Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
Reducing bone density and bone quality with increasing propensity of skeletal fracture are the main symptoms of osteoporosis. Disruption of the fine balance between bone formation and resorption leads to this progressive condition, which affects 50% of women and 30% of men after the age of 50. Aging, reduced nutrient (vitamin D and calcium) uptake, suppressed production of estrogen, and primarily, the dysregulation of cytokine balance leads to the pathophysiology of the disease. Hence, immunomodulation of bone remodeling is tightly controlled by the cytokine profiles, epigenetic marks, and metabolic programs of the involved cells, thus playing a key role in the prognosis of osteoporosis. In this chapter, we highlight this intricate interplay between the immune system, the associated cytokines, and bone remodeling.
*Address all correspondence to: ritobrata@gmail.com
1. Introduction
In healthy people, bone is an active tissue that is constantly replenished, in great part thanks to the immune system. Numerous inflammatory bone disorders are connected to any imbalance between the processes of bone production and bone resorption. The immune system is crucial for both tissue growth and bone resorption. In addition to immune cells and cytokines, bone cells, such as osteoblasts (OBs), osteoclasts (OCs), and osteocytes (OSs), also have an impact on how immune cells function on a cellular level. As a result, the immune microenvironment plays a critical role in regulating the rate at which bones heal, mend, and regenerate.
OBs, OCs, OSs, and other fundamental components make up bone. As a dynamic organ, the skeletal system and immune system interact intricately, maintaining a balance between bone creation and bone resorption. In addition to coexisting in the bone marrow (BM) cavity, bone and immune cells also share a range of regulatory molecules and progenitor cells. The regulation of bone homeostasis by immune cells is known to be influenced by bone cells. Likewise, the proliferation and differentiation of immune cells is also modulated by bone cells. As a result, the term “osteoimmunology” was created to emphasize the interaction between the immunological and skeletal systems [1].
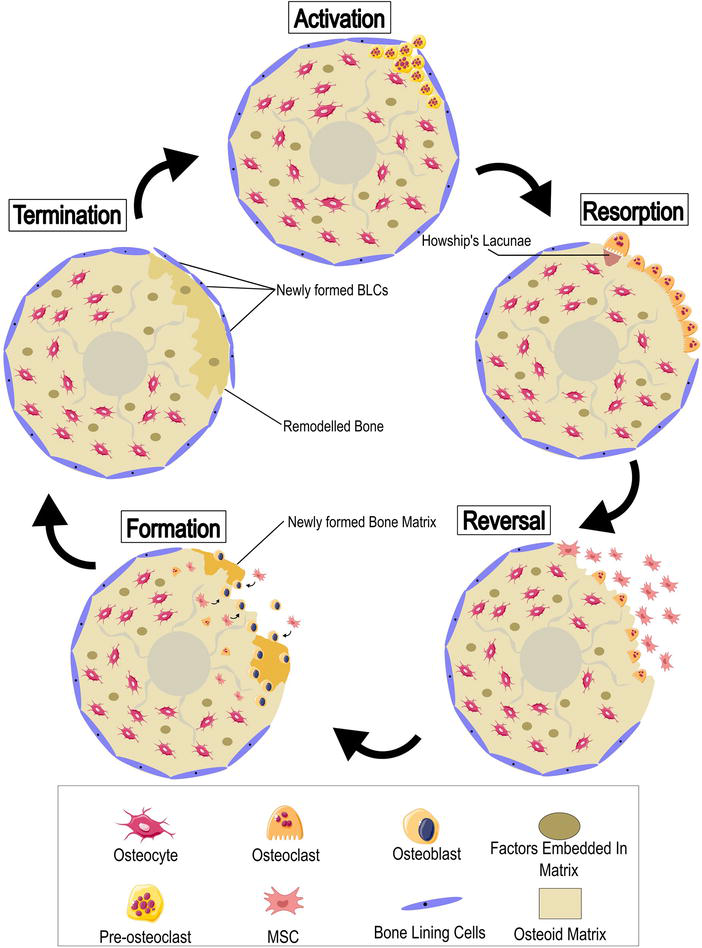
The skeleton is a very dynamic structure that changes throughout the course of a person’s life in order to maintain bone density and mineral homeostasis by continuously regenerating the bone matrix. The dynamic equilibrium that results from a succession of well-planned biological events—all of which are controlled by intricate interactions between the many cell types that are found in bone—is physiological bone remodeling. OBs and OCs are the primary cells involved in bone remodeling. Additionally, OSs, which operate as mechanical sensors, are crucial to the process of bone remodeling, whereas bone lining cells (BLCs) start the process by degrading the matrix. The five successive steps of the bone remodeling cycle are activation, resorption, reversal, formation, and termination [2].
An immune response is immediately triggered by tissue damage, and it typically lasts for days or even weeks. The two main effects of immune cells on bone regeneration are the catabolic effect of chronic inflammation during bone resorption and the anabolic effect of acute inflammation during bone regeneration. By encouraging the proliferation and osteoblastic differentiation of mesenchymal progenitor cells, acute inflammation, for instance, can result in the synthesis of chemokines that speed up bone formation during the early stages of bone fracture healing [3]. While an adaptive immune response is significant during late bone remodeling, an acute inflammatory response is essential at the start of bone regeneration. When the inflammatory response is under control, it can aid in bone regeneration; however, when the inflammation is out of control, it can be harmful to bone tissue. For an anabolic milieu that promotes bone growth and repair, the management of inflammation is essential.
This chapter delineates the immunological events leading to bone remodeling and the pathophysiology of osteoporosis. Detailed emphasis has been provided pertaining to the activity of immune cells leading to bone remodeling. Additionally, the chapter incorporates a separate section on the implication of bone generative and bone degenerative cytokines in bone remodeling. In the biological signal regulatory network, epigenetics primarily serves as a posttranscriptional controlling mechanism. The final section of the chapter discusses research developments on epigenetic mechanisms in the osteogenic differentiation and the pathogenesis of osteoporosis to provide a new direction for the treatment of diseases related to bone metabolism, considering the significant role of epigenetic mechanisms in the regulation of bone metabolism.
Our bones encompass a vast and intricate network of connective tissue, which provides mechanical support (skeletal system), protects our internal organs, and forms a dais for smoother functioning of numerous bodily functions. Although primarily formed of inorganic calcium and phosphate, it is a site of living cells, tissue, and a complex network of highly active metabolic processes [4]. Despite being hard and resilient, bones can be exceptionally adaptive and can restructure themselves under varying mechanical forces and differential loading [5].
Bone remodeling can be defined as the spatiotemporally regulated physiological phenomenon, which involves de- and re-construction of bone matrix to ensure precise turnover and proper distribution of osteogenic tissue throughout the body of the organism. Bone resorption and formation are tightly conjoined to ensure that under homeostatic conditions, there is minimal loss of bone mass in a healthy individual [5]. Bone remodeling assists in cleaning up the circulating blood by absorbing the toxic chemicals (like drugs), radioactive debris, heavy metals, etc. by temporarily storing them in newly ossified matrix and slowly releasing it during the latter phases of de-construction [6]. Ossified tissue also serves as a reservoir of two very important materials, namely calcium and phosphorus, and bone remodeling helps in maintaining a homeostatic level between the blood and the bone [6].
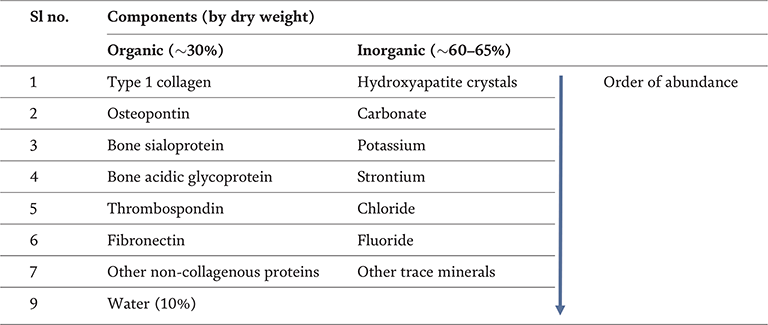
Bones, unlike other forms of connective tissue, consist of cells that make up the living organic matter, and the calcified matrix serves as the inorganic counterpart. The exact composition of the bone tissue is summarized in Table 1. Compact bones are involved in the bulk of skeletal structure supplying most attachment points for muscles and tendons, while cancellous bones are found along the inner layers of the compact bones and consist of trabeculae. These cancellous bones house the marrow, which is primarily involved in the metabolic and hematopoietic functions. Effective remodeling of the bone tissue requires the involvement of different types of cells, which together make up the most rudimentary functional structure called the BMU or basal multicellular unit [4] (Figure 1). BMUs primarily consist of BLCs, OBs, OSs, and OCs. At any given moment, there are about 1–2 million active BMUs engaged in remodeling, although they may be working asynchronously at different sites. Initial phases of bone remodeling ensure that BMUs can start excavating micro-tunnels along the poles of either the trabecular or cortical bones. This is followed by the bone resorption phase, which involves recruitment of giant multinucleate osteoclasts and decay of inner lining of the bone to form empty channels or tunnels called Howship’s lacunae [4]. In the latter part of remodeling process, the function of OCs is restricted in a manner resembling the reversal of their initial activity. Osteoprogenitors and post-osteoclastic cells are recruited to the site of remodeling [4], and they start reforming the lining (“cementing line”) of the lacunae, where new bone formation can take place. Additionally, the reabsorbed surfaces are cleaned up by the BLCs and resident macrophages. Finally, the differentiation of new osteoblasts from pre-osteoblastic mesenchymal stem cells takes place [8]. These osteoblasts restart ossification of the inner lining, as well as the mineralization of osteoid tissue that they have synthesized themselves. More importantly, tissue-resident immune cells, including B-cells, effector and regulatory T-cells, dendritic cells, natural killer cells, myeloid cells, etc. also impact the bone remodeling to maintain the homeostatic environment [9]. Thus, BMUs complete one cycle of bone remodeling, ensuring that refilling of the excavated channels occurs smoothly [4].
Table 1.
The different components of osteogenic tissue in order of their abundance [7].
Figure 1.
Process of bone remodeling and bone turnover mediated in five distinct stages, namely; activation, resorption, reversal, formation, and termination. Each of these stages presents with unique features that have been highlighted. The first phase involves disruption of the BLC layer and infiltration of pre-osteoclastic stem cells, which differentiate into OCs. These OCs then start degrading the bone. The reversal phase witnesses the restricted activity of the OCs with the simultaneous influx of MSCs, differentiating into OBs. The OBs dominate the formation phase and finally transition into termination phase, whereby the BLCs reform and the cycle of bone remodeling ends.
2.2 Role of bone remodeling in osteoporosis
The word osteoporosis can be viewed as an assembly of two parts namely, “osteo” denoting bone and “poros” meaning pores or holes. Literally, the word translates to “holes in the bones”. Hence, osteoporosis can be described as an assemblage of clinical conditions that primarily culminates into a significant decrease in bone density, skeletal deformity, and increased susceptibility to fractures. It is the most common form of bone disease in adults and over the age of 50 [10]. It is known to affect all genders and races, although females have higher disposition to develop this disease after menopause due to hormonal imbalance. This condition has no outright manifestations, and once detected, severely hampers the quality of life of the patients. In severe cases, it can result in other secondary complications and can even be the cause of mortality in patients. Affected individuals experience pain, inability to move freely, fractures from otherwise harmless impacts, and various physical restrictions.
Extensive bone remodeling occurs throughout the lifetime of an individual organism. As stated earlier, loss of osteogenic tissue is supplemented by the growth of new tissue, which ensures that there is no alteration in net bone mass [3]. A multitude of factors, including environmental, genetic, nutritional, hormonal, and physical strains, influence this bone formation and its shape. With aging, the degree of remodeling is affected and bone loss starts to occur. Various elements heighten this process of resorption over formation. This triggers fluctuations in the microarchitectural level, resulting in bone loss over time. Loss of individual trabecular bone plates structurally weakens the skeletal framework with significantly diminished bone density [8]. Evidently, this elevates the risk of fractures, which is further exacerbated by other age-related declines in bodily functions.
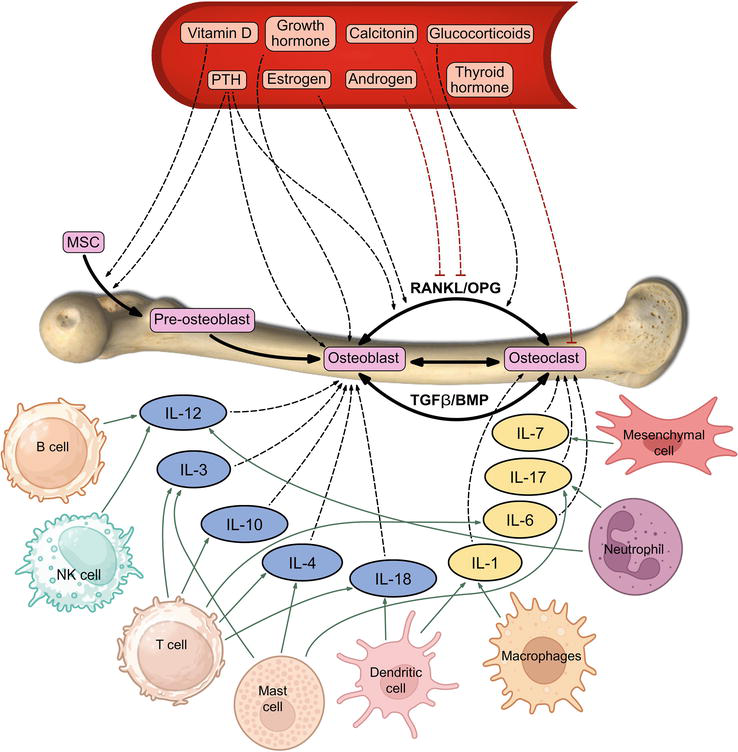
The coordinated process of bone formation and degradation is regulated by various local and systemic factors in a well-synchronized manner. Apart from age, lifestyle, diet, and genetic background, there are plethora of factors that regulate this harmonious cycle. While parathyroid hormone, estrogen, progesterone, and androgen are the hormones, vitamin D (Vit D) and calcium form the major micronutrients to regulate the concert of the bone remodeling process through paracrine, autocrine, or endocrine actions. Several growth factors and cytokines also play a pivotal role in the bone remodeling cycle (Figure 2).
Figure 2.
The complex process of bone remodeling is governed by various cytokines, micronutrients, and hormonal factors. Immune cells, such as dendritic cells, macrophages, neutrophils, mesenchymal cells, and T cells, secrete cytokines, including IL-1, IL-6, IL-17, IL-7, etc., which play a crucial role in initiating the process of bone degeneration. Contrastingly, some cytokines such asIL-12, IL-3, IL-10, IL-4, IL-18 etc. contribute to osteoblastic differentiation.
Apart from these factors, the immune system significantly impacts the regeneration and repair of osteogenic tissue. The ability of bone tissue to regenerate is determined by the immune microenvironment’s role in bone tissue healing, repair, and regeneration. Disruption of this well-synchronized process aids in the development of several degenerative diseases, such as osteoporosis and osteoarthritis. Thus, immunologic modulations can be attributed to the maturation of osteoporosis, and the varied functions of the different immune cells, in this regard, can be correctly and collectively termed as “immunoporosis” [11, 12]. The detailed discussion on the role of hormonal factors, micronutrients, and growth factors are beyond the scope of this chapter. Here, our focus is on the immunoregulation of bone remodeling process and how it affects immunoporosis [12]. A comprehensive overview has been given in Table 2.
The mechanism of bone remodeling is a fine balance between bone regeneration and degeneration, which has been regulated by diverse factors. The cells present in the bone microenvironment, such as bone marrow stromal cells, OBs, OCs, macrophages, and T lymphocytes produce these peptide factors, which engages with several cell signaling pathway to directly or indirectly modulate the osteoclastogenesis and osteoblastogenesis processes. Even though most of the cytokines have biphasic impacts, we can still group them into two different classes: (a) bone regenerative cytokines and (b) bone degenerative cytokines. In the subsequent sections, we discuss them in more detail.
4.1 Bone regenerative cytokines
4.1.1 IL-3
Activated T lymphocytes, mast cells, and OBs producing this cytokine share its βc subunit of receptor with GM-CSF and IL-5 receptors. IL-3 inhibits osteoclastogenesis through different pathways. This cytokine blocks the RANKL-dependent NF-κB nuclear translocation process. It inhibits the phosphorylation of IκB of the RANKL-induced pathway in pre-OC cells, which prevents the further development of the OCs [36]. IL-3 also downregulates the expression of TNFRs (TNFR1 and TNFR2) on OCs to suppress TNF-dependent OC differentiation and bone degeneration process [37, 38]. The mRNA and protein levels of PU.1, c-FOS, and c-Fms also get downregulated substantially by IL-3, which, in turn, suppresses the maturation of bone monocytes and OC precursor cells [39, 40, 41]. It directly targets the OB-specific genes, such as alkaline phosphatase, collagen type-I, OCN, OPN, and some transcription factors, such as Runx-2 and OSX. This cytokine also targets JAK/STAT signaling pathway to promote OBs differentiation [42]. IL-3 has the capability to enhance RANKL expression, independent of OPG expression [13]. This cytokine governs two functional variants of RANKL by decreasing the activity of metalloproteinases, encompassing ADAM10, ADAM17, ADAM19, and MMP3, to lessen the expression of soluble RANKL and enhance the expression of membrane-bound RANKL. This modulation is carried out through the JAK2/STAT5 signaling pathway, thus reinstating the reduced RANKL-OPG ratio in adult mice [13].
4.1.2 IL-4
Th2 cells predominantly secrete this pleiotropic cytokine that acts as an immunomodulatory cytokine but also as a potent inhibitor of osteoclastogenesis. IL-4 also targets the phosphorylation of IκB to inhibit NF-κB translocation in a STAT6-dependent manner, which directly inhibits both OCs maturity and bone resorption activity [14]. Like IL-3, it also blocks RANKL and TNF-α-induced osteoclastogenesis via the MAPK signaling pathway [43]. IL-4 also suppresses transcription factors nuclear factor activated T cells c1 (NFATC1) of RANKL-induced signaling pathway through STAT6, which further inhibits OCs formation [44]. The endothelial cells present in the bone’s vascular system release osteogenic cytokines and hormones that regulate the process of bone development, remodeling, and repair [45]. IL-4 and its closely associated counterpart, IL-13, can indirectly impede the formation of OCs by activating the STAT6 pathway. This activation prompts endothelial cells to release the hormone OPG, which is known for its role in safeguarding bone health [17]. IL-4 is also indirectly involved in inducing osteoblastogenesis. RANKL, in the presence of M-CSF, induces OCs formation, but in the presence of GM-CSF and IL-4, the very same cocktail helps to induce the TNF-α converting enzyme, which further sheds the M-CSF receptor and stimulates the monocyte to differentiate into dendritic cells rather than OC cells [46].
4.1.3 IL-10
Th2 and Treg cells are the main sources of this anti-inflammatory cytokine. It acts as a direct inhibitor of the RANKL-OPG-RANK axis [15]. This cytokine induces OPG expression, which, in turn, blocks soluble RANKL from binding with RANK and further inhibits OCs differentiation [47]. IL-10 also suppresses RANKL-induced OCs maturation and calcium signaling through triggering receptors expressed on myeloid cells 2 [TREM-2] transcriptional inhibition [48]. IL-10 also suppresses NFATC1 expression through inhibition of c-Fos, and c-Jun nuclear translocation [49]. Apart from the details mentioned earlier, the reduced expression of IL-10 cytokines in individuals with osteoporosis serves as additional confirmation of its anti-osteoclastogenic characteristics [50]. Even studies with IL-10-deficient mice show lower bone mass, reduced bone formation, and increased bone fracture [51]. Regarding osteogenic differentiation, IL-10 restrains bone marrow osteogenic function by impeding bone mineralization in mouse bone marrow cells. Moreover, it hampers the production of bone proteins such as ALP, type I collagen, and OCN [52].
4.1.4 IL-11
Another important player in bone remodeling, this cytokine is also known as adipogenesis inhibitory factor [AGIF] [53]. It can modulate the osteoclastogenesis process in both RANKL-dependent and independent pathways [54]. Research has shown that mice with IL-11 overexpression exhibit thickened bone cortex, increased bone mass, and enhanced bone strength; while the IL-11R knockout mice are characterized by impaired bone formation and increased adipose in bone marrow. This study indicates the positive role of IL-11 in osteoblastogenesis [55]. Compared to this, additional studies have also claimed that IL-11 can also promote osteogenesis and inhibit adipogenesis through the WNT signaling pathway [3].
4.1.5 IL-19
This cytokine belongs to the IL-10 family as well, and it operates in a comparable fashion to its counterpart, IL-10. It keeps the OCs in their precursor state by targeting and suppressing NF-κB and p38MAPK activation and c-Fos expression [19]. IL-19 also blocks OC-specific RANKL pathways. Furthermore, it can enhance the release of osteoclastogenic cytokines such as IL-1β, IL-6, and TNF-α in osteoporosis conditions. However, little study has been done on how IL-19 affects osteoporosis [56].
4.1.6 IL-12
Functioning as an anti-osteoclastogenic cytokine, it hinders NFATC1 to impede osteoclastogenesis driven by RANKL [57]. IL-12 also stimulates FASL-dependent apoptosis in OB cells [16]. Along with IL-18, IL-12 also induces an OC apoptosis mechanism, which reveals the anabolic role of this cytokine in the bone remodeling procedure.
4.1.7 IL-18
IL-18 is a pro-inflammatory cytokine that operates in synergy with TNF-α and IL-12 and is responsible for stimulating the generation of nitric oxide. This, in turn, expedites the apoptosis process in osteoclast cells [18]. Furthermore, it stimulates T cells to produce IFN-γ and GM-CSF for inhibiting OC maturation indirectly [58]. Studies conducted using mice that have undergone ovariectomy (OVX), display that administering IL-18 binding protein (IL-18BP), which acts against IL-18, results in the restoration of bone loss [59]. Moreover, the observation of reduced IL-18BP levels in the serum of women with osteoporosis further suggests the role of IL-18 as a cytokine that promotes bone regeneration [56].
4.1.8 IL-13
Th2-specific anti-osteoclastogenic cytokine IL-13 acts in a very similar way to IL-4. It triggers the production of OPG through STAT6 activation to inhibit OCs proliferation [17]. Both IL-13 and IL-4 are capable of blocking OB cyclooxygenase-2 (COX-2)-dependent prostaglandin synthesis, which, in turn, inhibits bone resorption [60].
4.1.9 IL-27
IL-27 is a member of the IL-6/12 cytokine family. This bone-regenerating cytokine blocks RANKL-dependent c-Jun and NFATc1 expression via MAPK and NF-κB pathway. It also suppresses NFATc action by downregulating TREM2 expression [61]. Additionally, it inhibits OCs maturation in c-Fos-dependent manner [20]. IL-27 suppresses RORγt-dependent IL-17 differentiation and induces IL-10-producing Treg cell formation by activating Egr2 while inhibiting OBs apoptosis in a similar way [62]. Additionally, the findings on female osteoporosis patients suggest that the blood levels of the RANKL repressor Id2 got increased and that Egr2 and IL-27 levels dropped in comparison, demonstrating the anabolic role of IL-17 in the bone regeneration process [62].
4.1.10 IL-29
IL-29 is a family member of the interferon family, which activates the JAK/STAT signaling pathway. Dendritic cell producing this cytokine inhibits OC formation and bone degeneration both in vivo and in vitro [21]. Similar to other bone-regenerating cytokines, it also has RANKL-dependent OC maturation and blocks NF-κB activation, and NFATC1 translocation [63].
4.1.11 IL-33
IL-33, belonging to the IL-1 family, possesses osteoprotective capabilities unlike IL-1. It suppresses osteoclastogenesis by at least three mechanisms. IL-33 inhibits RANKL-dependent NFATC1 expression via the ST2 receptor pathway, which, in turn, suppresses osteoclast formation [64]. In a very similar way, it induces OCs apoptosis by increasing BAX, Fas, and FASL expression [23]. An in vivo study has confirmed that IL-33 stimulates the mRNA expression of bone regenerative cytokines such as IL-4, IL-10, IL-13, and GM-CSF [65]. It also synergistically acts with Vit D in the repression of the OCs maturation [66]. Research has also suggested that mice exhibiting elevated levels of TNF-α can experience recovery through IL-33 treatment [67]. Moreover, IL-33 encourages OBs activity, facilitates the deposition of matrix minerals, and decreases the levels of sclerostin mRNA in primary OBs subjected to prolonged ascorbate treatment [68]. According to a recent study, postmenopausal women with osteoporosis have significantly lower IL-33 levels than the healthy controls [69].
4.1.12 IL-35
This anti-inflammatory and immunosuppressive cytokine belongs to the IL-12 family, which directly inhibits OCs formation, TNF-induced osteoclastogenesis and bone resorption. IL-35 suppresses NFATC1, c-Fos, and TRAP via the NF-κB and MAPK pathways. By activating JAK1/STAT1, this action not only prevents the development of OCs produced by TNF but also promotes apoptosis [24]. IL-35 also shows an inhibitory effect on RANKL and M-CSF-induced Th17/IL-17 axis-regulated OC formation and bone degeneration [70]. Osteoporosis occurs due to the imbalance of bone and adipogenesis. MSCs have the ability to differentiate either in OBs or adipocyte cells. Research has found that IL-33 stimulates MSC differentiation toward OBs formation by inducing the expression of OB-specific factors, such as β-catenin and Axin2. It also suppresses the apoptosis of MSCs and differentiation into adipocyte cells [71]. In contrast, IL-33 was also found to induce phosphorylation of osteoclast-specific signaling molecules such as Syk, phospholipase Cγ2, Gab2, MAP kinases, TAK-1, and NF-κB and transcription factors such as TRAF6, NFATc1, c-Fos, C-Src, histone-K, and calcitonin receptor, which leads to bone resorption [72].
4.2 Bone degenerative cytokines:
4.2.1 TNF
TNF family, consisting of TNF-α and TNF-β, plays a significant bidirectional role in bone erosion. TNF stimulates OCs development, even via RANKL/RANK-independent axis. TNF-α initiates the activation of stromal cells and OBs. This cascade indirectly leads to the upregulation of RANK in OC precursors and subsequently promotes the process of OC formation through M-CSF [73]. Instead of activating the RANK/RANKL axis, TNF-α can induce OCs differentiation through the sequential activation of NF-κβ, p50/52, c-Fos, and NFATC1 [74, 75]. TNF-α can also trigger IL-1β production by pre-OC cells, which, in turn, stimulate differentiation of OC cells [76]. Tumor necrosis factor receptor-associated factors (TRAFs) play a significant role in bone remodeling. They possess the ability to initiate OCs formation in response to TNF. Notably, among these factors, TRAF6 holds the capacity to influence and regulate the process of osteoclastogenesis [77]. Some studies have demonstrated that RANKL might trigger the degradation process of TRAF3 to enhance the OCs differentiation process in TRAF-independent manner [78]. A lower concentration of TNF stimulates osteoblastogenesis, while a higher concentration strictly inhibits it. TNF also inhibits both bone regenerating factors IGF-1 and RUNX2 [79].
4.2.2 IL-1
The bone, degenerative cytokine IL-1 comprises of IL-1α and IL-1β. This class of cytokines induces osteoclastogenesis by activating OCs differentiation, multinucleation, and survival [80]. IL-1β has both direct and indirect effect on the bone remodeling process. It can indirectly boost the TNF-mediated osteoclastogenesis process in addition to directly upregulating RANKL synthesis to improve OC differentiation [81]. Through a process involving the elevation of M-CSF and PGE2 expression, as well as the concurrent downregulation of OPG expression in OBs, IL-1 has the capacity to induce the production of OCs-like cells (OLCs) [82]. IL-1 could trigger OCs differentiation in bone marrow-derived macrophages (BMMs) through a pathway that does not rely on the RANKL/RANK interaction. This occurs via IL-1/IL-1R1 signaling and involves the activation of genes associated with osteogeneses such as NF-κB, JNK, p38, ERK, and the microphthalmia transcription factor (MITF), which, in turn, induces the differentiation of OCs [27]. However, this mechanism does not increase the expression of c-Fos or NFATc1, which both must remain at a specific baseline level for this process to occur [27]. IL-1 not only induces OCs formation but also suppresses OCs maturation and has an impact on the bone fracture healing process [27].
4.2.3 IL-6
Secreted by OBs, bone marrow stromal cells, OCs, macrophages, and T cells, this pleiotropic cytokine transmits its signal through IL-6R, which is shared by a distinctive set of factors such as IL-11, oncostatin M (OSM), leukemia inhibitory factor (LIF), cardiotrophin (CT-1), ciliary neurotrophic factor (CNT)] cardiotropin such as cytokine factor 1 (CLCF1), neuropoietin (NP), IL-27, and humanin [28]. IL-6 can indirectly promote osteoclastogenesis by activating RANKL, which is produced by stromal and osteoblast cells, through JAK-mediated activation of STAT3 [83]. Study in murine model demonstrates that excessive IL-6 expression can result in increased OC development and decreased bone mass [84]. Another study using a mouse model lacking in IL-6 demonstrates an increase in tartrate-resistant acid phosphatase (TRAP), positive OCs cells, and elevated ALP activity in OBs [85]. Another experiment with IL-6-deficient mice also shows increased apoptosis in OC cells [86]. This study has revealed the osteoclastogenic regulatory role of IL-6. However, research has also reported the suppressive role of IL-6 on OCs formation through the suppression of the RANK signaling pathway-driven degradation of IκB and the initiation of JNK activation [87]. The role of IL-6 in bone remodeling depends not only on its own concentration but also on the presence of different concentrations of RANKL. IL-6/IL-6R mediates its suppression regulation process through NF-κB, ERK, and JNK [88]. Additionally, IL-6 knock-out mice experiments have demonstrated that genes controlled by OBs, such as Runx2 and Col1A1, are upregulated following ovariectomy, while OCs-related genes, such as TRAP, MMP9, and CTSK, are found to be downregulated [89]. The osteoporosis mouse model has shown that the IL-6/STAT3 pathway suppresses β-catenin activity and leads to increased inflammation reaction in the bone microenvironment, while the recovery from the disease progression has been achieved by using an anti-IL-6 antibody [90]. Furthermore, IL-6 prompts an elevation in the expression of TLR2, TLR4, IL-1, and TNF-α within BMSCs. This, in turn, triggers activation of the AKT pathway and subsequently leads to the suppression of Setd7 expression [91]. IL-6 also targets the activation of JAK/STAT, SHP2/MEK2, SHP2/AKT signaling pathway in OBs and induce their differentiation [92]. Interestingly, IL-6 has also been found to activate STAT3, signaling which promotes osteoclastogenic differentiation in an autocrine/paracrine feedback loop [91].
4.2.4 IL-7
In response to the IL-1 and TNF-α, OBs and stromal cells produce another IL-2 superfamily cytokine, IL-7. This cytokine indirectly promotes osteoclastogenesis by triggering T lymphocytes to produce RANKL and TNF-α [29]. Studies with IL-7 transgenic mice have shown lower bone mass and reduced osteoclastogenesis [93]. Additionally, it has been observed that the quantity of T lymphocytes affects the function of IL-7 in the bone microenvironment [29]. Research has substantiated that the lack of estrogen, post-OVX leads to increased IL-7 levels. This, in turn, encourages the thymus-dependent differentiation of bone marrow-derived progenitor cells and the thymus-independent peripheral expansion of mature T cells. Consequently, this process enhances the development of T lymphocytes and triggers bone loss [94]. Apart from T lymphocyte development, IL-7 is also involved in B lymphocyte development, which also plays a major role in bone resorption [95]. Recent studies propose that IL-7/IL-7R might oversee the distinct mechanisms involving CTSK, NFATc1, and MMP9, along with the phosphorylation events of p38 and Akt [96]. This is accomplished through the activation of the c-Fos/c-Jun pathway, thereby leading to an augmentation in the count of OCs and the extent of bone resorption in macrophages that are stimulated by RANKL [97]. Another study has suggested that IL-7 also has an anti-osteoclastogenic role in the presence of CSF-1 and RANKL [96]. However, additional details are not yet available.
4.2.5 IL-15
IL-15 is another member of the IL-2 superfamily. This cytokine also performs its role in a very similar way to IL-17. IL-15 exerts an indirect stimulating influence on the creation of OCs, acting in synergy with RANKL to primarily trigger OCs formation. This is primarily accomplished by activating the extracellular signal-regulated kinase (ERK) pathway, which enables this collaborative influence [31]. Another study of bone marrow and OBs co-culture has shown that IL-5 treatment may trigger OBs apoptosis [98].
4.2.6 IL-34
Like M-CSF, this cytokine also binds to the CSF-1R and triggers OCs regeneration and differentiation in the very same way as IL-34. In the presence of RANKL, this cytokine amplifies the process of osteoclastogenesis [99]. Few studies have elucidated the role of this cytokine in bone remodeling. One such research investigation revealed that IL-34 facilitates the growth and specialization of bone marrow macrophages into osteoclasts. This is achieved through an elevation in NFATc1 expression, the stimulation of p-STAT3 expression, and the suppression of Smad7 expression, all in the absence of M-CSF [34]. Furthermore, another research study reported that a low dosage of IL-34 enhances fracture healing via the PI3K/AKT and Erk pathways, yet it does not affect osteoclast formation in vitro [32].
4.2.7 IL-17
The secretion of this pro-inflammatory cytokine by Th17 cells has a pivotal function in bone immunology. A study involving mice with ovariectomy (OVX) demonstrated elevated IL-17 expression alongside an increased count of Th17 cells. This elevation fosters the production of osteoclastogenic cytokines, including TNF-α, IL-6, and RANKL, within osteoblast cells. Consequently, this process contributes to bone loss [32]. Estrogen or IL-17 treatment leads to a significant amount of bone loss, where anti-IL-17 antibody administration enhances the activity of FOXO1 and ATF4 to increase osteoblastogenesis [100]. In humans, the level of IL-17 increases after menopause and shows a significantly lower amount of minerals in bone [101]. In addition to stimulating the expression of RANKL, TNF-α, IL-1, IL-6, and IL-8 cytokines in osteoclasts (OCs), IL-17 also triggers autophagy in OC precursors. This, in turn, leads to the formation of OCs via JNK signaling, showing an association with lower concentrations [102]. However, research also shows the dose-dependent effect of IL-17 in bone remodeling. Increased concentration of IL-17 suppresses OCs differentiation and downregulates the MMP-9 and histone-K expression to inhibit matrix protein hydrolysis during bone resorption [103]. IL-17 triggers the upregulation of M-CSF and RANKL expression in hMSCs, facilitating the progression of OCs formation in both in vitro and in vivo [104]. Research has shown that IL-17 shows a positive effect on early OBs differentiation but an inhibitory effect on osteoblastic calcification [105]. In vitro, elevated levels of IL-17 prompt OBs impairment through the NLRP3 inflammatory vesicle pathway. This instigates the liberation of IL-1 and RANKL, further disturbing the equilibrium of bone metabolism [106].
4.2.8 IL-20
IL-20 is another IL-10 family cytokine, highly expressed in osteoporosis patient serum compared to normal patients. Treatment with anti-IL-20 antibody leads to the suppression of M-CSF and RANKL-induced OCs formation in vitro [33]. IL-20 triggers osteoclastic signals such as NF-κB, TRAF6, STAT3, NFATC1, and c-Fos. This cytokine has the capability to induce the expression of RANKL in both OBs and OCs [33]. Study has shown that IL-20 could hinder the viability and maturation of OBs by increasing the expression of sclerostin while decreasing the levels of OSX, RUNX2, and OPG [107].
4.2.9 IL-22
Th22 cell producing this cytokine is another member of IL-10 family and helps to defense and wound healing of intestinal epithelium and skin tissue. Research has indicated the elevated levels of IL-22 in the blood serum. The observation has also revealed that instead of IL-17 alone, IL-22 plays an immense role in osteoclast differentiation through MAPK phosphorylation [108].
4.2.10 IL-23
This cytokine induces osteoclastic proliferation by differentiating naïve Th cells into Th17 cells, which produce osteoclastogenic IL-17, to further bone degeneration [109]. This cytokine can also induce OCs formation by upregulating RANKL expression in the bone marrow and CD4+ T cells [35]. Nonetheless, the impact of IL-23 on bone within a living organism is also subject to debate. This uncertainty arises from a particular study’s findings, which suggest that IL-23 indirectly impedes osteoclast development in vitro through a CD4+ T lymphocyte-dependent mechanism that is further contingent on the dosage employed [110].
The immune system, which functions as a protective barrier against invasive diseases and bodily harm, is made up of a variety of cell types. The immune system is divided into two main sections: the innate immune system, which provides rapid protection, and the adaptive immune system, which is highly specific to antigens. Upon tissue damage, the initial responders are predominantly innate immune cells like neutrophils, macrophages, and dendritic cells. As the tissue damage continues, specific adaptive immune cells such as T and B cells, which are tailored to the antigens involved, are drawn to the site. Acute inflammation governs the bone regeneration phase, while chronic inflammation regulates the bone resorption phase within the process of bone remodeling. Given below are the immune cells that are pivotal in bone biology.
5.1 Neutrophils
Neutrophil is the prevailing form of polymorphonuclear phagocyte cell, typically being the initial innate immune cell to arrive at the site of injury [111]. These cells combat pathogens through phagocytosis and by generating reactive oxygen species (ROS) via the synthesis of granzyme B and perforins [112].
Numerous clinical and animal experiments have implicated the involvement of neutrophils at the bone erosion site. The increased level of inflammatory cytokines and elevated number of macrophages at the bone lesions site indicates the effect of neutrophils during the bone healing mechanism [113]. A recent study underscores the fact that neutrophils produce an emergency extracellular matrix-rich fibronectin to trigger the fracture healing process [114]. In contrast, some studies have reported the involvement of neutrophils in osteoclastogenesis. Recent research has reported that TLR4-activated neutrophils can produce a higher amount of cell membrane RANKL (m-RANKL), which is different from CD3+ lymphocyte-producing soluble RANKL [115]. The activated neutrophils’ mRANKL affects both OCs and their mononuclear precursors, transforming them into fully developed and operational OCs that play a role in bone resorption [116]. Moreover, it has been noted that neutrophils augment the differentiation of OCs induced by vitamin D3 in co-culture setups involving bone marrow cells and OBs [117]. Additionally, neutrophils stimulate the breakdown of OPG, leading to increased osteoclastogenesis in bone marrow cells [118]. To summarize, neutrophils balance bone homeostasis during early phase of bone injury.
5.2 Macrophages
Macrophages are recognized as the initial precursors of OCs. Circulating macrophages started to reside within the organs to become tissue-resident macrophages (TRM), such as osteocytes in bone. Distinct macrophage populations exist within the bone structure, including bone marrow macrophages (BMMs), OCs, and osteal macrophages, often referred to as “osteomacs.” Osteomacs are observed near the OBs, offering anabolic assistance to these cells in terms of bone mineralization and the process of bone formation by releasing bone regenerating cytokines such as BMP-2, BMP-4, TGF-B1 [119]. Osteal macrophages play a major role in bone mineralization [120].
When exposed to a defined array of stimuli, immature macrophages can be categorized into two main classes: M1 macrophages, which are generally pro-inflammatory, and M2 macrophages, known for their anti-inflammatory properties [121]. Studies have shown that during early phase of bone injury, M1 macrophages can transdifferentiate into OCs followed by stimulated pro-inflammation [122]. An investigation using both the RAW 264.7 murine macrophage cell line and bone marrow-derived macrophages (BMDMs) highlights the fact that M1 macrophages induced by lipopolysaccharide (LPS) and IFN-γ, notably hinder RANKL-induced osteoclastogenesis by releasing IFN-γ and IL-12 cytokines when compared to M2 macrophages [123]. RANKL induces the differentiation of BMMs into M1 macrophages, which, in turn, targets M2 macrophage-specific factors OPN and RUNX2 for bone regeneration [124]. Subsequent research has indicated that IL-4 and IL-13 can elevate the proportion of M2 macrophages, leading to substantial improvement in bone regeneration during the ossification phase [125]. In contrast, experiments have been performed with OVX osteoporotic mice in the absence of estrogen. This experiment reveals that RANKL actively stimulates the differentiation of M2 macrophages into osteoclasts. This also indicates the protective role of estrogen over M2 macrophages against RANKL stimulation [126]. Collectively, these investigations highlight the significance of the plasticity and diversity of macrophages, positioning them as vital contributors to the preservation of bone homeostasis.
5.3 Dendritic cells
A surface covered with MHC-II and characterized by professional antigen-presenting cells serves as a link connecting the adaptive and innate immune responses. Recent emerging data proposes that dendritic cells (DCs) stimulate inflammation-driven osteoclastogenesis and amplify bone loss by serving as precursors to OCs, which have been supported by the whole genome sequencing data [127]. Both OCs and DCs exhibit the RANK receptor on their surfaces. This receptor not only facilitates DC interactions with T cells but also triggers the initiation of the NF-κB developmental signaling pathway, ultimately culminating in the process of osteoclastogenesis [128]. Under in vitro condition, DCs undergo trans-differentiation into OCs when exposed to M-CSF and RANKL. It has been noted that in comparison to OCs derived from monocytes, OC generation and fusion from DCs occur at a quicker pace [129]. However, a recent study has documented that interferon-λ1 (IFN-λ1) derived from DCs hinders the differentiation of osteoclasts by obstructing the NF-κB signaling pathway and the formation of the NLRP3 inflammasome. Moreover, the introduction of the IFN-λ1 monoclonal antibody was shown to reverse this impact [21]. DCs experience an extension of their lifespan and display increased levels of the cytokines IL-7 and IL-15 in conditions characterized by a lack of estrogen. Synchronized action of these cytokines triggers the production of pro-inflammatory cytokines IL-17A and TNF-α by T cells in antigen-independent way. These pro-inflammatory cytokines elicit the bone lesions in osteoporotic circumstances. This suggests the indirect influence of DCs in influencing bone health during estrogen deficiency [130]. T cell regulation influences the trans-differentiation process from DCs to OCs by controlling the secretion of IFN-γ. This interferon obstructs TRAF6, thereby hindering RANKL signaling. This obstruction leads to the inhibition of osteoclast maturation and activation [131]. Additionally, investigations also suggest that Th17 cells also play a role in governing the transformation from DCs to OCs. Moreover, under certain conditions, DCs have been observed to produce the anti-osteoporotic cytokine TGF-β [132]. To sum up, these studies indicate that dendritic cells perform a remarkable role in the regulation of osteoclastogenesis.
5.4 B cells
Following antigen stimulation, B cells undergo proliferation and differentiation into plasma cells. Emerging studies have implicated the role of B cells in regulation of the RANKL/OPG axis. This study has also revealed that B cells are one of the major sources of RANKL, which exacerbates bone loss in cases of ovariectomy-induced bone loss [133]. RA mice model study reveals that B cells directly suppress OBs differentiation by triggering the expression of OB inhibitors, such as CCL3 and TNF through ERK and NF-κB signaling pathways [134]. Moreover, another study revealed that RANKL stimulates B cell proliferation and subsequently function as supportive cells for osteoclasts [135]. In an inflammatory setting, activated T cells interact with B cells through the CD40/CD40L signaling pathway and stimulate OPG production, exerting a beneficial impact on the regeneration of bone tissue [136]. Additionally, TGF-β stimulates B cells to generate OPG, implying that B cells impede the process of osteoclast differentiation [137]. Recently, another subset of B cells, known as immunosuppressive B regulatory cells (Bregs) have been discovered. These cells have been shown to play immense role in the bone remodeling process [138]. The Bregs majorly produce IL-10 and IL-35. One study co-related the suppressed IL-10 levels along with low number of Bregs in osteoporotic mice model [139]. Additionally, IL-35 produced by these Bregs not only helps in B cell differentiation but also represses bone degeneration via induction of OPG along with suppressing RANKL production [139]. To conclude, the involvement of B cells in bone regeneration remains a subject of debate, and their precise function might be contingent on the specific pathological conditions present locally.
5.5 T cells
Originated from hematopoietic stem cells and matured at the thymus, T cells play immunoregulatory role through the production of various types of cytokines. While in the resting state, T cells can restrain bone resorption by osteoclasts. However, upon activation and the expression of RANKL, they participate in the creation of OCs, consequently amplifying bone resorption [131]. IFN-γ establishes an inverse relationship between T cell activation and the process of bone resorption. This is achieved as IFN-γ intervenes with RANKL-RANK signaling and triggers the degradation of the RANK adaptor protein, specifically TRAF6. TRAF6 acts as an intermediary protein that instigates the survival and differentiation of osteoclasts by activating downstream signaling pathways through the interaction of RANKL and RANK [131]. T cells are classified into two primary subclasses: CD8+ T cells and CD4+ T cells. Within this group, CD4+ T cells encompass diverse subsets that perform a dual function: ensuring immune protection and participating in bone remodeling.
CD8+ T cells are well known for their cytotoxic activity against invading pathogens. CD8+ T cells also play protective role in bone remodeling process by profoundly suppressing osteoclastogenesis. Studies have revealed that CD8+ T cells do not exert a regulatory effect on osteoblastogenesis in an OPG-dependent manner, as evidenced by the fact that treatment with OPG does not result in any alteration of outcomes [140]. Administration of anabolic PTH treatment to mice is observed to notably elevate the synthesis of Wnt10b by CD8+ T cells found in the bone marrow. This, in turn, triggers the activation of canonical Wnt signaling within pre-osteoblasts. This finding underscores the pivotal role of T cells in the mechanism of anabolic PTH’s effects [141]. Mice devoid of T cells display reduced Wnt signaling in pre-osteoblasts, resulting in compromised osteoblastic commitment, proliferation, differentiation, and lifespan. These collective effects ultimately lead to an attenuated anabolic response within trabecular bone and an inability to enhance bone strength [142].
CD4+ T cells regulate their immune-regulatory effect through cytokines secretion. It comprises various subsets, including Th1, Th2, Th9, Th17, TFh, and Th22 cells. IL-4 impedes osteoclastogenesis by suppressing the NFATc1 factor responsible for osteoclast differentiation. Furthermore, IL-4 hinders bone resorption by obstructing NF-κB and Ca2+ signaling in mature osteoclasts [143]. IFN-γ possesses the ability to directly inhibit osteoclastogenesis; however, it can also stimulate the generation of osteoclasts by inducing the expression of RANKL on activated T cells. Furthermore, the direct impact of IFN-γ on osteoclastogenesis varies depending on the stage of osteoclast differentiation [144]. RORγt-expressing Th17 cells play stimulating role in osteoclastogenesis primarily by generating IL-17. This cytokine acts on osteoclast precursors to stimulate RANK production, and it also induces the expression of RANKL in stromal cells and osteoblasts [145]. In vitro study has also revealed that Treg cells directly enhance the functionality of osteoblasts and enhance the osteogenic differentiation capacity of osteoblast precursor cells, such as bone marrow mesenchymal stem cells (BMMSCs) [146]. Additional investigations have demonstrated that Treg cells interact with CD8+ T cells to increase the production of WNT10b, which exerts its effects on stromal cells and osteoblasts, thereby promoting bone formation [147]. Th9 cells, characterized by the expression of PU.1, release IL-9, which influences the expression of various genes related to osteoclastogenesis, including matrix metalloproteinases (MMPs) [148]. IL-9 secretion substantially amplifies the differentiation of osteoclasts. A study noted that Th22 cells contribute to the differentiation of osteoclasts by releasing the IL-22 cytokine. This cytokine triggers the expression of NFATc1 in CD14+ monocytes, leading to exacerbated bone degradation in patients with rheumatoid arthritis (RA) [108]. Moreover, another study implies that IL-22, by increasing the expression of RANKL, prompts the differentiation of osteoclasts and induces bone resorption in the subchondral bone of temporomandibular joint osteoarthritis (TMJ-OA) and in cases of periodontitis [149]. To conclude, through several subsets, T cells play pivotal role in bone remodeling process.
Epigenetic regulation involves heritable changes in the DNA or gene loci to alter their expression pattern without any modification in the sequence of the corresponding DNA segments. Epigenetic modifications are essential in establishing cell fates, genomic imprinting, and differentiation of a particular cell type from their respective precursors. Any abnormality in such control mechanisms severely affects the cells and gives rise to various diseases including, autoimmune, somatic, metabolic, and even malignant tumors leading to cancer [150]. Recruitment and differentiation of these cells involve several growth factors and secreted cytokines, which induce several intracellular pathways and interaction of several transcription factors. Bone remodeling being such a critical process, is very tightly controlled by such epigenetic regulatory mechanisms [150]. In recent times, much-needed research has highlighted the importance of epigenetic regulatory mechanisms involved in bone remodeling. Studies delineating the roles of these genomic changes have garnered a lot of traction, and it has been shown that the differentiation of OBs and OCs is significantly dependent on epigenetic regulatory mechanisms [150]. Epigenetic alterations can be broadly classified into DNA modifications, histone modifications, chromatin remodeling, and transcription of noncoding RNAs. Methylation and hydroxy methylation of DNA are well known for repressing certain gene loci [151]. In contrast, histone tail modifications including acetylation, phosphorylation, ubiquitination, ribosylation, etc. critically regulate expression of a wide array of genes by both activating and repressing them [152] along with aiding in DNA repair. Chromatin-remodelers constitute three fundamental classes of proteins namely, readers, writers, and erasers. These proteins act in unison to control the expression pattern of a large variety of transcription factors, which, in turn, regulate the fates of the involved cells [153]. Detailing each of these processes is not possible within the finite limits of this chapter. Hence, the following sections will primarily focus on summarizing the augmentations of histone proteins and their modulation of bone remodeling.
6.1 Histone modifications
Histones constitute a class of small proteins that provide structural support to chromosomes and help in compacting the genomic material in a eukaryotic cell. The histone proteins are also responsible for regulating the expression of genes by harboring several marks called histone modifications. This enables the modified histone proteins to directly interfere with the compaction of chromatin and recruit other protein complexes to alter their target gene transcription. Modifications involve addition of specific chemical groups to exposed residues on the N-terminal tails of these histones.
6.1.1 Histone acetylation
Acetylation of histone tails primarily occurs at the ε-amino group of the lysine residues. This transfer reaction is catalyzed by histone acetyltransferases or HATs. Acetylation of histones results in the opening of the chromatin due to removal of positive charges from the histone tail and making the regulatory regions more accessible to the TF complexes, which generally act as gene activation marks. They can also serve as DNA repair signals. Several studies have indicated that class I HATs, such as p300/CBP, are recruited to the promoters of numerous pro-osteoblastic genes, such as RUNX2, and OSX among others [154, 155]. In one such study, Kim et al. observed that the vitamin D receptor facilitated the recruitment of CBP and p300 to the promoters of CYP and OPN genes during exposure to 1, 25-dihydroxyvitamin D3 [155]. This recruitment subsequently results in the enhanced acetylation of histone H4 in osteoblasts. p300/CBP-associated factor (PCAF/KAT2B) is an additional histone acetyltransferase (HAT) that plays a crucial role in the process of osteogenic differentiation [155]. PCAF expression gets upregulated following osteogenic induction mediated by Smad signaling resulting in an increased expression of BMP pathway genes through the augmentation of histone H3K9 acetylation [154]. Recent genome-wide association studies have examined the chromatin landscape during the process of osteogenesis from MSCs, which display a significant increase in the levels of H3K9Ac, H4K5Ac, and H3K27Ac marks globally [156]. Similar to osteoblast differentiation, osteoclastogenesis is also influenced by histone acetylation. One study indicates that targeted pharmacological suppression of Bromodomain and PHD finger-containing protein significantly hinders the process of RANKL-induced differentiation, namely in the formation of osteoclasts responsible for bone resorption [157].
HDACs also control the epigenetic landscape by reversing the effects of acetylation. Evidently, the expression of osteoblast-related genes is decreased by HDAC1, HDAC2, HDAC3, and HDAC8 [158]. HDAC1 is indeed recruited to the promoter regions of OSX and OCN, leading to the suppression of osteoblast development [159]. Similarly, HDAC3 and RUNX2 are found to interact and result in the repression of the OCN promoter, leading to the subsequent inhibition of osteoblast development [160]. HDAC3 is also found to control the expression of important osteogenic factors, such as RUNX2 and Col1a [160]. Sirtuin 1 (SIRT1) (another class of HDACs), has been found to enhance the differentiation of BMSCs into osteoblasts. Moreover, SIRT1 can facilitate the deacetylation of β-catenin, disrupting the Wnt/β-catenin pathway and resulting in its increased localization within the nucleus and upregulate osteoblast function [47].
6.1.2 Histone methylation
Methylation of histone takes place on several basic residues, including lysine, histidine, and arginine. Major methylation marks can be found on various H3 and H4 residues, including H3K4, H3K9, H3R17, H3K79, H4R3, H4K20, and H4K59. Methylation marks on histones can act as both transcriptional activators (H3K4, H3K79, H3K36, and H3R17), and silencers (H3K9, H3K27, and H3K20). Similar to what is observed during histone acetylation, methylation of histones is also carried out by histone methyl transferases or HMTs. Studies have found that H3K36 tri-methylation increases the interaction of RUNX2 and p300, leading to upregulation of osteogenic genes [158]. Recent studies have revealed interesting insights about additional histone methylation marks that can control the osteoblastic differentiation form BDMSCs. Levels of H3K4me3 and H3K36me3 are found to be increased significantly on a global scale [161]. Corresponding decrease on repressive methylation marks, such as H3K27me3, is also evident [161]. The essentiality of multipotent development of MSCs has been determined through the identification of active histone marks, including, H3K4me1, H3K4me3, and H3K36me3 during osteoblastic differentiation [156]. Another important HMT, EZH2 is shown to promote H3K27 tri-methylation of the promoters of key regulatory genes such as RUNX2, OPN, OSC, MX1, FHL-1, etc. and suppress osteoclastogenesis [162]. Similarly, PRMT5 (another methyl transferase) is found to suppress osteoblastic differentiation [163]. It is responsible for the global methylation of arginine residues on H3R8 and H4R3. Blocking this symmetric demethylation enhances osteogenic differentiation of MSCs, indicating the importance of PRMT5 activity on fate specification of MSCs during osteogenesis [163]. During osteoclastogenesis, EZH2 is found to positively boost the osteoclast activity by downregulating IRF8 (a known negative regulator). The IRF8 gene promoter has increased repressive H3K27me3 marks via recruitment of EZH2 by RANKL [164]. In contrast, DOT1L methyl transferase is found to suppress osteoclast formation by methylating H3K79 [165].
Just as HMTs, histone demethylases are crucial in maintaining the right balance to control the important cell specification fates. Enzymes like JMJD3 (KDM6B) demethylate H3K27 to upregulate key genes, such as RUNX2, OCX, OSN, etc. to boost osteoblast differentiation [166]. Interestingly, JMJD3 also endorses osteoclast formation by upregulating NFATC1 gene in T-cells via restricting repressive histone marks at its promoter [167]. Another demethylase, JARID1B, was shown to demethylate RUNX2 promoter leading to suppressed osteoblastic lineage specification [168]. Thus, the critical balance between histone methylation and demethylation controls the lineage specification of both OBs and OCs to ensure smoother osteogenic turnover.
In addition to providing structural support and protecting interior organs, bones also play a number of metabolic roles, particularly in preserving the body’s mineral balance. When bone resorption outpaces bone creation, bone loss will take place and, in extreme situations, result in osteoporosis. The pathophysiology of aberrant bone metabolism and osteoporosis will be better understood via a thorough investigation of these epigenetic pathways. However, very little is now known about the epigenetics of anomalies in bone remodeling. Further, research is needed to understand the known epigenetic regulatory factors, mechanisms, and the connection between their downstream target genes and associated disorders. The mechanisms of epigenetic modification and histone alterations also need to be clarified. The clinical diagnosis and management of osteoporosis will be greatly enhanced by the discovery of specific biomarkers connected to the condition. A comprehensive epigenetic spectrum of healthy bone and bone illnesses based on the entire genome will be established thanks to the widespread application of epigenetic microarrays, high-throughput sequencing, and other emerging technologies. The biological underpinnings of the fundamental molecular mechanisms of bone remodeling and the delicate balance between anabolism and catabolism in bone tissue will be further revealed by additional study in this field, opening up new avenues for the diagnosis and treatment of common bone remodeling disorders.
1.Okamoto K, Nakashima T, Shinohara M, Negishi-Koga T, Komatsu N, Terashima A, et al. Osteoimmunology: The conceptual framework unifying the immune and skeletal systems. Physiological Reviews. 2017;97(4):1295-1349
2.Kenkre JS, Bassett J. The bone remodelling cycle. Annals of Clinical Biochemistry. 2018;55(3):308-327
3.Loi F, Cordova LA, Pajarinen J, Lin TH, Yao Z, Goodman SB. Inflammation, fracture and bone repair. Bone. 2016;86:119-130
4.Feng X, McDonald JM. Disorders of bone remodeling. Annual Review of Pathology. 2011;6:121-145
5.Rowe P, Koller A, Sharma S. Physiology, bone remodeling. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2023
6.Bonucci E, Ballanti P. Osteoporosis-bone remodeling and animal models. Toxicologic Pathology. 2014;42(6):957-969
7.Feng X. Chemical and biochemical basis of cell-bone matrix interaction in health and disease. Current Chemical Biology. 2009;3(2):189-196
8.Marion NW, Mao JJ. Mesenchymal stem cells and tissue engineering. Methods in Enzymology. 2006;420:339-361
9.Zhao E, Xu H, Wang L, Kryczek I, Wu K, Hu Y, et al. Bone marrow and the control of immunity. Cellular & Molecular Immunology. 2012;9(1):11-19
10.Sozen T, Ozisik L, Basaran NC. An overview and management of osteoporosis. European Journal of Rheumatology. 2017;4(1):46-56
11.Srivastava RK, Dar HY, Mishra PK. Immunoporosis: Immunology of osteoporosis-role of T cells. Frontiers in Immunology. 2018;9:657
12.Saxena Y, Routh S, Mukhopadhaya A. Immunoporosis: Role of innate immune cells in osteoporosis. Frontiers in Immunology. 2021;12:687037
13.Singh K, Piprode V, Mhaske ST, Barhanpurkar-Naik A, Wani MR. IL-3 differentially regulates membrane and soluble RANKL in osteoblasts through metalloproteases and the JAK2/STAT5 pathway and improves the RANKL/OPG ratio in adult mice. Journal of Immunology. 2018;200(2):595-606
14.Abu-Amer Y. IL-4 abrogates osteoclastogenesis through STAT6-dependent inhibition of NF-kappaB. The Journal of Clinical Investigation. 2001;107(11):1375-1385
15.Moore KW, de Waal MR, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annual Review of Immunology. 2001;19:683-765
16.Nagata N, Kitaura H, Yoshida N, Nakayama K. Inhibition of RANKL-induced osteoclast formation in mouse bone marrow cells by IL-12: Involvement of IFN-gamma possibly induced from non-T cell population. Bone. 2003;33(4):721-732
17.Stein NC, Kreutzmann C, Zimmermann SP, Niebergall U, Hellmeyer L, Goettsch C, et al. Interleukin-4 and interleukin-13 stimulate the osteoclast inhibitor osteoprotegerin by human endothelial cells through the STAT6 pathway. Journal of Bone and Mineral Research. 2008;23(5):750-758
18.Kitaura H, Fujimura Y, Yoshimatsu M, Kohara H, Morita Y, Aonuma T, et al. IL-12- and IL-18-mediated, nitric oxide-induced apoptosis in TNF-alpha-mediated osteoclastogenesis of bone marrow cells. Calcified Tissue International. 2011;89(1):65-73
19.Tsubaki M, Takeda T, Matsuda T, Yamamoto Y, Higashinaka A, Yamamoto K, et al. Interleukin 19 suppresses RANKL-induced osteoclastogenesis via the inhibition of NF-kappaB and p38MAPK activation and c-Fos expression in RAW264.7 cells. Cytokine. 2021;144:155591
20.Furukawa M, Takaishi H, Takito J, Yoda M, Sakai S, Hikata T, et al. IL-27 abrogates receptor activator of NF-kappa B ligand-mediated osteoclastogenesis of human granulocyte-macrophage colony-forming unit cells through STAT1-dependent inhibition of c-Fos. Journal of Immunology. 2009;183(4):2397-2406
21.Chen Y, Wang Y, Tang R, Yang J, Dou C, Dong Y, et al. Dendritic cells-derived interferon-lambda1 ameliorated inflammatory bone destruction through inhibiting osteoclastogenesis. Cell Death & Disease. 2020;11(6):414
22.Yan H, Dong M, Liu X, Shen Q, He D, Huang X, et al. Multiple myeloma cell-derived IL-32gamma increases the immunosuppressive function of macrophages by promoting indoleamine 2,3-dioxygenase (IDO) expression. Cancer Letters. 2019;446:38-48
23.Lima IL, Macari S, Madeira MF, Rodrigues LF, Colavite PM, Garlet GP, et al. Osteoprotective effects of IL-33/ST2 link to osteoclast apoptosis. The American Journal of Pathology. 2015;185(12):3338-3348
24.Peng M, Wang Y, Qiang L, Xu Y, Li C, Li T, et al. Interleukin-35 inhibits TNF-alpha-induced osteoclastogenesis and promotes apoptosis via shifting the activation from TNF receptor-associated death domain (TRADD)-TRAF2 to TRADD-Fas-associated death domain by JAK1/STAT1. Frontiers in Immunology. 2018;9:1417
25.Takayanagi H, Sato K, Takaoka A, Taniguchi T. Interplay between interferon and other cytokine systems in bone metabolism. Immunological Reviews. 2005;208:181-193
26.Sims NA, Jenkins BJ, Nakamura A, Quinn JM, Li R, Gillespie MT, et al. Interleukin-11 receptor signaling is required for normal bone remodeling. Journal of Bone and Mineral Research. 2005;20(7):1093-1102
27.Kim JH, Jin HM, Kim K, Song I, Youn BU, Matsuo K, et al. The mechanism of osteoclast differentiation induced by IL-1. Journal of Immunology. 2009;183(3):1862-1870
28.Rose-John S. Interleukin-6 family cytokines. Cold Spring Harbor Perspectives in Biology. 2018;10(2):a028415
29.Toraldo G, Roggia C, Qian WP, Pacifici R, Weitzmann MN. IL-7 induces bone loss in vivo by induction of receptor activator of nuclear factor kappa B ligand and tumor necrosis factor alpha from T cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(1):125-130
30.Kopesky P, Tiedemann K, Alkekhia D, Zechner C, Millard B, Schoeberl B, et al. Autocrine signaling is a key regulatory element during osteoclastogenesis. Biology Open. 2014;3(8):767-776
31.Okabe I, Kikuchi T, Mogi M, Takeda H, Aino M, Kamiya Y, et al. IL-15 and RANKL play a synergistically important role in osteoclastogenesis. Journal of Cellular Biochemistry. 2017;118(4):739-747
32.Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: A new candidate in the pathogenesis of osteoporosis. PLoS One. 2012;7(9):e44552
33.Hsu YH, Chen WY, Chan CH, Wu CH, Sun ZJ, Chang MS. Anti-IL-20 monoclonal antibody inhibits the differentiation of osteoclasts and protects against osteoporotic bone loss. The Journal of Experimental Medicine. 2011;208(9):1849-1861
34.Cheng X, Wan QL, Li ZB. AG490 suppresses interleukin-34-mediated osteoclastogenesis in mice bone marrow macrophages. Cell Biology International. 2017;41(6):659-668
35.Chen L, Wei XQ, Evans B, Jiang W, Aeschlimann D. IL-23 promotes osteoclast formation by up-regulation of receptor activator of NF-kappaB (RANK) expression in myeloid precursor cells. European Journal of Immunology. 2008;38(10):2845-2854
36.Khapli SM, Mangashetti LS, Yogesha SD, Wani MR. IL-3 acts directly on osteoclast precursors and irreversibly inhibits receptor activator of NF-kappa B ligand-induced osteoclast differentiation by diverting the cells to macrophage lineage. Journal of Immunology. 2003;171(1):142-151
37.Yogesha SD, Khapli SM, Wani MR. Interleukin-3 and granulocyte-macrophage colony-stimulating factor inhibits tumor necrosis factor (TNF)-alpha-induced osteoclast differentiation by down-regulation of expression of TNF receptors 1 and 2. The Journal of Biological Chemistry. 2005;280(12):11759-11769
38.Yogesha SD, Khapli SM, Srivastava RK, Mangashetti LS, Pote ST, Mishra GC, et al. IL-3 inhibits TNF-alpha-induced bone resorption and prevents inflammatory arthritis. Journal of Immunology. 2009;182(1):361-370
39.Oh J, Lee MS, Yeon JT, Choi SW, Kim HS, Shim H, et al. Inhibitory regulation of osteoclast differentiation by interleukin-3 via regulation of c-Fos and Id protein expression. Journal of Cellular Physiology. 2012;227(5):1851-1860
40.Gupta N, Barhanpurkar AP, Tomar GB, Srivastava RK, Kour S, Pote ST, et al. IL-3 inhibits human osteoclastogenesis and bone resorption through downregulation of c-Fms and diverts the cells to dendritic cell lineage. Journal of Immunology. 2010;185(4):2261-2272
41.Lee J, Seong S, Kim JH, Kim K, Kim I, Jeong BC, et al. STAT5 is a key transcription factor for IL-3-mediated inhibition of RANKL-induced osteoclastogenesis. Scientific Reports. 2016;6:30977
42.Barhanpurkar AP, Gupta N, Srivastava RK, Tomar GB, Naik SP, Joshi SR, et al. IL-3 promotes osteoblast differentiation and bone formation in human mesenchymal stem cells. Biochemical and Biophysical Research Communications. 2012;418(4):669-675
43.Wei S, Wang MW, Teitelbaum SL, Ross FP. Interleukin-4 reversibly inhibits osteoclastogenesis via inhibition of NF-kappa B and mitogen-activated protein kinase signaling. The Journal of Biological Chemistry. 2002;277(8):6622-6630
44.Cheng J, Liu J, Shi Z, Xu D, Luo S, Siegal GP, et al. Interleukin-4 inhibits RANKL-induced NFATc1 expression via STAT6: A novel mechanism mediating its blockade of osteoclastogenesis. Journal of Cellular Biochemistry. 2011;112(11):3385-3392
45.te Velde AA, Huijbens RJ, Heije K, de Vries JE, Figdor CG. Interleukin-4 (IL-4) inhibits secretion of IL-1 beta, tumor necrosis factor alpha, and IL-6 by human monocytes. Blood. 1990;76(7):1392-1397
46.Hiasa M, Abe M, Nakano A, Oda A, Amou H, Kido S, et al. GM-CSF and IL-4 induce dendritic cell differentiation and disrupt osteoclastogenesis through M-CSF receptor shedding by up-regulation of TNF-alpha converting enzyme (TACE). Blood. 2009;114(20):4517-4526
47.Liu D, Yao S, Wise GE. Effect of interleukin-10 on gene expression of osteoclastogenic regulatory molecules in the rat dental follicle. European Journal of Oral Sciences. 2006;114(1):42-49
48.Park-Min KH, Ji JD, Antoniv T, Reid AC, Silver RB, Humphrey MB, et al. IL-10 suppresses calcium-mediated costimulation of receptor activator NF-kappa B signaling during human osteoclast differentiation by inhibiting TREM-2 expression. Journal of Immunology. 2009;183(4):2444-2455
49.Mohamed SG, Sugiyama E, Shinoda K, Taki H, Hounoki H, Abdel-Aziz HO, et al. Interleukin-10 inhibits RANKL-mediated expression of NFATc1 in part via suppression of c-Fos and c-Jun in RAW264.7 cells and mouse bone marrow cells. Bone. 2007;41(4):592-602
50.Ma X, Zhu X, He X, Yi X, Jin A. The Wnt pathway regulator expression levels and their relationship to bone metabolism in thoracolumbar osteoporotic vertebral compression fracture patients. American Journal of Translational Research. 2021;13(5):4812-4818
51.Dresner-Pollak R, Gelb N, Rachmilewitz D, Karmeli F, Weinreb M. Interleukin 10-deficient mice develop osteopenia, decreased bone formation, and mechanical fragility of long bones. Gastroenterology. 2004;127(3):792-801
52.de Waal MR, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: An autoregulatory role of IL-10 produced by monocytes. The Journal of Experimental Medicine. 1991;174(5):1209-1220
53.Kawashima I, Ohsumi J, Mita-Honjo K, Shimoda-Takano K, Ishikawa H, Sakakibara S, et al. Molecular cloning of cDNA encoding adipogenesis inhibitory factor and identity with interleukin-11. FEBS Letters. 1991;283(2):199-202
54.Kudo O, Sabokbar A, Pocock A, Itonaga I, Fujikawa Y, Athanasou NA. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone. 2003;32(1):1-7
55.Takeuchi Y, Watanabe S, Ishii G, Takeda S, Nakayama K, Fukumoto S, et al. Interleukin-11 as a stimulatory factor for bone formation prevents bone loss with advancing age in mice. The Journal of Biological Chemistry. 2002;277(50):49011-49018
57.Amcheslavsky A, Bar-Shavit Z. Interleukin (IL)-12 mediates the anti-osteoclastogenic activity of CpG-oligodeoxynucleotides. Journal of Cellular Physiology. 2006;207(1):244-250
58.Udagawa N, Horwood NJ, Elliott J, Mackay A, Owens J, Okamura H, et al. Interleukin-18 (interferon-gamma-inducing factor) is produced by osteoblasts and acts via granulocyte/macrophage colony-stimulating factor and not via interferon-gamma to inhibit osteoclast formation. The Journal of Experimental Medicine. 1997;185(6):1005-1012
59.Mansoori MN, Shukla P, Kakaji M, Tyagi AM, Srivastava K, Shukla M, et al. IL-18BP is decreased in osteoporotic women: Prevents inflammasome mediated IL-18 activation and reduces Th17 differentiation. Scientific Reports. 2016;6:33680
60.Onoe Y, Miyaura C, Kaminakayashiki T, Nagai Y, Noguchi K, Chen QR, et al. IL-13 and IL-4 inhibit bone resorption by suppressing cyclooxygenase-2-dependent prostaglandin synthesis in osteoblasts. Journal of Immunology. 1996;156(2):758-764
61.Kalliolias GD, Zhao B, Triantafyllopoulou A, Park-Min KH, Ivashkiv LB. Interleukin-27 inhibits human osteoclastogenesis by abrogating RANKL-mediated induction of nuclear factor of activated T cells c1 and suppressing proximal RANK signaling. Arthritis and Rheumatism. 2010;62(2):402-413
62.Shukla P, Mansoori MN, Kakaji M, Shukla M, Gupta SK, Singh D. Interleukin 27 (IL-27) alleviates bone loss in estrogen-deficient conditions by induction of early growth response-2 gene. The Journal of Biological Chemistry. 2017;292(11):4686-4699
63.Peng Q, Luo A, Zhou Z, Xuan W, Qiu M, Wu Q, et al. Interleukin 29 inhibits RANKL-induced osteoclastogenesis via activation of JNK and STAT, and inhibition of NF-kappaB and NFATc1. Cytokine. 2019;113:144-154
64.Kiyomiya H, Ariyoshi W, Okinaga T, Kaneuji T, Mitsugi S, Sakurai T, et al. IL-33 inhibits RANKL-induced osteoclast formation through the regulation of Blimp-1 and IRF-8 expression. Biochemical and Biophysical Research Communications. 2015;460(2):320-326
65.Saleh H, Eeles D, Hodge JM, Nicholson GC, Gu R, Pompolo S, et al. Interleukin-33, a target of parathyroid hormone and oncostatin m, increases osteoblastic matrix mineral deposition and inhibits osteoclast formation in vitro. Endocrinology. 2011;152(5):1911-1922
66.De Martinis M, Ginaldi L, Sirufo MM, Bassino EM, De Pietro F, Pioggia G, et al. IL-33/vitamin D crosstalk in psoriasis-associated osteoporosis. Frontiers in Immunology. 2020;11:604055
67.Ohori F, Kitaura H, Ogawa S, Shen WR, Qi J, Noguchi T, et al. IL-33 inhibits TNF-alpha-induced Osteoclastogenesis and bone resorption. International Journal of Molecular Sciences. 2020;21(3):1130
68.Schulze J, Bickert T, Beil FT, Zaiss MM, Albers J, Wintges K, et al. Interleukin-33 is expressed in differentiated osteoblasts and blocks osteoclast formation from bone marrow precursor cells. Journal of Bone and Mineral Research. 2011;26(4):704-717
69.Ginaldi L, De Martinis M, Saitta S, Sirufo MM, Mannucci C, Casciaro M, et al. Interleukin-33 serum levels in postmenopausal women with osteoporosis. Scientific Reports. 2019;9(1):3786
70.Zhang H, Li Y, Yuan L, Yao L, Yang J, Xia L, et al. Interleukin-35 is involved in angiogenesis/bone remodeling coupling through T helper 17/Interleukin-17 Axis. Frontiers in Endocrinology. 2021;12:642676
71.Li Y, Wang X, Lu J. Interleukin-35 promote osteogenesis and inhibit adipogenesis: Role of Wnt/beta-catenin and PPARgamma signaling pathways. Inflammation. 2023;46(2):522-533
72.Mun SH, Ko NY, Kim HS, Kim JW, Kim DK, Kim AR, et al. Interleukin-33 stimulates formation of functional osteoclasts from human CD14(+) monocytes. Cellular and Molecular Life Sciences. 2010;67(22):3883-3892
73.Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. The Journal of Experimental Medicine. 2000;191(2):275-286
74.Yamashita T, Yao Z, Li F, Zhang Q, Badell IR, Schwarz EM, et al. NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. The Journal of Biological Chemistry. 2007;282(25):18245-18253
75.Matsuo K, Galson DL, Zhao C, Peng L, Laplace C, Wang KZ, et al. Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. The Journal of Biological Chemistry. 2004;279(25):26475-26480
76.Yao Z, Xing L, Qin C, Schwarz EM, Boyce BF. Osteoclast precursor interaction with bone matrix induces osteoclast formation directly by an interleukin-1-mediated autocrine mechanism. The Journal of Biological Chemistry. 2008;283(15):9917-9924
77.Mizukami J, Takaesu G, Akatsuka H, Sakurai H, Ninomiya-Tsuji J, Matsumoto K, et al. Receptor activator of NF-kappaB ligand (RANKL) activates TAK1 mitogen-activated protein kinase kinase kinase through a signaling complex containing RANK, TAB2, and TRAF6. Molecular and Cellular Biology. 2002;22(4):992-1000
78.Yao Z, Lei W, Duan R, Li Y, Luo L, Boyce BF. RANKL cytokine enhances TNF-induced osteoclastogenesis independently of TNF receptor associated factor (TRAF) 6 by degrading TRAF3 in osteoclast precursors. The Journal of Biological Chemistry. 2017;292(24):10169-10179
79.Gilbert L, He X, Farmer P, Boden S, Kozlowski M, Rubin J, et al. Inhibition of osteoblast differentiation by tumor necrosis factor-alpha. Endocrinology. 2000;141(11):3956-3964
80.Polzer K, Joosten L, Gasser J, Distler JH, Ruiz G, Baum W, et al. Interleukin-1 is essential for systemic inflammatory bone loss. Annals of the Rheumatic Diseases. 2010;69(1):284-290
81.Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL. IL-1 mediates TNF-induced osteoclastogenesis. The Journal of Clinical Investigation. 2005;115(2):282-290
82.Tanabe N, Maeno M, Suzuki N, Fujisaki K, Tanaka H, Ogiso B, et al. IL-1 alpha stimulates the formation of osteoclast-like cells by increasing M-CSF and PGE2 production and decreasing OPG production by osteoblasts. Life Sciences. 2005;77(6):615-626
83.Palmqvist P, Persson E, Conaway HH, Lerner UH. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. Journal of Immunology. 2002;169(6):3353-3362
84.De Benedetti F, Rucci N, Del Fattore A, Peruzzi B, Paro R, Longo M, et al. Impaired skeletal development in interleukin-6-transgenic mice: A model for the impact of chronic inflammation on the growing skeletal system. Arthritis and Rheumatism. 2006;54(11):3551-3563
85.Lazzaro L, Tonkin BA, Poulton IJ, McGregor NE, Ferlin W, Sims NA. IL-6 trans-signalling mediates trabecular, but not cortical, bone loss after ovariectomy. Bone. 2018;112:120-127
86.Liu H, Feng W, Yimin CJ, Lv S, Hasegawa T, et al. Histological evidence of increased osteoclast cell number and asymmetric bone resorption activity in the tibiae of Interleukin-6-deficient mice. The Journal of Histochemistry and Cytochemistry. 2014;62(8):556-564
87.Yoshitake F, Itoh S, Narita H, Ishihara K, Ebisu S. Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of NF-kappaB signaling pathways. The Journal of Biological Chemistry. 2008;283(17):11535-11540
88.Feng W, Liu H, Luo T, Liu D, Du J, Sun J, et al. Combination of IL-6 and sIL-6R differentially regulate varying levels of RANKL-induced osteoclastogenesis through NF-kappaB, ERK and JNK signaling pathways. Scientific Reports. 2017;7:41411
89.Zhu S, He H, Gao C, Luo G, Xie Y, Wang H, et al. Ovariectomy-induced bone loss in TNFalpha and IL6 gene knockout mice is regulated by different mechanisms. Journal of Molecular Endocrinology. 2018;60(3):185-198
90.Li X, Zhou ZY, Zhang YY, Yang HL. IL-6 contributes to the defective osteogenesis of bone marrow stromal cells from the vertebral body of the glucocorticoid-induced osteoporotic mouse. PLoS One. 2016;11(4):e0154677
91.Xie Z, Tang S, Ye G, Wang P, Li J, Liu W, et al. Interleukin-6/interleukin-6 receptor complex promotes osteogenic differentiation of bone marrow-derived mesenchymal stem cells. Stem Cell Research & Therapy. 2018;9(1):13
92.Kaneshiro S, Ebina K, Shi K, Higuchi C, Hirao M, Okamoto M, et al. IL-6 negatively regulates osteoblast differentiation through the SHP2/MEK2 and SHP2/Akt2 pathways in vitro. Journal of Bone and Mineral Metabolism. 2014;32(4):378-392
93.Salopek D, Grcevic D, Katavic V, Kovacic N, Lukic IK, Marusic A. Increased bone resorption and osteopenia are a part of the lymphoproliferative phenotype of mice with systemic over-expression of interleukin-7 gene driven by MHC class II promoter. Immunology Letters. 2008;121(2):134-139
94.Ryan MR, Shepherd R, Leavey JK, Gao Y, Grassi F, Schnell FJ, et al. An IL-7-dependent rebound in thymic T cell output contributes to the bone loss induced by estrogen deficiency. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(46):16735-16740
95.Miller JP, Izon D, DeMuth W, Gerstein R, Bhandoola A, Allman D. The earliest step in B lineage differentiation from common lymphoid progenitors is critically dependent upon interleukin 7. The Journal of Experimental Medicine. 2002;196(5):705-711
96.Zhao JJ, Wu ZF, Yu YH, Wang L, Cheng L. Effects of interleukin-7/interleukin-7 receptor on RANKL-mediated osteoclast differentiation and ovariectomy-induced bone loss by regulating c-Fos/c-Jun pathway. Journal of Cellular Physiology. 2018;233(9):7182-7194
97.Lee SK, Kalinowski JF, Jastrzebski SL, Puddington L, Lorenzo JA. Interleukin-7 is a direct inhibitor of in vitro osteoclastogenesis. Endocrinology. 2003;144(8):3524-3531
98.Takeda H, Kikuchi T, Soboku K, Okabe I, Mizutani H, Mitani A, et al. Effect of IL-15 and natural killer cells on osteoclasts and osteoblasts in a mouse coculture. Inflammation. 2014;37(3):657-669
99.Chen Z, Buki K, Vaaraniemi J, Gu G, Vaananen HK. The critical role of IL-34 in osteoclastogenesis. PLoS One. 2011;6(4):e18689
100.Dixit M, Singh KB, Prakash R, Singh D. Functional block of IL-17 cytokine promotes bone healing by augmenting FOXO1 and ATF4 activity in cortical bone defect model. Osteoporosis International. 2017;28(7):2207-2220
101.Bhadricha H, Patel V, Singh AK, Savardekar L, Patil A, Surve S, et al. Increased frequency of Th17 cells and IL-17 levels are associated with low bone mineral density in postmenopausal women. Scientific Reports. 2021;11(1):16155
102.Lubberts E, van den Bersselaar L, Oppers-Walgreen B, Schwarzenberger P, Coenen-de Roo CJ, Kolls JK, et al. IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-kappa B ligand/osteoprotegerin balance. Journal of Immunology. 2003;170(5):2655-2662
103.Kitami S, Tanaka H, Kawato T, Tanabe N, Katono-Tani T, Zhang F, et al. IL-17A suppresses the expression of bone resorption-related proteinases and osteoclast differentiation via IL-17RA or IL-17RC receptors in RAW264.7 cells. Biochimie. 2010;92(4):398-404
104.Huang H, Kim HJ, Chang EJ, Lee ZH, Hwang SJ, Kim HM, et al. IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells: Implications for bone remodeling. Cell Death and Differentiation. 2009;16(10):1332-1343
105.Wang Z, Tan J, Lei L, Sun W, Wu Y, Ding P, et al. The positive effects of secreting cytokines IL-17 and IFN-gamma on the early-stage differentiation and negative effects on the calcification of primary osteoblasts in vitro. International Immunopharmacology. 2018;57:1-10
106.Lei L, Sun J, Han J, Jiang X, Wang Z, Chen L. Interleukin-17 induces pyroptosis in osteoblasts through the NLRP3 inflammasome pathway in vitro. International Immunopharmacology. 2021;96:107781
107.Hsu YH, Chiu YS, Chen WY, Huang KY, Jou IM, Wu PT, et al. Anti-IL-20 monoclonal antibody promotes bone fracture healing through regulating IL-20-mediated osteoblastogenesis. Scientific Reports. 2016;6:24339
108.Miyazaki Y, Nakayamada S, Kubo S, Nakano K, Iwata S, Miyagawa I, et al. Th22 cells promote osteoclast differentiation via production of IL-22 in rheumatoid arthritis. Frontiers in Immunology. 2018;9:2901
109.Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. The Journal of Experimental Medicine. 2006;203(12):2673-2682
110.Quinn JM, Sims NA, Saleh H, Mirosa D, Thompson K, Bouralexis S, et al. IL-23 inhibits osteoclastogenesis indirectly through lymphocytes and is required for the maintenance of bone mass in mice. Journal of Immunology. 2008;181(8):5720-5729
111.Nathan C. Neutrophils and immunity: Challenges and opportunities. Nature Reviews. Immunology. 2006;6(3):173-182
112.Wagner C, Iking-Konert C, Denefleh B, Stegmaier S, Hug F, Hansch GM. Granzyme B and perforin: Constitutive expression in human polymorphonuclear neutrophils. Blood. 2004;103(3):1099-1104
113.Bastian OW, Koenderman L, Alblas J, Leenen LP, Blokhuis TJ. Neutrophils contribute to fracture healing by synthesizing fibronectin+ extracellular matrix rapidly after injury. Clinical Immunology. 2016;164:78-84
114.Kovtun A, Bergdolt S, Wiegner R, Radermacher P, Huber-Lang M, Ignatius A. The crucial role of neutrophil granulocytes in bone fracture healing. European Cells & Materials. 2016;32:152-162
115.Chakravarti A, Raquil MA, Tessier P, Poubelle PE. Surface RANKL of Toll-like receptor 4-stimulated human neutrophils activates osteoclastic bone resorption. Blood. 2009;114(8):1633-1644
116.Quinn JM, Neale S, Fujikawa Y, McGee JO, Athanasou NA. Human osteoclast formation from blood monocytes, peritoneal macrophages, and bone marrow cells. Calcified Tissue International. 1998;62(6):527-531
117.Sugisaki R, Miyamoto Y, Yoshimura K, Sasa K, Kaneko K, Tanaka M, et al. Possible involvement of elastase in enhanced osteoclast differentiation by neutrophils through degradation of osteoprotegerin. Bone. 2020;132:115216
118.Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Science Translational Medicine. 2014;6(229):229ra40
119.Blom AB, van Lent PL, Holthuysen AE, van der Kraan PM, Roth J, van Rooijen N, et al. Synovial lining macrophages mediate osteophyte formation during experimental osteoarthritis. Osteoarthritis and Cartilage. 2004;12(8):627-635
120.Chang MK, Raggatt LJ, Alexander KA, Kuliwaba JS, Fazzalari NL, Schroder K, et al. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. Journal of Immunology. 2008;181(2):1232-1244
121.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity. 2014;41(1):14-20
122.Lassus J, Salo J, Jiranek WA, Santavirta S, Nevalainen J, Matucci-Cerinic M, et al. Macrophage activation results in bone resorption. Clinical Orthopaedics and Related Research. 1998;352:7-15
123.Yamaguchi T, Movila A, Kataoka S, Wisitrasameewong W, Ruiz Torruella M, Murakoshi M, et al. Proinflammatory M1 macrophages inhibit RANKL-induced osteoclastogenesis. Infection and Immunity. 2016;84(10):2802-2812
124.Huang R, Wang X, Zhou Y, Xiao Y. RANKL-induced M1 macrophages are involved in bone formation. Bone Research. 2017;5:17019
125.Schlundt C, El Khassawna T, Serra A, Dienelt A, Wendler S, Schell H, et al. Macrophages in bone fracture healing: Their essential role in endochondral ossification. Bone. 2018;106:78-89
126.Dou C, Ding N, Zhao C, Hou T, Kang F, Cao Z, et al. Estrogen deficiency-mediated M2 macrophage osteoclastogenesis contributes to M1/M2 ratio alteration in ovariectomized osteoporotic mice. Journal of Bone and Mineral Research. 2018;33(5):899-908
127.Gallois A, Lachuer J, Yvert G, Wierinckx A, Brunet F, Rabourdin-Combe C, et al. Genome-wide expression analyses establish dendritic cells as a new osteoclast precursor able to generate bone-resorbing cells more efficiently than monocytes. Journal of Bone and Mineral Research. 2010;25(3):661-672
128.Santiago-Schwarz F, Anand P, Liu S, Carsons SE. Dendritic cells (DCs) in rheumatoid arthritis (RA): Progenitor cells and soluble factors contained in RA synovial fluid yield a subset of myeloid DCs that preferentially activate Th1 inflammatory-type responses. Journal of Immunology. 2001;167(3):1758-1768
129.Rivollier A, Mazzorana M, Tebib J, Piperno M, Aitsiselmi T, Rabourdin-Combe C, et al. Immature dendritic cell transdifferentiation into osteoclasts: A novel pathway sustained by the rheumatoid arthritis microenvironment. Blood. 2004;104(13):4029-4037
130.Cline-Smith A, Axelbaum A, Shashkova E, Chakraborty M, Sanford J, Panesar P, et al. Ovariectomy activates chronic low-grade inflammation mediated by memory T cells, which promotes osteoporosis in mice. Journal of Bone and Mineral Research. 2020;35(6):1174-1187
131.Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. 2000;408(6812):600-605
132.Jin Y, Wi HJ, Choi MH, Hong ST, Bae YM. Regulation of anti-inflammatory cytokines IL-10 and TGF-beta in mouse dendritic cells through treatment with Clonorchis sinensis crude antigen. Experimental & Molecular Medicine. 2014;46(1):e74
133.Onal M, Xiong J, Chen X, Thostenson JD, Almeida M, Manolagas SC, et al. Receptor activator of nuclear factor kappaB ligand (RANKL) protein expression by B lymphocytes contributes to ovariectomy-induced bone loss. The Journal of Biological Chemistry. 2012;287(35):29851-29860
134.Sun W, Meednu N, Rosenberg A, Rangel-Moreno J, Wang V, Glanzman J, et al. B cells inhibit bone formation in rheumatoid arthritis by suppressing osteoblast differentiation. Nature Communications. 2018;9(1):5127
135.Fujiwara Y, Piemontese M, Liu Y, Thostenson JD, Xiong J, O'Brien CA. RANKL (receptor activator of NFkappaB ligand) produced by osteocytes is required for the increase in B cells and bone loss caused by estrogen deficiency in mice. The Journal of Biological Chemistry. 2016;291(48):24838-24850
136.Konnecke I, Serra A, El Khassawna T, Schlundt C, Schell H, Hauser A, et al. T and B cells participate in bone repair by infiltrating the fracture callus in a two-wave fashion. Bone. 2014;64:155-165
137.Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nature Medicine. 1996;2(10):1132-1136
138.Sapra L, Bhardwaj A, Mishra PK, Garg B, Verma B, Mishra GC, et al. Regulatory B cells (Bregs) inhibit osteoclastogenesis and play a potential role in ameliorating ovariectomy-induced bone loss. Frontiers in Immunology. 2021;12:691081
139.Srivastava RK, Sapra L. The rising era of “Immunoporosis”: Role of immune system in the pathophysiology of osteoporosis. Journal of Inflammation Research. 2022;15:1667-1698
140.Choi Y, Woo KM, Ko SH, Lee YJ, Park SJ, Kim HM, et al. Osteoclastogenesis is enhanced by activated B cells but suppressed by activated CD8(+) T cells. European Journal of Immunology. 2001;31(7):2179-2188
141.Terauchi M, Li JY, Bedi B, Baek KH, Tawfeek H, Galley S, et al. T lymphocytes amplify the anabolic activity of parathyroid hormone through Wnt10b signaling. Cell Metabolism. 2009;10(3):229-240
142.Bedi B, Li JY, Tawfeek H, Baek KH, Adams J, Vangara SS, et al. Silencing of parathyroid hormone (PTH) receptor 1 in T cells blunts the bone anabolic activity of PTH. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(12):E725-E733
143.Yamada A, Takami M, Kawawa T, Yasuhara R, Zhao B, Mochizuki A, et al. Interleukin-4 inhibition of osteoclast differentiation is stronger than that of interleukin-13 and they are equivalent for induction of osteoprotegerin production from osteoblasts. Immunology. 2007;120(4):573-579
144.Gao Y, Grassi F, Ryan MR, Terauchi M, Page K, Yang X, et al. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. The Journal of Clinical Investigation. 2007;117(1):122-132
145.Noonan K, Marchionni L, Anderson J, Pardoll D, Roodman GD, Borrello I. A novel role of IL-17-producing lymphocytes in mediating lytic bone disease in multiple myeloma. Blood. 2010;116(18):3554-3563
146.Lei H, Schmidt-Bleek K, Dienelt A, Reinke P, Volk HD. Regulatory T cell-mediated anti-inflammatory effects promote successful tissue repair in both indirect and direct manners. Frontiers in Pharmacology. 2015;6:184
147.Tyagi AM, Yu M, Darby TM, Vaccaro C, Li JY, Owens JA, et al. The microbial metabolite butyrate stimulates bone formation via T regulatory cell-mediated regulation of WNT10B expression. Immunity. 2018;49(6):1116-31 e7
148.Kar S, Gupta R, Malhotra R, Sharma V, Farooque K, Kumar V, et al. Interleukin-9 facilitates osteoclastogenesis in rheumatoid arthritis. International Journal of Molecular Sciences. 2021;22(19):10397
149.Monasterio G, Budini V, Fernandez B, Castillo F, Rojas C, Alvarez C, et al. IL-22-expressing CD4(+) AhR(+) T lymphocytes are associated with RANKL-mediated alveolar bone resorption during experimental periodontitis. Journal of Periodontal Research. 2019;54(5):513-524
150.Xu F, Li W, Yang X, Na L, Chen L, Liu G. The roles of epigenetics regulation in bone metabolism and osteoporosis. Frontiers in Cell and Development Biology. 2020;8:619301
151.Kiselev IS, Kulakova OG, Boyko AN, Favorova OO. DNA methylation As an epigenetic mechanism in the development of multiple sclerosis. Acta Naturae. 2021;13(2):45-57
152.Alaskhar Alhamwe B, Khalaila R, Wolf J, von Bulow V, Harb H, Alhamdan F, et al. Histone modifications and their role in epigenetics of atopy and allergic diseases. Allergy, Asthma and Clinical Immunology. 2018;14:39
153.Biswas S, Rao CM. Epigenetic tools (the writers, the readers and the erasers) and their implications in cancer therapy. European Journal of Pharmacology. 2018;837:8-24
154.Zhang P, Liu Y, Jin C, Zhang M, Lv L, Zhang X, et al. Histone H3K9 acetyltransferase PCAF is essential for osteogenic differentiation through bone morphogenetic protein signaling and may Be involved in osteoporosis. Stem Cells. 2016;34(9):2332-2341
155.Kim S, Shevde NK, Pike JW. 1,25-Dihydroxyvitamin D3 stimulates cyclic vitamin D receptor/retinoid X receptor DNA-binding, co-activator recruitment, and histone acetylation in intact osteoblasts. Journal of Bone and Mineral Research. 2005;20(2):305-317
156.Meyer MB, Benkusky NA, Sen B, Rubin J, Pike JW. Epigenetic plasticity drives adipogenic and osteogenic differentiation of marrow-derived mesenchymal stem cells. The Journal of Biological Chemistry. 2016;291(34):17829-17847
157.Meier JC, Tallant C, Fedorov O, Witwicka H, Hwang SY, van Stiphout RG, et al. Selective targeting of bromodomains of the bromodomain-PHD fingers family impairs osteoclast differentiation. ACS Chemical Biology. 2017;12(10):2619-2630
158.Yi SJ, Lee H, Lee J, Lee K, Kim J, Kim Y, et al. Bone remodeling: Histone modifications as fate determinants of bone cell differentiation. International Journal of Molecular Sciences. 2019;20(13):3147
159.Lee HW, Suh JH, Kim AY, Lee YS, Park SY, Kim JB. Histone deacetylase 1-mediated histone modification regulates osteoblast differentiation. Molecular Endocrinology. 2006;20(10):2432-2443
160.Feigenson M, Shull LC, Taylor EL, Camilleri ET, Riester SM, van Wijnen AJ, et al. Histone deacetylase 3 deletion in mesenchymal progenitor cells hinders long bone development. Journal of Bone and Mineral Research. 2017;32(12):2453-2465
161.Hakelien AM, Bryne JC, Harstad KG, Lorenz S, Paulsen J, Sun J, et al. The regulatory landscape of osteogenic differentiation. Stem Cells. 2014;32(10):2780-2793
162.Hemming S, Cakouros D, Isenmann S, Cooper L, Menicanin D, Zannettino A, et al. EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem Cells. 2014;32(3):802-815
163.Kota SK, Roening C, Patel N, Kota SB, Baron R. PRMT5 inhibition promotes osteogenic differentiation of mesenchymal stromal cells and represses basal interferon stimulated gene expression. Bone. 2018;117:37-46
164.Fang C, Qiao Y, Mun SH, Lee MJ, Murata K, Bae S, et al. Cutting edge: EZH2 promotes osteoclastogenesis by epigenetic silencing of the negative regulator IRF8. Journal of Immunology. 2016;196(11):4452-4456
165.Gao Y, Ge W. The histone methyltransferase DOT1L inhibits osteoclastogenesis and protects against osteoporosis. Cell Death & Disease. 2018;9(2):33
166.Ohguchi H, Harada T, Sagawa M, Kikuchi S, Tai YT, Richardson PG, et al. KDM6B modulates MAPK pathway mediating multiple myeloma cell growth and survival. Leukemia. 2017;31(12):2661-2669
167.Yang D, Okamura H, Nakashima Y, Haneji T. Histone demethylase Jmjd3 regulates osteoblast differentiation via transcription factors Runx2 and osterix. The Journal of Biological Chemistry. 2013;288(47):33530-33541
168.Rojas A, Aguilar R, Henriquez B, Lian JB, Stein JL, Stein GS, et al. Epigenetic control of the bone-master Runx2 gene during osteoblast-lineage commitment by the histone demethylase JARID1B/KDM5B. The Journal of Biological Chemistry. 2015;290(47):28329-28342
Written By
Sayantee Hazra, Shagnik Chattopadhyay and Ritobrata Goswami
Submitted: 30 August 2023Reviewed: 15 September 2023Published: 20 November 2023
 Open access peer-reviewed chapter
Open access peer-reviewed chapter