromin deficiency and extracellular matrix cooperate to increase transforming potential through FAK-dependent signaling. Cancers (Basel). 2021; 13 :2329. DOI: 10.3390/cancers13102329 - 19.
Le LQ, Parada LF. Tumor microenvironment and neurofibromatosis type I: Connecting the GAPs. Oncogene. 2007; 26 :4609-4616. DOI: 10.1038/sj.onc.1210261 - 20.
Akshintala S, Baldwin A, Liewehr DJ, Goodwin A, Blakeley JO, Gross AM, et al. Longitudinal evaluation of peripheral nerve sheath tumors in neurofibromatosis type 1: Growth analysis of plexiform neurofibromas and distinct nodular lesions. Neuro-Oncology. 2020; 22 :1368-1378. DOI: 10.1093/neuonc/noaa053 - 21.
Plotkin SR, Bredella MA, Cai W, Kassarjian A, Harris GJ, Esparza S, et al. Quantitative assessment of whole-body tumor burden in adult patients with neurofibromatosis. PLoS One. 2012; 7 :e35711. DOI: 10.1371/journal.pone.0035711 - 22.
Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, et al. Activity of Selumetinib in Neurofibromatosis type 1-related plexiform Neurofibromas. The New England Journal of Medicine. 2016; 375 :2550-2560. DOI: 10.1056/NEJMoa1605943 - 23.
Fisher MJ, Blakeley JO, Weiss BD, Dombi E, Ahlawat S, Akshintala S, et al. Management of neurofibromatosis type 1-associated plexiform neurofibromas. Neuro-Oncology. 2022; 24 :1827-1844. DOI: 10.1093/neuonc/noac146 - 24.
Guha D, Davidson B, Nadi M, Alotaibi NM, Fehlings MG, Gentili F, et al. Management of peripheral nerve sheath tumors: 17 years of experience at Toronto Western hospital. Journal of Neurosurgery. 2018; 128 :1226-1234. DOI: 10.3171/2017.1.JNS162292 - 25.
Yoshinaga A, Tsuge I, Yoshinaga D, Ogino S, Sakamoto M, Saito S, et al. Reduction of intraoperative bleeding in diffuse plexiform Neurofibroma resection using the LigaSure vessel sealing system. Plastic and Reconstructive Surgery. 2021; 148 :344e-346e. DOI: 10.1097/PRS.0000000000008163 - 26.
Canavese F, Krajbich JI. Resection of plexiform neurofibromas in children with neurofibromatosis type 1. Journal of Pediatric Orthopedics. 2011; 31 :303-311. DOI: 10.1097/BPO.0b013e31820cad77 - 27.
Needle MN, Cnaan A, Dattilo J, Chatten J, Phillips PC, Shochat S, et al. Prognostic signs in the surgical management of plexiform neurofibroma: The Children's Hospital of Philadelphia experience, 1974-1994. The Journal of Pediatrics. 1997; 131 :678-682. DOI: 10.1016/s0022-3476(97)70092-1 - 28.
Ikuta K, Nishida Y, Sakai T, Koike H, Ito K, Urakawa H, et al. Surgical treatment and complications of deep-seated nodular plexiform Neurofibromas associated with Neurofibromatosis type 1. Journal of Clinical Medicine. 2022; 11 :5695. DOI: 10.3390/jcm11195695 - 29.
Robertson KA, Nalepa G, Yang FC, Bowers DC, Ho CY, Hutchins GD, et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: A phase 2 trial. The Lancet Oncology. 2012; 13 :1218-1224. DOI: 10.1016/S1470-2045(12)70414-X - 30.
Weiss B, Widemann BC, Wolters P, Dombi E, Vinks A, Cantor A, et al. Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas: A neurofibromatosis clinical trials consortium phase II study. Neuro-Oncology. 2015; 17 :596-603. DOI: 10.1093/neuonc/nou235 - 31.
Jakacki RI, Dombi E, Potter DM, Goldman S, Allen JC, Pollack IF, et al. Phase I trial of pegylated interferon-alpha-2b in young patients with plexiform neurofibromas. Neurology. 2011; 76 :265-272. DOI: 10.1212/WNL.0b013e318207b031 - 32.
Gross A, Bishop R, Widemann BC. Selumetinib in plexiform Neurofibromas. The New England Journal of Medicine. 2017; 376 :1195. DOI: 10.1056/NEJMc1701029 - 33.
Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ, et al. Selumetinib in children with inoperable plexiform Neurofibromas. The New England Journal of Medicine. 2020; 382 :1430-1442. DOI: 10.1056/NEJMoa1912735 - 34.
Gross AM, Glassberg B, Wolters PL, Dombi E, Baldwin A, Fisher MJ, et al. Selumetinib in children with neurofibromatosis type 1 and asymptomatic inoperable plexiform neurofibroma at risk for developing tumor-related morbidity. Neuro-Oncology. 2022; 24 :1978-1988. DOI: 10.1093/neuonc/noac109 - 35.
Espirito Santo V, Passos J, Nzwalo H, Carvalho I, Santos F, Martins C, et al. Selumetinib for plexiform neurofibromas in neurofibromatosis type 1: A single-institution experience. Journal of Neuro-Oncology. 2020; 147 :459-463. DOI: 10.1007/s11060-020-03443-6 - 36.
Cacchione A, Fabozzi F, Carai A, Colafati GS, Baldo GD, Rossi S, et al. Safety and efficacy of Mek inhibitors in the treatment of plexiform Neurofibromas: A retrospective study. Cancer Control. 2023; 30 . DOI: 10.1177/10732748221144930 - 37.
Baldo F, Grasso AG, Cortellazzo Wiel L, Maestro A, Trojniak MP, Murru FM, et al. Selumetinib in the treatment of symptomatic intractable plexiform Neurofibromas in Neurofibromatosis type 1: A prospective case series with emphasis on side effects. Paediatric Drugs. 2020; 22 :417-423. DOI: 10.1007/s40272-020-00399-y - 38.
Ronsley R, Hounjet CD, Cheng S, Rassekh SR, Duncan WJ, Dunham C, et al. Trametinib therapy for children with neurofibromatosis type 1 and life-threatening plexiform neurofibroma or treatment-refractory low-grade glioma. Cancer Medicine. 2021; 10 :3556-3564. DOI: 10.1002/cam4.3910 - 39.
Guerreiro Stucklin AS, Mueller S. Opportunities for the treatment of NF1-associated low-grade gliomas: How to decide on the best treatment options for patients? Neuro-Oncology. 2020; 22 :1415-1416. DOI: 10.1093/neuonc/noaa201 - 40.
Weiss BD, Wolters PL, Plotkin SR, Widemann BC, Tonsgard JH, Blakeley J, et al. NF106: A Neurofibromatosis clinical trials consortium phase II trial of the MEK inhibitor Mirdametinib (PD-0325901) in adolescents and adults with NF1-related plexiform Neurofibromas. Journal of Clinical Oncology. 2021; 39 :797-806. DOI: 10.1200/JCO.20.02220 - 41.
Fisher MJ, Shih CS, Rhodes SD, Armstrong AE, Wolters PL, Dombi E, et al. Cabozantinib for neurofibromatosis type 1-related plexiform neurofibromas: A phase 2 trial. Nature Medicine. 2021; 27 :165-173. DOI: 10.1038/s41591-020-01193-6 - 42.
Klesse LJ, Jordan JT, Radtke HB, Rosser T, Schorry E, Ullrich N, et al. The use of MEK inhibitors in Neurofibromatosis type 1-associated tumors and Management of Toxicities. The Oncologist. 2020; 25 :e1109-e1116. DOI: 10.1634/theoncologist.2020-0069 - 43.
de Blank P, Fouladi M, Huse JT. Molecular markers and targeted therapy in pediatric low-grade glioma. Journal of Neuro-Oncology. 2020; 150 :5-15. DOI: 10.1007/s11060-020-03529-1 - 44.
Hanzlik E, Archambault B, El-Dairi M, Schroeder K, Patel MP, Lipp ES, et al. Use of Trametinib in children and young adults with progressive low-grade glioma and Glioneuronal tumors. Journal of Pediatric Hematology/Oncology. 2022; 45 :464-470. DOI: 10.1097/MPH.0000000000002598 - 45.
Somatilaka BN, Sadek A, McKay RM, Le LQ. Malignant peripheral nerve sheath tumor: Models, biology, and translation. Oncogene. 2022; 41 :2405-2421. DOI: 10.1038/s41388-022-02290-1 - 46.
Kimura M, Kamata Y, Matsumoto K, Takaya H. Electron microscopical study on the tumor of von Recklinghausen's neurofibromatosis. Acta Pathologica Japonica. 1974; 24 :79-91. DOI: 10.1111/j.1440-1827.1974.tb00809.x - 47.
Parsons CM, Canter RJ, Khatri VP. Surgical management of neurofibromatosis. Surgical Oncology Clinics of North America. 2009; 18 :175-196. DOI: 10.1016/j.soc.2008.08.009 - 48.
Wolkenstein P, Zeller J, Revuz J, Ecosse E, Leplege A. Quality-of-life impairment in neurofibromatosis type 1: A cross-sectional study of 128 cases. Archives of Dermatology. 2001; 137 :1421-1425. DOI: 10.1001/archderm.137.11.1421 - 49.
Huson SM, Harper PS, Compston DA. Von Recklinghausen neurofibromatosis. A clinical and population study in south-East Wales. Brain. 1988; 111 (Pt 6):1355-1381. DOI: 10.1093/brain/111.6.1355 - 50.
Grobmyer SR, Reith JD, Shahlaee A, Bush CH, Hochwald SN. Malignant peripheral nerve sheath tumor: Molecular pathogenesis and current management considerations. Journal of Surgical Oncology. 2008; 97 :340-349. DOI: 10.1002/jso.20971 - 51.
Brambullo T, Biffoli B, Scortecci L, Messana F, Vindigni V, Bassetto F. Antibiotic prophylaxis in plastic surgery: From systematic review to operative algorithm. World Journal of Plastic Surgery. 2022; 11 :24-36. DOI: 10.52547/wjps.11.2.24 - 52.
Friedrich RE, Korf B, Funsterer C, Mautner VF. Growth type of plexiform neurofibromas in NF1 determined on magnetic resonance images. Anticancer Research. 2003; 23 :949-952 - 53.
Korf BR. Malignancy in neurofibromatosis type 1. The Oncologist. 2000; 5 :477-485. DOI: 10.1634/theoncologist.5-6-477 - 54.
Littlewood AH, Stilwell JH. The vascular features of plexiform neurofibroma with some observations on the importance of pre-operative angiography and the value of pre-operative intra-arterial embolisation. British Journal of Plastic Surgery. 1983; 36 :501-506. DOI: 10.1016/0007-1226(83)90140-6 - 55.
Santoro A, Tursz T, Mouridsen H, Verweij J, Steward W, Somers R, et al. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: A randomized study of the European Organization for Research and Treatment of cancer soft tissue and bone sarcoma group. Journal of Clinical Oncology. 1995; 13 :1537-1545. DOI: 10.1200/JCO.1995.13.7.1537 - 56.
Vauthey JN, Woodruff JM, Brennan MF. Extremity malignant peripheral nerve sheath tumors (neurogenic sarcomas): A 10-year experience. Annals of Surgical Oncology. 1995; 2 :126-131. DOI: 10.1007/BF02303627 - 57.
Hruban RH, Shiu MH, Senie RT, Woodruff JM. Malignant peripheral nerve sheath tumors of the buttock and lower extremity. A study of 43 cases. Cancer. 1990; 66 :1253-1265. DOI: 10.1002/1097-0142(19900915)66:6<1253::aid-cncr2820660627>3.0.co;2-r - 58.
Anghileri M, Miceli R, Fiore M, Mariani L, Ferrari A, Mussi C, et al. Malignant peripheral nerve sheath tumors: Prognostic factors and survival in a series of patients treated at a single institution. Cancer. 2006; 107 :1065-1074. DOI: 10.1002/cncr.22098 - 59.
Garozzo D. Peripheral nerve tumors in neurofibromatosis 1: An overview on management and indications for surgical treatment in our experience. Neurology India. 2019; 67 :S38-S44. DOI: 10.4103/0028-3886.250697 - 60.
Marjanska A, Galazka P, Wysocki M, Styczynski J. New Frontiers in therapy of peripheral nerve sheath tumors in patients with Neurofibromatosis type 1: Latest evidence and clinical implications. Anticancer Research. 2020; 40 :1817-1831. DOI: 10.21873/anticanres.14136 - 61.
Reilly KM, Kim A, Blakely J, Ferner RE, Gutmann DH, Legius E, et al. Neurofibromatosis type 1-associated MPNST state of the science: Outlining a research agenda for the future. Journal of the National Cancer Institute. 2017; 109 :djx124. DOI: 10.1093/jnci/djx124 - 62.
Ahlawat S, Fayad LM, Khan MS, Bredella MA, Harris GJ, Evans DG, et al. Current whole-body MRI applications in the neurofibromatoses: NF1, NF2, and schwannomatosis. Neurology. 2016; 87 :S31-S39. DOI: 10.1212/WNL.0000000000002929 - 63.
Valeyrie-Allanore L, Ismaili N, Bastuji-Garin S, Zeller J, Wechsler J, Revuz J, et al. Symptoms associated with malignancy of peripheral nerve sheath tumours: A retrospective study of 69 patients with neurofibromatosis 1. The British Journal of Dermatology. 2005; 153 :79-82. DOI: 10.1111/j.1365-2133.2005.06558.x - 64.
Lavrador JP, Lascelles K, Ferner R, Thomas N, Zebian B. Neurofibroma of the cervical spine in infants. Child's Nervous System. 2016; 32 :2277-2279. DOI: 10.1007/s00381-016-3253-5 - 65.
van Sandick JW, van Coevorden F. Plexiform neurofibroma with intraspinal extension. Journal of the American College of Surgeons. 2002; 195 :572. DOI: 10.1016/s1072-7515(02)01299-1 - 66.
Leonard JR, Ferner RE, Thomas N, Gutmann DH. Cervical cord compression from plexiform neurofibromas in neurofibromatosis 1. Journal of Neurology, Neurosurgery, and Psychiatry. 2007; 78 :1404-1406. DOI: 10.1136/jnnp.2007.121509 - 67.
Wang T, Yin H, Han S, Yang X, Wang J, Huang Q, et al. Malignant peripheral nerve sheath tumor (MPNST) in the spine: A retrospective analysis of clinical and molecular prognostic factors. Journal of Neuro-Oncology. 2015; 122 :349-355. DOI: 10.1007/s11060-015-1721-5 - 68.
Pollack IF, Colak A, Fitz C, Wiener E, Moreland M, Mulvihill JJ. Surgical management of spinal cord compression from plexiform neurofibromas in patients with neurofibromatosis 1. Neurosurgery. 1998; 43 :248-255; discussion 255-246. DOI: 10.1097/00006123-199808000-00038 - 69.
Nguyen R, Kluwe L, Fuensterer C, Kentsch M, Friedrich RE, Mautner VF. Plexiform neurofibromas in children with neurofibromatosis type 1: Frequency and associated clinical deficits. The Journal of Pediatrics. 2011; 159 (652–655):e652. DOI: 10.1016/j.jpeds.2011.04.008 - 70.
Ross AL, Panthaki Z, Levi AD. Surgical management of a giant plexiform neurofibroma of the lower extremity. World Neurosurgery. 2011; 75 :754-757. DOI: 10.1016/j.wneu.2010.09.030 - 71.
Kim A, Stewart DR, Reilly KM, Viskochil D, Miettinen MM, Widemann BC. Malignant peripheral nerve sheath tumors state of the science: Leveraging clinical and biological insights into effective therapies. Sarcoma. 2017; 2017 :7429697. DOI: 10.1155/2017/7429697 - 72.
Sobczuk P, Teterycz P, Czarnecka AM, Switaj T, Kosela-Paterczyk H, Kozak K, et al. Malignant peripheral nerve sheath tumors - outcomes and prognostic factors based on the reference center experience. Surgical Oncology. 2020; 35 :276-284. DOI: 10.1016/j.suronc.2020.09.011 - 73.
Kruft S, von Heimburg D, Reill P. Treatment of irreversible lesion of the radial nerve by tendon transfer: Indication and long-term results of the merle d'Aubigne procedure. Plastic and Reconstructive Surgery. 1997; 100 :610-616; discussion 617-618. DOI: 10.1097/00006534-199709000-00010 - 74.
Friedman JM. Epidemiology of neurofibromatosis type 1. American Journal of Medical Genetics. 1999; 89 :1-6 - 75.
Chaker-Margot M, Werten S, Dunzendorfer-Matt T, Lechner S, Ruepp A, Scheffzek K, et al. Structural basis of activation of the tumor suppressor protein neurofibromin. Molecular Cell. 2022; 82 (1288–1296):e1285. DOI: 10.1016/j.molcel.2022.03.011 - 76.
Dunzendorfer-Matt T, Mercado EL, Maly K, McCormick F, Scheffzek K. The neurofibromin recruitment factor Spred1 binds to the GAP related domain without affecting Ras inactivation. Proceedings of the National Academy of Sciences of the United States of America. 2016; 113 :7497-7502. DOI: 10.1073/pnas.1607298113 - 77.
Ko JM, Sohn YB, Jeong SY, Kim HJ, Messiaen LM. Mutation spectrum of NF1 and clinical characteristics in 78 Korean patients with neurofibromatosis type 1. Pediatric Neurology. 2013; 48 :447-453. DOI: 10.1016/j.pediatrneurol.2013.02.004 - 78.
Welti S, Fraterman S, D'Angelo I, Wilm M, Scheffzek K. The sec14 homology module of neurofibromin binds cellular glycerophospholipids: Mass spectrometry and structure of a lipid complex. Journal of Molecular Biology. 2007; 366 :551-562. DOI: 10.1016/j.jmb.2006.11.055 - 79.
Stowe IB, Mercado EL, Stowe TR, Bell EL, Oses-Prieto JA, Hernandez H, et al. A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Genes & Development. 2012; 26 :1421-1426. DOI: 10.1101/gad.190876.112 - 80.
Ferner RE. The neurofibromatoses. Practical Neurology. 2010; 10 :82-93. DOI: 10.1136/jnnp.2010.206532 - 81.
Gutmann DH, Blakeley JO, Korf BR, Packer RJ. Optimizing biologically targeted clinical trials for neurofibromatosis. Expert Opinion on Investigational Drugs. 2013; 22 :443-462. DOI: 10.1517/13543784.2013.772979 - 82.
Kim A, Dombi E, Tepas K, Fox E, Martin S, Wolters P, et al. Phase I trial and pharmacokinetic study of sorafenib in children with neurofibromatosis type I and plexiform neurofibromas. Pediatric Blood & Cancer. 2013; 60 :396-401. DOI: 10.1002/pbc.24281 - 83.
Kim A, Lu Y, Okuno SH, Reinke D, Maertens O, Perentesis J, et al. Targeting refractory sarcomas and malignant peripheral nerve sheath tumors in a phase I/II study of Sirolimus in combination with Ganetespib (SARC023). Sarcoma. 2020; 2020 :5784876. DOI: 10.1155/2020/5784876 - 84.
Parker VER, Keppler-Noreuil KM, Faivre L, Luu M, Oden NL, De Silva L, et al. Safety and efficacy of low-dose sirolimus in the PIK3CA-related overgrowth spectrum. Genetics in Medicine. 2019; 21 :1189-1198. DOI: 10.1038/s41436-018-0297-9 - 85.
Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nature Medicine. 2016; 22 :497-505. DOI: 10.1038/nm.4082 - 86.
Yamauchi M, Barker TH, Gibbons DL, Kurie JM. The fibrotic tumor stroma. The Journal of Clinical Investigation. 2018; 128 :16-25. DOI: 10.1172/JCI93554 - 87.
Messersmith L, Krauland K. Neurofibroma. Treasure Island (FL): StatPearls; 2023 - 88.
Jiang C, McKay RM, Le LQ. Tumorigenesis in neurofibromatosis type 1: Role of the microenvironment. Oncogene. 2021; 40 :5781-5787. DOI: 10.1038/s41388-021-01979-z - 89.
Parrinello S, Noon LA, Harrisingh MC, Wingfield Digby P, Rosenberg LH, Cremona CA, et al. NF1 loss disrupts Schwann cell-axonal interactions: A novel role for semaphorin 4F. Genes & Development. 2008; 22 :3335-3348. DOI: 10.1101/gad.490608 - 90.
Mazuelas H, Magallon-Lorenz M, Fernandez-Rodriguez J, Uriarte-Arrazola I, Richaud-Patin Y, Terribas E, et al. Modeling iPSC-derived human neurofibroma-like tumors in mice uncovers the heterogeneity of Schwann cells within plexiform neurofibromas. Cell Reports. 2022; 38 :110385. DOI: 10.1016/j.celrep.2022.110385 - 91.
Wu J, Williams JP, Rizvi TA, Kordich JJ, Witte D, Meijer D, et al. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell. 2008; 13 :105-116. DOI: 10.1016/j.ccr.2007.12.027 - 92.
Choi K, Komurov K, Fletcher JS, Jousma E, Cancelas JA, Wu J, et al. An inflammatory gene signature distinguishes neurofibroma Schwann cells and macrophages from cells in the normal peripheral nervous system. Scientific Reports. 2017; 7 :43315. DOI: 10.1038/srep43315 - 93.
Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002; 296 :920-922. DOI: 10.1126/science.1068452 - 94.
Mayes DA, Rizvi TA, Cancelas JA, Kolasinski NT, Ciraolo GM, Stemmer-Rachamimov AO, et al. Perinatal or adult Nf1 inactivation using tamoxifen-inducible PlpCre each cause neurofibroma formation. Cancer Research. 2011; 71 :4675-4685. DOI: 10.1158/0008-5472.CAN-10-4558 - 95.
Prada CE, Jousma E, Rizvi TA, Wu J, Dunn RS, Mayes DA, et al. Neurofibroma-associated macrophages play roles in tumor growth and response to pharmacological inhibition. Acta Neuropathologica. 2013; 125 :159-168. DOI: 10.1007/s00401-012-1056-7 - 96.
Yang FC, Chen S, Clegg T, Li X, Morgan T, Estwick SA, et al. Nf1+/− mast cells induce neurofibroma like phenotypes through secreted TGF-beta signaling. Human Molecular Genetics. 2006; 15 :2421-2437. DOI: 10.1093/hmg/ddl165 - 97.
Mirsky R, Woodhoo A, Parkinson DB, Arthur-Farraj P, Bhaskaran A, Jessen KR. Novel signals controlling embryonic Schwann cell development, myelination and dedifferentiation. Journal of the Peripheral Nervous System. 2008; 13 :122-135. DOI: 10.1111/j.1529-8027.2008.00168.x - 98.
Roger E, Martel S, Bertrand-Chapel A, Depollier A, Chuvin N, Pommier RM, et al. Schwann cells support oncogenic potential of pancreatic cancer cells through TGFbeta signaling. Cell Death & Disease. 2019; 10 :886. DOI: 10.1038/s41419-019-2116-x - 99.
Armstrong AE, Rhodes SD, Smith A, Chen S, Bessler W, Ferguson MJ, et al. Early administration of imatinib mesylate reduces plexiform neurofibroma tumor burden with durable results after drug discontinuation in a mouse model of neurofibromatosis type 1. Pediatric Blood & Cancer. 2020; 67 :e28372. DOI: 10.1002/pbc.28372