Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
Due to Evans et al. investigation, it is known that 26% of patients with NF II type involve spinal cord or nervous tissue tumors in the spinal column area. Although so many patients with NF have spinal tumors, not everyone has the potential to the neurological progressive symptoms due to spinal cord compression and not always tumors lead to invalidation of patients with neurofibromatosis. Unfortunately, a lot of surgeons consider that tumors required surgery and very often decide to remove a maximum number of tumors. Neurofibromas has different structural and growth patterns. Most of them has a fusiform type of growing. Sometimes surgeon needs to remove tumor with incoming nerve root with the risk of neurological deficit, or to perform intralesional debulking of soft tumor component, which occasionally leads to continued tumor growth. Furthermore, the decision and the sequence of the tumors removal rely on the leading cause of patient deficits. First of all symptomatic intracranial tumors should be removed, then intradural lesions. Certainly, patients with symptomatic spinal tumor need individual approach to the neurofibromatosis aspect. Decision-making needs to be accepted by neuroradiologist, neurologist, and neurosurgeon by looking at all sides of CNS imaging. So, it is important to make an algorithm for all doctors who try to treat patients with neurofibromatosis.
“AXIS” Private Spine Neurosurgery Hospital, Moscow, The Russian Federation
Yuri M. Poluektov
Burdenko Neurosurgical Institute, Moscow, Russia
Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow, Russian Federation
*Address all correspondence to: vkorolishin@gmail.com
1. Introduction
Accordingly, our experience and literature date, there are a lot of patients with neurofibromatosis in the population, who need surgeon attention. Treatment of these patients’ categories required very special access.
This chapter is devoted to important aspects of the diagnosis and treatment of patients with severe symptoms of neurofibromatosis. A small percentage of these patients suffer from manifestations of neurofibromatosis associated with the growth of tumors along the spinal cord. Often tumors compress the spinal cord or only threaten to compress the spinal cord. In both cases, it is necessary to differentiate the risks of treatment and the threat of a tumor, then offer the patient a solution that will be as effective as it is safe.
The prevalence of neurofibromatosis type 1 (NF1) varies depending on population density and the level of the country’s medical service development and is approximately 1 case per 3000 people [1].
NF1 is characterized by an autosomal dominant inheritance pattern. Key mutations in the NF1 gene affect the regulation of neurofibromin, which is structurally similar to negative regulators of the RAS proto-oncogene pathway. Thus, dysfunction of neurofibromin leads to hyperactivation of the RAS cascade, leading to uncontrolled cell growth mediated through [2].
The defining features of the disease include multiple neurofibromas and peripheral nerve tumors that affect the cranial and peripheral nerves, causing the compression of the spinal cord. In addition, chiasmatic gliomas are common in the pediatric population (up to 20% of all patients with confirmed neurofibromatosis) [3], gliomas of the brainstem, and spinal cord. Among adult patients with NF1, low-grade gliomas are widespread, and the overall risk of developing malignancies is 10–50 times higher than in the general population. The most common intramedullary tumor in NF1 is astrocytoma [4, 5].
NF 1, most frequently, manifests with dermatological symptoms, which include café-au-lait spots and cutaneous neurofibromas. Large studies focused on the frequency of the disease manifestation with various signs have not been conducted.
Thus, the true prevalence of the simultaneous occurrence of tumors of the CNS (central nervous system) and PNS (peripheral nervous system) was not carried out. Also, the incidence of multilevel CNS and PNS lesions in the general population of patients with NF1 has not been evaluated. However, our experience and the experience of a number of researchers suggest that multilevel lesions of the spinal cord occur most often, while one or several tumors can be symptomatic at the same time [6].
The overall cumulative risk of malignancy in patients with NF1 is up to 40% by the age of 50. That is, 2–5 times higher than in the general population. The risk of CNS malignant tumors developing is 40% higher, and the risk of developing MPNST is 1000 times higher [1].
2.2 Diagnostics
The manifestations of the disease can be very heterogeneous and depend on the age of the patient. The first symptoms that can be identified after birth are a cafe-au-lait spot on the skin, dysplasia of the long bones and orbital region, development of plexiform neurofibromas. Then, in early childhood, patients have an increased risk of developing chiasma and optic nerve gliomas, ADHD (Attention deficit hyperactivity disorder), and disorders associated with delayed psychoemotional development. In adolescence, the disease is often manifested by musculoskeletal deformities (including scoliosis), dermal and paraspinal neurofibromas, and brainstem gliomas. Monitoring of these patients should be systemic and aimed at early detection and regular control of the tumor growth of critical localization (threatening the life and health of patients). Regular checkups allow to detect MPNST and high-grade gliomas at an early stage [1].
Patients suspected of having NF needs regular monitoring, depending on the symptoms and growth rate of the lesions, these patients are suggested to undergo a full body MRI once a year/5 years, or a CNS MRI with contrast followed by monitoring for 2 years every 4 months and then 1 once a year up to 5 years (in case of CNS tumors).
2.3 Genetic testing
We recommend genetic testing to be performed at any age in individuals who meet the NF criteria. In addition, patients with an unusual NF1 phenotype, such as multilevel extramedullary tumors, should be tested (even in the absence of large NF1 criteria).
The global diagnostic algorithm implies observation by different specialists from birth (dermatologist, ophthalmologist, neurologist, psychiatrist), and the approach to neuroimaging and other examinations is determined by the manifestation of the disease in each case.
2.4 Imaging
It is believed that more than 50% of patients with NF1 have tumors of the internal organs, and therefore panMRI may be the method of choice after the symptoms manifestation. To detect intracranial or spinal lesions, it is recommended to perform MRI (1.5 T and higher) with i.v. contrasting. We recommend the MRI of entire neuroaxis regardless of the expected level of tumor location.
Full-body CT and full-spine X-ray in two projections can be recommended for progressive musculoskeletal deformity for orthopedic correction planning.
PET with FDG cannot be recommended as a screening method, however, when MPNST is suspected, it is the optimal method of investigation. In addition, PET can be used to determine the source of biopsy material in the case of progressive multiple tumor growth.
For neurofibromatosis type 2 (NF2), the prevalence does not exceed 1 in 300,000 people. NF2 also has an autosomal dominant inheritance pattern and is caused by a mutation in the NF2 gene. This gene encodes a 595-amino acid protein called merlin or SCH, most similar to the exrin-readixin-moesin protein system, these are membrane-cytoskeleton scaffold proteins (i.e., linking actin filaments to the cell membrane or membrane glycoproteins) that act as critical regulators of contact-dependent proliferation inhibition and maintain the normal organization of the cytoskeleton by modulating cell motility, attachment, remodeling, and proliferation, thus regulating cell growth and development (tumor-suppressing function). Merlin regulates proliferation by acting through several cascades that include Ras GAP/Raf proteins (which regulate the MEK/ERK pathway) and PI3K/AKT proteins (which regulate the mTOR pathway) [7].
3.2 Diagnostics
NF2 is characterized by the development of vestibular schwannomas (VS); schwannoma of other cranial, spinal, and cutaneous nerves; intracranial and spinal meningiomas and/or other tumors of the central nervous system (eg ependymoma and astrocytoma). The most common intramedullary tumor is ependymoma.
Multilevel and multiple lesions are a hallmark of NF2 (MISME syndrome (Multiple Inherited Schwannomas, Meningiomas, and Ependymomas)). Multiple meningiomas are the second most important diagnostic criterion and occur in approximately 50% of cases [8].
3.3 Genetic testing
We recommend that genetic testing be performed at any age in individuals who meet the criteria for neurofibromatosis. In addition, patients with an unusual NF2 phenotype, such as multilevel extramedullary tumors and or extended intramedullary tumors, should be tested [9].
Otherwise, the diagnostic algorithm is indistinguishable from that for NF1. One of the main challenges for the management of such patients is the preservation of hearing in progressive bilateral vestibular schwannomas. The use of PET-CT as a diagnostic method fades into the background and can be used as part of preoperative planning for the removal or biopsy of CNS tumors. MPNST is uncharacteristic of NF2.
4. Treatment regimen for symptomatic spinal tumors in patients with neurofibromatosis
Extramedullary tumors often cause a decrease in the quality of life and cause disability for patients with neurofibromatosis. Dysfunction of the pelvic organs and impaired tissue trophism indirectly associated with tumors along the spinal cord can lead to life-threatening situations. Therefore, treatment must be timely. When meeting patients with NF, the clinician is faced with the problem of identifying the main and concomitant symptoms.
Treatment options include methods of local and systemic treatment of patients with NF. Local treatment includes surgery and radiation therapy. In recent years, drug therapy has shown significant efficacy against certain types of tumors.
The need for surgical treatment usually arises when growing tumors cause clinical symptoms. The most common is pain syndrome. If the tumor grows into the intervertebral foramen, along the nerve root, the pain is caused by stretching of the sensitive fascicle and has a burning and shooting pattern along the entire innervated dermatome, which increases with a change in body position. Local pain is usually associated with local tumor growth due to the involvement of the meninges of the spinal cord. At the stage when the tumor compresses the spinal cord, a syndrome of conduction disturbance is followed by paresis and dysfunction of the pelvic organs.
The priority clinical task is to determine the level of damage to the nervous system. So the removal of tumors nerve sheath tumor can significantly disrupt the function of nerve structures due to possible damage to the nerves adjacent to the tumor, which are the source of tumor growth.
In our opinion, an erroneous tactic is the removal of all detected neurofibromas and the attempt of the most radical resection of tumors leads to an aggravation of the neurological deficit and worsens the survival prognosis of patients with neurofibromatosis. Only tumors that threaten the patient’s life or lead to an irreparable loss of function (paresis, dysfunction of the pelvic organs) are subject to removal.
Neurofibromatosis is a pathology that reduces the patient’s quality of life throughout life, so surgical treatment, no matter how necessary it would seem, should not be risky for the patient’s life or health. That is, the operation should not carry more risks than the disease itself.
This principle is dominant in choosing the optimal method of treating patients with neurofibromatosis and, in our opinion, is the only true one.
4.1 Extramedullary tumors
4.1.1 Neurofibroma
Neurofibromas are more common in patients with phacomatosis. According to our observations, neurofibroma is the most common cause of pain and more often leads to dysfunction of nerve structures in patients with NF.
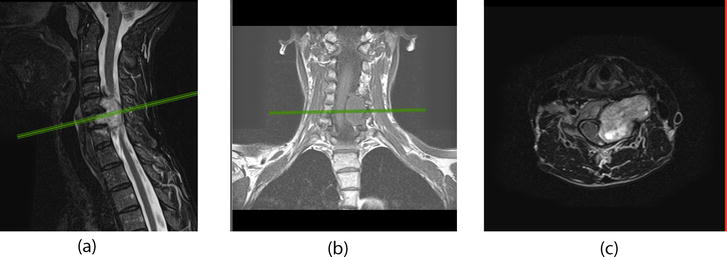
Neurofibroma can be located symmetrically or asymmetrically in the area of the spinal canal or with an offset towards the intervertebral foramen and can be large. Often, neurofibroma has a confluent type of growth, namely, in the area of one localization there may be several nerves affected by neurofibroma, connected in the plexus area, each of which individually is a source of tumor growth and together they represent a large conglomerate of tumor tissue. Figure 1 presents the patient with severe spinal cord compression by a dumbbell nervous tumor on the cervical level. In neurofibromatosis, both the first and second types, the phenomenon of widespread growth of neurofibromas is noted. However, this type of growth is more characteristic of the first type. Men are more likely to get sick, the tumor is more often located intradurally or intra- extradurally and grows like an hourglass. Relatively more common at the level of the cervical region.
Figure 1.
Contrast-enhanced MRI of the cervical spine of a patient with an intra-extradural dumbbell tumor that compresses the spinal cord and extends extravertebral, adjacent to the vertebral and carotid arteries. (a) Sagittal view (T2W + contrast enhancement); (b) coronal view (T1W) and (c) axial view (T2W + contrast enhancement).
Surgical treatment is the most optimal tactic since tumor resection is more effective in terms of local control compared to radiation therapy and intratumoral total resection with preservation of the tumor capsule. More often than in other departments, neurofibromas are detected at the level of the cervical spine and are associated with a higher number of relapses due to the frequent absence of clear boundaries and, therefore, the impossibility of total removal.
Surgical treatment should be safe for the patient, that is, focus on solving the main problem: reducing symptoms or preventing symptoms while maintaining the quality of life. Surgical approaches should be adequate to the size of the pathological formation. Our experience allows us to recommend a minimally invasive method for removing an intradural tumor or tumors at different levels. To create the surgeon’s comfort and reduce the risks of surgical trauma to adjacent nerve structures, the method is used to remove tumors of no more than 2 segments located laterally or centrally-laterally. If the tumor is located bilaterally or circularly relative to the spinal cord, it is desirable to use the posterior median approach with laminectomy. Laminectomy should be carried out taking into account the prevention of postoperative tissue edema. For minimally invasive surgical approaches, tube and telescopic retractors are used to view the bottom of the wound and dilate soft tissues. Analysis of the results of large series of operations suggests that operations are performed faster and with a lower risk of postoperative complications such as postoperative CSF leaking, intermuscular hematoma, soft tissue suture failure, superficial infection, or soft tissue infection. In addition, due to the absence of the need to skeletonize a large surface of the spine, the incision area hurts less, and the speed of the operation increases. So, the operation time is reduced from 40 minutes to 60 minutes according to various sources.
4.1.2 Schwannoma
Spinal schwannoma in neurofibromatosis is somewhat more common than in the general population, but the true prevalence remains unknown. In the general population, the overall incidence rate is 0.24 (95% CI 0.23–0.24) per 100,000 people. The highest prevalence of this pathology occurs at the age of 65–74 years, however, with neurofibromatosis, the life expectancy of patients is much lower, and therefore, it can be assumed that a large percentage of patients in the early age groups belong to patients with neurofibromatosis [10].
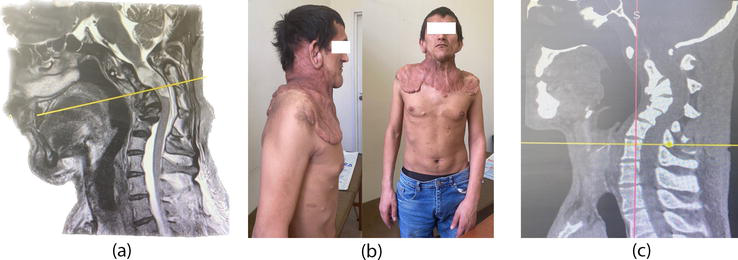
Schwannomas tend to grow intradurally and asymmetrically. Tumors grow slowly and rarely cause clinical symptoms until the size of the tumor is more than a quarter of the diameter of the spinal canal. Patients most often complain of local pain and pain in the extremity associated with a change in body position (Figure 2). If you do not conduct an examination and do not follow the path of local treatment, the patient feels weakness in the legs/arms within a year from the onset of symptoms, depending on the localization. Surgical treatment aims to reduce clinical symptoms and reduce the risk of tumor re-growth. In the event that the tumor is large, the method of choice will be intratumoral resection with the division of the tumor into separate fragments, followed by its removal without residue. Tumors that are localized in the region of the spinal canal from the craniovertebral junction to the sacrum become symptomatic when the spinal cord or nerve root is compressed.
Figure 2.
MRI of the cervical spine with contrast enhancement in the sagittal projection: Dorsolateral intra-extradural tumor (schwannoma) with severe spinal compression (a). Patient appearance (b). CT 3D of patient cervical spine (c).
Sridhar et al. proposed a universal classification scheme for spinal schwannomas. It can be used to unify the approach to tumor classification and optimize surgical approaches (Table 1) [11].
Type
Classification
Ia
Intraspinal, intradural tumor of <2 vertebral segments in length
Ib
Intraspinal, extradural tumor of <2 vertebral segments in length
II
Intraspinal tumor of >2 vertebral segments in length (giant tumor)
III
Intraspinal tumor with extension into nerve root foramen
IVa
Intraspinal tumor with extraspinal extension (dumbbell tumors) < 2.5 cm
IVb
Intraspinal tumor with extraspinal extension (dumbbell tumors) > 2.5 cm (giant tumor)
V
Tumor with erosion into the vertebral bodies, lateral and posterior extensions into myofascial planes (giant invasive tumor)
Table 1.
Classification of benign nerve sheath tumors [11].
Total surgical removal with or without nerve root preservation is the gold standard of surgical treatment. Another treatment option for patients with small asymptomatic neoplasms is radiosurgery (CyberKnife) [12].
The risk of recurrence in surgical treatment is extremely small and does not exceed 6%. The main risk factor for local recurrence is Subtotal resection of the tumor, high risk of proliferative activity, and malignant histology of the tumor. Radiotherapy is a promising treatment for disease recurrence [13].
4.1.3 Meningiomas
Meningiomas occur in about half of patients with type II NF and often occur in multiple regions of the spine. In the study, it was found that cranial meningiomas correlate with mortality in patients with this disease (the risk of death is 2.5 times higher than in patients without intracranial meningiomas. Similar studies have not been conducted in patients with spinal meningiomas [14].
For type I NF, meningioma is not a common pathology, but multiple CNS lesions may occur [15].
The optimal method for diagnosing meningiomas is MRI with IV contrast. Without contrast, meningiomas are visible on T2WI. Calcifications are usually seen on MRI as areas of void. Occasionally tumors may be isointense on T1WI and T2WI.
An additional diagnostic method is CT. The tumor is visualized as a homogeneous, hyperintense mass with a wide attachment to the dura. On non-contrast series, it has a density of 60-70H. In addition, CT allows you to see calcifications, which allows you to make a more accurate diagnosis and simplifies intraoperative marking. For cranial meningiomas, CT angiography is an important study, as it provides comprehensive information about the tumor blood supply. In spinal surgery, this method is used extremely rarely.
The histological classifications of meningiomas can be found in more detail in the corresponding guidelines. When planning surgical treatment, the localization of meningioma relative to the spinal cord is of great importance, so they can be divided into several main localizations:
Dorsal
Ventral
Lateral
Ventrolateral
Dorsolateral
In percentage terms, the incidence of meningiomas of various localizations very much depended on the specific sample, but most often tumors were found in the thoracic spine, less often in the cervical, and extremely rarely in the lumbar [16]. Menigiomas are often associated with other tumors. Currently, local treatment is the most optimal tactic.
Most meningiomas are slow-growing benign tumors. With cranial asymptomatic formations of small size, you can choose expectant tactics, however, when it comes to multilevel lesions, it is necessary to immediately begin treatment. The effectiveness of irradiation for the treatment of benign neoplasms is still unproven, however, we suggest that in the presence of one or more small meningiomas that do not cause neurological symptoms (accidental findings), radiosurgical treatment can be performed.
Surgery remains the gold standard for the treatment of meningiomas. Removal of a meningioma should begin with the separation of the tumor matrix from the site of attachment, trying to preserve the tumor capsule, which is partially formed by the arachnoid membrane, as much as possible. The tumor matrix is usually vascularized. The tactics of removal depend on the localization of the tumor matrix. Dorsal and dorsolateral meningiomas. You should know that immediately under the dura mater is the tumor matrix. Therefore, the incision of the membranes must be carried out with extreme caution, starting with the outer layer of the dura mater. Removal of the neoplasm in a single block with such localization is not always possible. In some cases, to achieve radicality, it becomes necessary to excise part of the membranes along with the tumor. Especially in the case of germination of meningioma of the outer leaf of the dura mater.
6.1 Ventrolateral and lateral meningioma
The matrix, as a rule, is located in the root funnel zone. Often, the posterior roots of the spinal cord are, stretched on the surface of the spinal cord, and in most cases, it is possible to dislocate the tumor without damaging them. If the posterior root interferes with the removal of the tumor, it can be cut so as not to expose the spinal cord to unnecessary trauma. After the complete separation of the matrix from the inner layer of the DM, the tumor can be removed en bloc. This reduces the likelihood of tumor cells spreading along the CSF pathways.
6.2 Ventral meningioma
Removal of a tumor of this localization can be carried out by slicing, using not only microsurgical instruments but also an ultrasonic aspirator. Reducing the volume of the tumor avoids unnecessary traction of the spinal cord during removal.
6.3 Meningiomas with petrificates
The petrification site is resected along with the inner leaf of the dura mater. If the removal of the petrified part of the DM is associated with spinal cord traction, the affected DM is not removed in favor of subsequent radiation therapy.
6.3.1 Examination and follow up
With total tumor resection, it is possible to conduct dynamic monitoring with contrast MRI at intervals of 3, 6, and 12 months during the first year, then every 6 months for 2 years, then another 3 years with an interval of 12 months and every 5 years during life. If continued growth is detected, radiation therapy in the amount of 50–55 Gy is recommended [17].
For subtotal resection, the first MRI is recommended after 1–1.5 months followed by radiotherapy (the role of prophylactic radiotherapy remains controversial due to the continuing risk of recurrence) [16, 17, 18, 19].
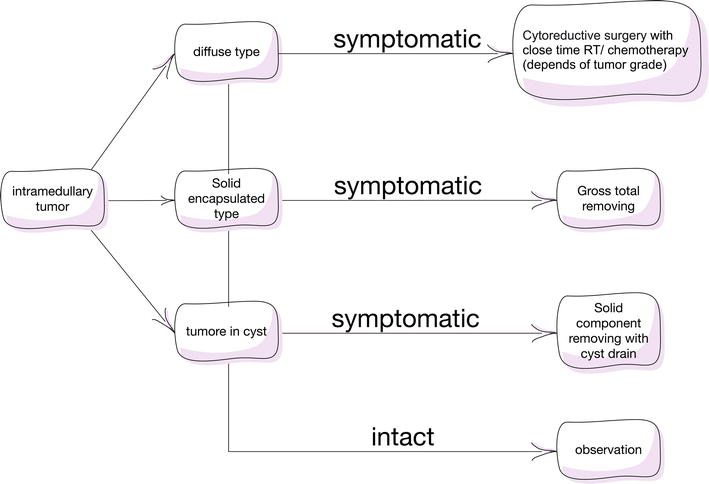
7. Intramedullary spinal cord tumors (IMSCT) associated with neurofibromatosis
Intramedullary tumors of the spinal cord are rare, mostly benign tumors, formed mainly from glial and ependymal cells, as well as cells of a different histological nature. Intramedullary tumors can lead to severe neurological deficits in the form of motor and sensory disorders, decrease, and loss of conduction function, which, in turn, leads to a significant decrease in the patient’s quality of life or death.
With NF I and type II, the risk of developing intramedullary tumors increases. For NF I type, astrocytomas are the most characteristic, while for NF II - epindymomas. It should be noted that intramedullary neoplasms are an extremely rare disease, its prevalence, according to various sources, does not exceed 0.3 per 100,000 population. According to M. Lee, Kuschel et al., the incidence of NF in patients with OMI is about 2.5%–3.9% [20, 21]. Data analysis of patients with NF D. Samartzis, M. Baser, D. Evans - IMSCT (ependymoma, pilocytic astrocytoma) occurs in 19 and 2.5% of cases, respectively.
The true prevalence in the population requires further screening studies. The survival of patients with intramedullary tumors varies depending on the degree of their malignancy, the five-year survival of such patients ranges from 28 to 90%. Gliomas account for up to 80% of all intramedullary tumors, among them the most common are astrocytomas (60–70%) and ependymomas. (30–40)%. Astrocytomas have a higher incidence in children than in adults, while ependymomas are more common in adults than in children. The sex-age characteristic of prevalence depends on the sample [22]. In 2011, R. Scott et al. published information on the treatment of 55 patients with ependymomas in NF-2. It is known that the risk of developing intramedullary tumors is higher in NF, but reliable statistics are not available at the moment. It is known that the risk of developing malignant neoplasms of the central nervous system directly correlates with the age of patients [1].
According to the available data, 20% of IMSCT cases lead to clinical manifestations and 13% have active growth without clinical symptoms. All this leads to the disability of patients. It is difficult to determine the degree of necessity and justification for intervention, especially in patients without clinical manifestations.
The gold standard for the treatment of intramedullary tumors is microsurgical removal using neuromonitoring [23]. The first stage of the operation is surgical access to the dura mater (DM), which is opened linearly. Then, access to the tumor is made through the posterior median sulcus (if the tumor is localized in the thickness of intact tissues; in some cases, a part of the tumor is located on the surface of the spinal cord and is visualized immediately upon opening the DM). The tumor is removed using microsurgical instruments under an operating microscope and neurophysiological monitoring. Tumor is removed within the morphologically unchanged brain tissue. The degree of surgical aggression is limited in the direction where the pronounced decrease in motor-evoked potentials occurs.
Treatment regimen is defined by the sum of clinical and imaging data. Recommendations for treatment are given by a neurosurgeon, a neurologist, and a radiation therapist.
In our opinion, the participation of a neurologist is necessary, due to the fact that patients with neurofibromatosis always have competing symptoms. Clinically intact intramedullary tumors need local treatment only if there is evidence of continued tumor growth.
Astrocytomas are predominantly benign intramedullary tumors characterized by bad delimitation from the healthy tissue of the spinal cord. Astrocytoma occurs most frequently in adult patients and is the second most common intramedullary tumor (30–35% of all spinal cord tumors and up to 60% in childhood; 0.5% of all CNS tumors. Tumors are more often located in the region of the thoracic spinal cord (67%), followed by the cervical region (30%) and lumbar thickening and cone (3%).
In childhood whole spinal cord can undergo tumor transformation. In adults, a total lesion spreading is extremely rare. In rare cases when we see an adult with total spinal cord involvement we can assume the long-term persistence of the pathology. Cervico- medullary location is common for children and young people either.
According to the literature, 70–80% of tumors have the first and second degrees of malignancy (grade I, II), that is includes a group of low-grade tumors, 15–25% are high-grade (Grade III), about 5% are tumors of the fourth degree of malignancy (glioblastoma) (Grade IV). Thus, 20–30% of tumors are classified as high-grade gliomas [22].
Almost 20% of these lesions are associated with the formation of syringomyelitic cysts. The main surgical challenge is to find the tumor’s “dissection plane” (the ability to separate the tumor from healthy tissue) for safe and radical removal. In patients with NF1, astrocytoma is most common in young men and rarely undergoes malignant transformation.
Generally, astrocytomas are found accentrically during routine imaging. Early symptoms are usually mild, leading to long and ineffective treatment. On MRI, a cystic component and poor contrast uptake are the most typical signs. Tumors can be visualized on T2W and T1-contrast series. The main imaging challenge is to differentiate tumor with demyelination and myelopathy. CT can be helpful to exclude ossified thoracic herniations resulting in myelopathy. The most significant sign is thevisualisation of the “mass effect” on MRI series (spinal cord diameter in the axial projection is increased in comparison to the neighboring area).
8.1 Nuances of surgery
There is no unified algorithm for the treatment of intramedullary astrocytoma. Surgical treatment of astrocytoma in patients with neurofibromatosis is complicated. Till that time when the symptoms appear, it is possible to discuss alternative therapy versus surgery. But the manifestation of symptoms often associates with big size of the tumor. And if the tumor was indicated in that stage, the surgery is not avoidance. The main problem of astrocytoma surgery is the absence of a transition zone in the region of the growing place – total tumor removal is impossible. So, facts and literature data indicate that surgical treatment is not absolute and does not predict good results. In the rule, as the main method of astrocytoma surgery is maximal cytoreduction or debulking. This method is the most effective for quickly increasing symptom redaction. An intact tumor in a patient with neurofibromatosis requires only observation, but if the tumor grows, radiation therapy is necessary.
Usually, the posterior approach applies. The millstone of approach is myelotomy through the posterior median sulcus. Good results depend on the quality and safety of myelotomy. Although identification of the midline is a difficult goal, it is possible by more wide laminectomy and the development of a healthy spinal cord area. It makes sure accurate midline identification and dissection with save of all vessels. In that case, if the tumor tissue is unidentified from spinal cord tissue, astrocytoma, as a rule, has poorly defined borders, and soft tumor tissue debulked by ultrasonic aspirator. This is the best method for reserving spaces inside the spinal cord creating. Some experts recommend debulking as treatment option if total tumor resection is associated with severe complications. It is desirable to leave a part of the tumor in order to avoid deterioration of the neurological status.
8.2 Examination and follow up
Contrast-enhanced MRI of the operation area is desirable to do within the first 24 hours, after 2 months, 6 months, 9 months, and 12 months, then, if there are no signs of progression, it is possible to continue monitoring at intervals of 6 months.
Ependymoma is the most spread intramedullary tumor in population. Ependymomas account for 60–70% of all spinal neuroepithelial tumors. The average age of the patients was 39 years; these tumors are found more often (about 60%) in men. Basically, intramedullary ependymoma grows from the cells of the central spinal cord canal and looks symmetrically along its walls. In 25–30% of patients, ependymomas may have asymmetric growth of varying severity and spread laterally in the spinal cord. Tumors that are large in diameter can push apart the thinned posterior columns and come to the surface of the spinal cord directly under the arachnoid. Extramedullary growth of tumor formations is extremely rare. Ependymomas are most often located in the cervical region of the spinal cord. Ependymomas of the upper cervical spinal cord segments may extend into the craniovertebral junction and reach the caudal fourth ventricle. Such an arrangement is rare. Ependymomas do not rise above McRay line, and only syringomyelic cysts located above the upper pole extend the spinal cord.
9.1 Nuances of surgery
Ependymomas are more often available for radical removal. It is possible, by course tumor growing into the central spinal canal. Several evidence demonstrate that more aggressive tumors have less connection with the surrounding healthy spinal cord tissue. Therefore, they have great potential for gross total removal.
At the same time, tumors with a high potential for malignancy (Grade III) are more often complicated by metastases.
The posterior approach through the median sulcus of the spinal cord is most preferred. If the tumor is tightly adjacent to healthy tissue of the spinal cord, it is preferable to perform intratumorally decompression as a first step. Then the tumor poles removing.
9.2 Observation and follow-up
Contrast-enhanced MRI of the operation area is desirable to do within the first 24 hours, after 2 months, 6 months, 9 months, and 12 months, then, if there are no signs of progression, it is possible to continue monitoring at intervals of 6 months.
Too often malignant and aggressive ependymoma (Grade II-III) are complicated by local tumor recurrence and metastatic spread along the spinal cord membranes.
So, if the local tumor recurrence/continued growth/spread tumor features or histological malignancy symptoms were detected – a radiologist’s participation is necessary. Follow-up includes postoperative craniospinal radiotherapy, regardless of whether continued growth or tumor recurrence after.
MPNST is a rare complication of neurofibromatosis and accounts for 5–10% of all soft tissue sarcomas [24]. Frequency of occurrence - 1 per 1 million population per year [25]. Approximately 50% of MPNST cases are complicated by metastases within 2 years of diagnosis [26]. The majority of MPNST arise from peripheral nerves and nervous plexuses (sciatic nerve, brachial plexos truncs, sacral plexus). The incidence of MPNST of intracranial localization in the world population is less than 5%. The most important etiological factors in the development of MPNST are neurofibromatosis (NF) type 1 and radiological treatment for other cancers in history. MPNST is developed in 8–13% of patients with type I NF during their life [27]. In 10% of cases of MPNST appearance was associated with radiotherapy [28]. According to various observations, malignancy of neurofibroma occurs approximately 15 years after its detection.
The WHO classification of histological types of MPNST:
mesenchymal
glandular
epithelioid
neuroepithelial
triton
Clinical manifestations depend on the location of the tumor. The rapid development of symptoms is characterized (within several months), severe pain, and neurological deficits of varying severity. There are two forms according to localization:
skin form
deep formation
Too long time of observation and avoidance of surgery leads to metastases appeare may occur in regional lymph nodes and internal organs (lungs, liver, bones, etc.). Metastasis is carried out both hematogenously and lymphogenously.
10.1 Nuances of surgery
The main treatment for the primary tumor is en block resection. The main goal of surgery is to remove tumor. It provides the best result in terms of local recurrences and distant metastases. Localization of the tumor is very important, be the course of the totality of surgical removal depends on the growing place and relative of the tumor to surrounding tissue. The boundaries of the tumor are poorly defined, so often removal is partial [29].
The modern surgical approach to the treatment of patients with locally aggressive (without distant metastases) types of MPNST involves a complete cut of tumor. Thus, amputation of the limb, or very aggressive en block resection of the tumor within the margin of surrounding tissues of 2.5–4 cm, has always been considered the main method of treating MPNST. Alternative treatment as adjuvant methods of radiation and chemotherapy is possible but limited. So the view on surgical treatment has now not radically changed. For example, the location of MPNST in the limbs allows planning a wide gross total resection while maintaining the function of the limb. At the same time, this cannot prevent local recurrences or generalization of the process, since even the presence of an adequate resection margin is not a guarantee of total tumor removal. So early lymphatic metastasis is the indication for amputation of limb. The defeat of regional lymph nodes is rarely detected. Unfortunately, amputation is a limited method if the tumor is located in or around the spine.
10.1.1 Radiation therapy
Radiation therapy has become an integral part of the control of local recurrence in most soft tissue sarcomas (Suh JS) and can be used as adjuvant treatment or intraoperatively. But last publications show unhelpful of radiation methods for tumor cells of MPNST as to local control as in general spread [16].
10.1.2 Chemotherapy
Systemic medication treatment is used in the stage of generalization of the process, but there is no convincing evidence of effectiveness [30, 31].
10.1.3 Prognosis
The median survival time for patients with MPNST is 32 months [31]. The median life expectancy from the time of diagnosis, according to some authors, is 3 months. The causes of death in patients are usually metastases to the lungs and liver. Distant metastases (target organs - pleura, lungs, liver, adrenal glands, pia mater, cerebral hemispheres) are detected within 2 years from the date of diagnosis in 63% of patients. Overall 2-year and 5-year survival rates are 57% and 39% with treatment [31]. The frequency of local recurrences of MPNST is 65%.
11. Patients with multifocal symptomatic tumors
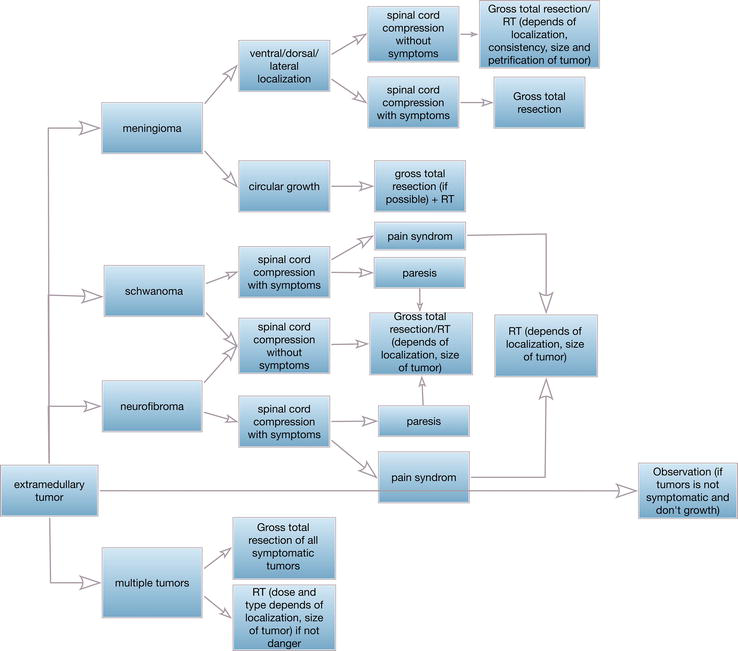
In patients with neurofibromatosis, meningiomas, and schwannomas appear with equal frequency. The sequence of surgical interventions strongly depends on the level of a symptomatic tumor on the spinal cord and brain tumors, in the presence of simultaneously symptomatic tumors at different levels, simultaneous surgery is not always preferable and a higher tumor is removed, or the one that causes the most threatening symptoms. Simultaneous operations are performed if the tumors are equally dangerous to the life or health of the patient. Presented lower algorithm allows to avoid risks (Figure 4).
Figure 4.
Extramedullary surgical treatment algorithm.
Conflict of interest
The authors declare no conflict of interest.
References
1.Gutmann DH et al. Neurofibromatosis type 1//Nature Reviews Disease Primers. – 2017. – Т. 3. – №. 1. – С. 1-17
2.Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Research. 2005;65:2755-2760
3.Listernick R et al. Optic gliomas in children with neurofibromatosis type 1//The Journal of Pediatrics. - 1989. - T. 114. - No. 5. - S. 788-792
4.Gutmann DH et al. Gliomas presenting after age 10 in individuals with neurofibromatosis type 1 (NF1) //Neurology. – 2002. – Т. 59. – №. 5. – С. 759-761
5.Strowd III RE et al. Histologically benign, clinically aggressive: Progressive non-optic pathway pilocytic astrocytomas in adults with NF1//American Journal of Medical Genetics Part A. – 2016. – Т. 170. – №. 6. – С. 1455-1461
6.Tonsgard JH. Clinical manifestations and management of neurofibromatosis type 1//Seminars in Pediatric Neurology. – WB Saunders, 2006. – Т. 13. – №. 1. – С. 2-7
7.Ruggieri M, Praticò AD, Evans DG. Diagnosis, management, and new therapeutic options in childhood neurofibromatosis type 2 and related forms//Seminars in Pediatric Neurology. – WB Saunders, 2015. – Т. 22. – №. 4. – С. 240-258
8.Martuza RL, Eldridge R. Neurofibromatosis 2: (bilateral acoustic Neurofibromatosis). The New England Journal of Medicine. 1988;318:684-688
9.Campian J, Gutmann DH. CNS tumors in neurofibromatosis//Journal of Clinical Oncology. – 2017. – Т. 35. – №. 21. – С. 2378
10.Tish S et al. The epidemiology of spinal schwannoma in the United States between 2006 and 2014//Journal of Neurosurgery: Spine. – 2019. – Т. 32. – №. 5. – С. 661-666
11.Sridhar K, Ramamurthi R, Vasudevan MC, et al. Giant invasive spinal schwannomas: Definition and surgical management. Journal of Neurosurgery. 2001;94:210-215
12.Konovalov AN, Golanov AV, Gorlachev GE, Kornienko VN, Trunin YY, Kotel'nikova TM, et al. Application of robotized system for radiosurgery CyberKnife for treatment of neurosurgical patients. Zhurnal Voprosy Neirokhirurgii Imeni N.N. Burdenko. 2012;76(1):3-12 (In Russ.)]
13.Fehlings MG et al. Risk factors for recurrence of surgically treated conventional spinal schwannomas: Analysis of 169 patients from a multicenter international database//Spine. – 2016. – Т. 41. – №. 5. – С. 390
14.Baser ME, Friedman JM, Aeschliman D, Joe H, Wallace AJ, Ramsden RT, et al. Predictors of the risk of mortality in neurofibromatosis 2. American Journal of Human Genetics. 2002;71:715-723
15.Goutagny S, Kalamarides M. Meningiomas and neurofibromatosis//Journal of Neuro-Oncology. – 2010. – Т. 99. – С. 341-347
16.Gottfried ON et al. Spinal meningiomas: Surgical management and outcome//Neurosurgical Focus. - 2003. - T. 14. - No. 6. - S. 1-7
17.Golanov AV et al. Stereotactic radiotherapy for spinal meningiomas and neurinomas//Problems of Neurosurgery. – 2015;78(1):3-11
18.Goldbrunner R et al. EANO guidelines for the diagnosis and treatment of meningiomas//The Lancet Oncology. – 2016. – Т. 17. – №. 9. – С. e383-e391
19.Yolcu YU et al. Trends in the utilization of radiotherapy for spinal meningiomas: Insights from the 2004-2015 National Cancer Database//Neurosurgical Focus. – 2019. – Т. 46. – №. 6. – С. E6
20.Kushel YV, Belova YD, Tekoev AR. Intramedullary spinal cord tumors and neurofibromatosis//Zhurnal Voprosy Neirokhirurgii Imeni NN Burdenko. – 2017;81(1):70-73. DOI: 10.17116/neiro201780770-73
21.Lee M, Rezai AR, Freed D, Epstein FJ. Intramedullary spinal cord tumors in neurofibromatosis. Neurosurgery. 1996;38(1):32-37
22.Samartzis D et al. Intramedullary spinal cord tumors: Part I—Epidemiology, pathophysiology, and diagnosis //Global Spine Journal. – 2015. – Т. 5. – №. 5. – С. 425-435
23.Hadley MN et al. Guidelines for the use of electrophysiological monitoring for surgery of the human spinal column and spinal cord//Neurosurgery. – 2017. – Т. 81. – №. 5. – С. 713-732
24.Woodruff JM. Malignant tumors of peripheral nerve sheath buttock and lower extremity a study of 43 cases. Cancer. 1990;66:1253-1265. DOI: 10.1002/1097;2-p
25.Angelov L, Guha A. Peripheral nerve Tumors. Neuro oncology foundation. In: Bernstein M, editor. Ms. Berger. New York: Theme Publishers; 2000. pp. 434-444 One
26.Karmody CS. Malignant schwannoma of the trigeminal nerve. Otolaryngology - Head and Neck Surgery. 1979;87:594-598
27.Brenner W, Gawad Ka FR, Hagel C, von Deimling A, de Wit M, Boucher R, et al. Prognostic value of PET FDG in patients with neurofibromatosis 1-type and malignant tumors of peripheral nerve sheath. European Journal of Nuclear Medicine and Molecular Imaging. 2006;11:1-5
28.Amin SA, Flanagan A, Patterson D, Lehovsky J. Radiation therapy - induced malignant tumors of the peripheral nerves of the caudal tail. Spine. 2004;29(21):E506-E509
29.Carli M, Ferrari A, Mattke A, Zanetti I, Casanova M, Bisogno G, et al. Pediatric malignant peripheral nerve sheath tumor: The Italian and German soft tissue sarcoma cooperative group. Journal of Clinical Oncology. 2005;23(33):8422-8430
30.Action DV, PR R, Hornicek J, Gebhardt MC, Mankin HJ. Survival data for patients with malignant schwannoma. Wedge Orthopedic Relative Research. 2004;426:69-73
31.Masaru F. Descended from Beppu g, Asanuma to Fuji K: A malignant tumor of the peripheral nerve sheath responds to chemotherapy. Journal of Bone and Joint Surgery (British). 2004;86(1):113-115
Written By
Vasiliy Korolishin and Yuri M. Poluektov
Submitted: 01 February 2023Reviewed: 03 February 2023Published: 25 April 2023
 Open access peer-reviewed chapter
Open access peer-reviewed chapter