Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
Angiogenesis is pivotal for tumor growth and the development of metastases. Thus, vascular endothelial growth factor (VEGF) plays an integral role in angiogenesis and is often overexpressed in most solid cancers making it an attractive target for treatment. As the use of VEGF inhibitors is expanding in cancer therapy, it is increasingly recognized that these medications may result in significant adverse renal toxicities, including hypertension, proteinuria, thrombotic microangiopathy, and other glomerular diseases. In this chapter, we will discuss the importance of VEGF signaling in maintaining renal integrity, the various classes of VEGF inhibitors, and the adverse effects that affect the kidneys.
Kaiser Permanente Mid-Atlantic States Internal Medicine Residency, USA
Shanshan Gustafson
Kaiser Permanente Mid-Atlantic States Internal Medicine Residency, USA
Tulsi Mehta
Mid-Atlantic Permanente Medical Group (MAPMG): Nephrology Department, USA
*Address all correspondence to: hoon.x.chang@kp.org
1. Introduction
Pathological angiogenesis plays a key role in the growth and metastasis of malignancies. This process is largely driven by vascular endothelial growth factor (VEGF) produced by tumor cells. This signal protein acts through the VEGF receptor that stimulates endothelial cells to proliferate and migrate [1]. This signaling pathway is a therapeutic target in the management of many solid organ cancers as well as various retinal diseases. VEGF pathway inhibitors are first-line agents in the treatment of advanced colorectal cancer, metastatic breast cancer, and metastatic renal cell carcinoma [2, 3]. Intravitreal injection of anti-VEGF agents is the first line to treat age-related macular degeneration and diabetic macular edema [4]. New drugs are continuously in development, and their therapeutic benefits, as well as adverse events, are a topic of investigation. Nephrotoxicity is reported in 10–20% of patients and can result in a wide range of abnormalities. Clinical manifestations of anti-VEGF treatment range from asymptomatic proteinuria to renal failure. In this chapter, we will focus on the effects of systemic and intravitreal administration of VEGF inhibitors on kidney function.
The vascular endothelial growth factor (VEGF) signaling pathway is one of the major pathways of angiogenesis. VEGF-A, which is the most well-studied isoform within the VEGF family, is a proangiogenic peptide that exerts mitogenic effects specifically on vascular endothelial cells, thus promoting angiogenesis and increasing vascular permeability. The expression of VEGF-A increases in response to hypoxia, oncogene activation, and cytokine stimulation [5, 6].
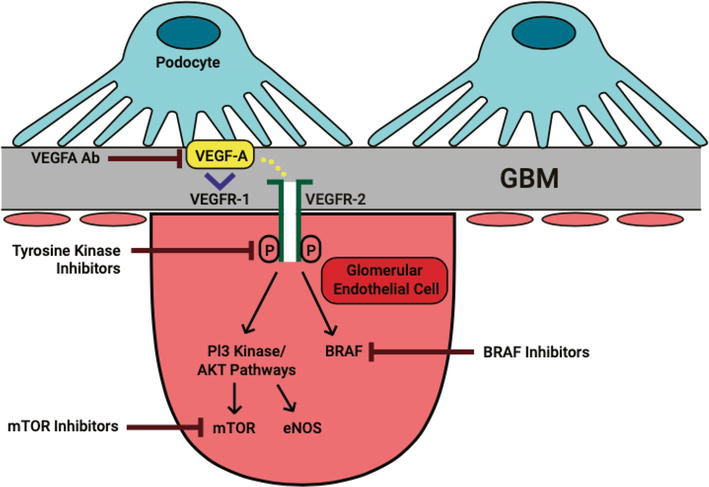
In the kidney, the VEGF-A is mainly expressed by glomerular podocytes and, to a lesser degree, by renal tubular cells [5, 6, 7, 8, 9]. The effect of the VEGF-A is mediated by its binding to receptor tyrosine kinases (RTKs) called VEGFR-1 and VEGFR-2 [5, 10]. The VEGFR1 has a higher affinity for VEGF-A compared to VEGFR-2 [11]; however, it has a less proangiogenic effect. In fact, the circulating soluble form of VEGFR1 (sVEGFR1) acts as a decoy receptor, inhibiting VEGF-A signaling [12]. VEGFR2 has been more extensively studied and is recognized as the most important receptor, responsible for the majority of VEGFA signaling (Figure 1).
Figure 1.
VEGFA-VEGFR2 signaling pathways and various therapeutic targets.
The VEGFR2 is mainly expressed on the surface of both endothelial cells and podocytes, although the presence of the receptors at the podocytes for autocrine effect is controversial. While some studies have established that VEGFR2 is expressed in adult mouse podocytes and glomerular endothelial cells [13, 14], others have failed to detect any expression of VEGFR2 in podocytes—suggesting that paracrine signaling may play a bigger role in VEGF-VEGFR signaling [15].
All RTKs, such as VEGFR2, share similar molecular structures with a ligand-binding site in the extracellular domain, a transmembrane region, and a cytoplasmic region that contains the protein kinase domain with an ATP-binding site [16]. The binding of VEGF-A to VEGFR2 leads to dimerization of the receptors and subsequent phosphorylation of tyrosine kinases in the cytoplasmic domain. Such interaction leads to activation of downstream signaling pathways such as the RAF/MAPK/ERK pathway, endothelial nitric oxide synthase (eNOS) pathway, and mammalian target of rapamycin (mTOR) pathway. While angiogenesis does not occur in normal adult kidneys, a delicate balance between proangiogenic and antiangiogenic factors is crucial for the normal structure and function of the glomerulus [8, 10].
The structural integrity at the renal capillary bed, consisting of endothelial cells, basement membrane, and the podocyte, is critical for maintaining proper glomerular filtration. The VEGF-mediated epithelial-endothelial crosstalk is an integral process for maintaining glomerular integrity [17, 18, 19].
Depending on the level of expression, VEGF-A can exert a varying range of effects. For example, multiple murine studies demonstrated that the knockout of Vegfa during embryogenesis is uniformly lethal at or before birth [20, 21]. In adult mice, Vegfa knockout mice can induce thrombotic microangiopathy (TMA) [18]. A clinical example of reduced VEGF-A signaling in the kidney is preeclampsia. Preeclampsia is a syndrome of pregnancy that is characterized by hypertension (HTN), proteinuria, and injury to various organs including the kidney. The renal histological features of preeclampsia involve the development of glomerular capillary endotheliosis. It has been found that patients with preeclampsia have increased levels of soluble VEGFR1/sFlt-1 and an overall decreased level of free VEGF and PlGF (placental growth factor), leading to disruption of the epithelial-endothelial crosstalk.
On the contrary, overexpression of VEGF-A and an unregulated increase in the downstream signaling pathway can lead to a different set of problems. A clinical example of renal overexpression of VEGF and its receptors is diabetic nephropathy. The increased VEGF signaling induces new vessel formation in the kidney, stimulates renal hypertrophy, and leads to proteinuria [6, 22].
3. Current drugs that target VEGF signaling pathway
Molecularly targeted therapies are the cornerstone of precision medicine and have greatly advanced the treatment of many cancers. Unlike traditional chemotherapy, these agents spare damage to healthy cells and can cause fewer side effects compared to conventional treatments. Pathological angiogenesis plays a key role in solid tumor growth and metastasis [23]. The VEGF pathway is one of the most potent promoters of angiogenesis and has become a key target in antiangiogenic therapies to treat various cancers. Numerous drug classes exist to inhibit VEGF and VEGFR signaling. Major classes include monoclonal antibodies (mAbs) against VEGF, receptor tyrosine kinase inhibitors (TKIs), and downstream inhibitors of the VEGF pathway [24]. The toxicity profiles associated with targeted inhibitors of the VEGF pathway differ from those seen with traditional cytotoxic chemotherapy. As a class, inhibitors of the VEGF pathway can cause damage to the glomerulus, tubules, and vasculature, causing various clinical outcomes such as hypertension, proteinuria, nephrotic syndrome, acute kidney injury, and electrolyte abnormalities.
3.1 Anti-VEGF MAB
Agents that inhibit VEGF-A activity through antibody binding include bevacizumab, ranibizumab, aflibercept, and ramucirumab. Bevacizumab and ranibizumab are monoclonal antibodies (mAbs) that inhibit angiogenesis by binding all isoforms of VEGF-A. Aflibercept is a recombinant soluble decoy receptor composed of extracellular binding domains of VEGFR1 and VEGFR-2 and the Fc portion of IgG that binds to VEGFRs. Like bevacizumab and ranibizumab, aflibercept binds all isoforms of VEGFA, but in addition, it binds related VEGFR1 ligands, VEGFB, and PIGF. Ramucirumab is a fully humanized mAB that specifically inhibits VEGFR-2. The renal toxicities associated with direct VEGF inhibition include hypertension and proteinuria. Bevacizumab was the first anti-VEGF drug introduced to clinical practice and is the focus of many clinical trials investigating anti-VEGF ligand blockade. Multiple meta-analyses have found a dose-dependent increase in the risk of hypertension and proteinuria in patients taking bevacizumab [25, 26]. Hypertension can occur any time after therapy initiation and may persist after prolonged treatment. Proteinuria is typically asymptomatic and decreases after treatment completion or dose reduction. Wu, et al. found that combination therapy of bevacizumab with chemotherapy was associated with a significantly increased risk for high-grade proteinuria (Grade 3 or above: urine protein ≥3.5 g/24 h or dipstick ≥4+ or nephrotic syndrome) and nephrotic syndrome compared to chemotherapy alone [27]. Multiple cases of biopsy-confirmed thrombotic microangiopathy (TMA) have been reported in patients treated with bevacizumab. In murine models, local genetic ablation of VEGF in kidney podocytes has been shown to lead to the development of microvascular injury and thrombotic microangiopathy [18]. Intravitreal administration of bevacizumab (off-label use), ranibizumab, and aflibercept are used to treat vision-threatening retinal diseases such as neovascular age-related macular degeneration, diabetic retinopathy, diabetic macular edema, and retinal vein occlusion [28]. Intravitreal injection of VEGF inhibitors has shown to cause significant depletion of circulating VEGF levels and can have systemic effects. Reizer et al. found that patients with and without preexisting hypertension both had significant elevations in blood pressure 3 weeks after intravitreal bevacizumab injection. Drug-induced hypertension resolved by week 6 for patients without baseline hypertension but persisted for those with preexisting hypertension and on the antihypertensive therapy [29].
3.2 Tyrosine kinase inhibitors of VEGF signaling
Tyrosine kinase inhibitors (TKIs) can interfere with various families of structurally similar RTKs, including VEGFR, platelet-derived growth factor (PDGF) receptor, epidermal growth factor (EGF) receptor, and fibroblast growth factor (FGF) receptor. They are commonly called multitargeted TKIs, and their effects depend on their specificities for different RTKs. TKIs directed at VEGFR include sorafenib, sunitinib, pazopanib, vandetanib, axitinib, regorafenib, cabozantinib, nintedanib, lenvatinib, dasatinib, and ponatinib. In contrast with anti-VEGF ligands and aflibercept, MTKIs are associated with increased risk for podocytopathies, including minimal change disease (MCN) and collapsing focal sclerosing glomerulosclerosis (cFSGN) [30]. MTKIs also result in dose-dependent increases in blood pressure. More potent MTKIs such as axitinib have shown higher rates of hypertension compared to sorafenib and sunitinib at the maximum tolerated doses [31, 32]. Proteinuria also depends on the specificity and potency of TKI. In a phase II trial of axitinib, a potent and specific VEGF-R TKI, 32% of patients (17 of 52) with renal cell carcinoma developed Grade 2 or higher proteinuria (measured by dipstick) and two patients had proteinuria >1 g per 24 hours.
3.3 Downstream inhibitors: b-Raf inhibitors, mTOR signaling, and eNOS inhibition
Angiogenesis can also be inhibited by targeting the downstream signaling pathways of VEGFR.
3.3.1 MAPK/ERK pathway
VEFG induces ERK1/2 signaling of the mitogen-activated protein kinase (MAPK) cascade. This pathway is important in regulating endothelial differentiation and proliferation. Novel chemotherapies targeting various portions of the MAPK/ERK pathway are currently in development. Vemurafenib and dabrafenib target B-Raf, an upstream component of the MEPK/ERK intracellular pathway, are approved agents for the management of melanoma. B-Raf inhibitors can lead to podocytopathy, slit diaphragm dysfunction, and subsequent proteinuria. A retrospective study by Muzet et al. found that 78% of patients (58 of 74) receiving vemurafenib for the treatment of metastatic melanoma developed AKI. Kidney biopsies were performed on three of the patients and showed tubular toxicity and interstitial fibrosis. Changes were chronic in two of the three cases [33]. In multiple studies, AKI on vemurafenib showed a male predominance [33, 34]. There is fewer data regarding the renal toxicity profile of dabrafenib. According to the US Food and Drug Administration (FDA) drug information packet, renal failure was present in 4% of patients (2 of 55) receiving a combination treatment of dabrafenib and trametinib (MEK inhibitor) for metastatic melanoma. Renal failure was typically associated with fever, chills, and dehydration and responded well to dose interruption and general supportive measures [35]. Hypophosphatemia has also been reported for both vemurafenib and dabrafenib [36].
3.3.2 eNOS pathway
Nitric oxide (NO) produced by endothelial cells is an essential molecule in modulating the proangiogenic properties of VEGF. VEGF activation of VEGFR-2 results in the activation of endothelial nitric oxide synthase (eNOS) and NO-induced vasodilation. Pharmacologic VEGFR blockade results in a reduction in downstream NO production, leading to reduced sodium excretion and endothelial injury, and vascular toxicities such as hypertension, thromboembolism, and pulmonary arterial hypertension [37].
3.3.3 mTOR inhibitors
The mammalian target of rapamycin (mTOR) of the phosphatidylinositol-3-kinase (Pi3K)/Akt signaling pathway is another important downstream RTK target. Inhibition of this pathway reduces VEGF-mediated angiogenesis and endothelial cell proliferation by both reducing VEGF synthesis and the VEGFR2-mediated signaling [38]. mTOR is also responsible for maintaining podocyte integrity via regulation of the autophagy [39]. mTOR inhibitors were initially developed as immunosuppressive agents and are still widely used in the setting of organ transplants. Currently, temsirolimus, ridaforolimus, and everolimus are approved for the treatment of various cancers such as renal cell carcinoma, neuroendocrine tumors, and hormone-receptor-positive breast cancer [40]. Everolimus is associated with proteinuria, and reported incidence varies widely between 3 and 36% [41]. Sirolimus is associated with de novo TMA in kidney transplant patients that developed in the absence of calcineurin inhibitors [39]. The proposed mechanism of nephrotoxicity of mTOR inhibitors involves the downregulation of VEGF signaling and disruption of the autophagic pathway (Table 1) [42].
4. Adverse effects of VEGF signaling inhibition related to renal function
4.1 Hypertension
Hypertension (HTN) is one of the most frequently reported adverse side effects related to therapeutic inhibition of VEGF signaling. Depending on the specific medication, the incidence and severity of hypertension may vary. For example, in one systematic review regarding Bevacizumab [26], the incidence of all-grade hypertension (Grade 1: asymptomatic, transient increase greater than 20 mmHg diastolic or greater than 150/100 mmHg; Grade 2 recurrent or persistent or symptomatic increase greater than 20 mmHg diastolic or greater than 150/100 mmHg) was as high as 32% in the low-dose group (Bevacizumab 3, 5, or 7.5 mg/kg/dose) and up to 36% in the high-dose group (Bevacizumab 10 or 15 mg/kg/dose). Interestingly, about 8.7 and 16% developed Grade 3 hypertension (defined as requiring more than one drug or more intensive therapy than previously) in the low-dose and high-dose groups, respectively. Among patients receiving the TKIs, the total incidence of all-grade or high-grade hypertension (Grade 3 or 4; Grade 4 is defined as a hypertensive crisis) was 23.0 and 4.4%, respectively [49].
The overall risk of hypertension was increased with combination therapy compared to monotherapy with antiangiogenesis therapy [56, 57]. As such, the development of HTN may lead to discontinuation of the drug and limited clinical use. Hypertension that develops in the setting of antiangiogenesis treatment is often a dose-dependent effect [26, 58, 59]. While the exact molecular processes for HTN related to VEGF inhibition remain elusive, several different mechanisms have been proposed.
First, the HTN from VEGF inhibition may be due to decreased levels of nitric oxide and prostacyclin, which are potent vasodilators [26, 59, 60, 61]. As briefly mentioned before, one of the downstream pathways of VEGF signaling is the eNOS pathway. VEGF-VEGFR activation stimulates the endothelial NO synthase via recruitment of phosphoinositide 3-kinase and mediates activation of Akt and eNOS and production of nitric oxide (NO) [59, 62]. The VIVA trial (VEGF in Ischemia for Vascular Angiogenesis) infused recombinant human VEGF, both intravenously and intra-coronary, and demonstrated a dose-dependent reduction in blood pressure [63]. Therefore, antagonism of VEGF is considered high risk for hypertension as it could lead to subsequent downregulation of the eNOS pathway, causing less NO production and elevation in vascular resistance. In addition to the direct vasodilatory effect, NO also participates in tubuloglomerular feedback, pressure natriuresis, and sodium balance [64, 65].
Another explanation is the dose-dependent release of a potent vasoconstrictor, endothelin 1 (ET-1), that is associated with VEGF inhibition [66, 67, 68]. TKIs, such as regorafenib and sunitinib, were associated with increased circulating endothelin-1 levels and subsequent elevations in the blood pressure. ET-1 exerts its vasoconstrictor effect via interactions with ETA/ETB receptors. Kappers et al. were able to demonstrate that the HTN from the VEGF inhibition can be reversed to the pretreatment level with the administration of ETA/ETB receptor blocker, tezosentan.
Vascular rarefaction, which is the decreased capillary density, has also been proposed as a contributor to VEGF-inhibition-related HTN. Because microvessels (arterioles and capillaries) are important contributors of peripheral vascular resistance, disruption of this may lead to hypertension. Several studies have shown that vascular rarefactions, either functional or structural rarefactions, are associated with the use of VEGF inhibitors [69, 70, 71].
Other proposed mechanisms also exist; however, the current evidence is conflicting. For example, it has been proposed that VEGF inhibition may reduce renal fractional excretion of sodium (FENa) and cause subsequent volume-dependent hypertension. The sodium reabsorption occurs via the sodium chloride co-transporter (NCC) in the distal tubule and the epithelial sodium channel (ENaC) in the collecting duct, respectively. It was hypothesized that inhibition of VEGFR2, which is expressed throughout the distal tubule and collecting duct [72], would cause increased ENaC and NCC expression and lead to decreased diuresis, natriuresis, and increased sodium reabsorption. While one study was able to demonstrate the proposed effect [73], the other study was not able to show a significant difference in the amount of ENaC and NCC expression in the VEGF inhibition group [74].
Although there have been speculations that VEGF inhibition may exaggerate the severity of angiotensin II-dependent hypertension, the HTN from VEGF inhibition was associated with reductions in renin mRNA expression [59, 68] and urinary aldosterone excretion [59]. Suppression of renin may be an early compensatory response to the increase in BP caused by VEGF signaling inhibition.
Interestingly, it has been suggested that the development of hypertension is linked to the treatment efficacy of the VEGF inhibition, especially in patients with renal cell carcinoma and osteosarcoma [2, 62, 75, 76, 77]. Given the improved clinical outcomes without significant HTN-associated adverse events, HTN can potentially serve as an efficacy biomarker for antiangiogenesis therapies.
4.2 Proteinuria
Proteinuria is another common adverse effect that is associated with VEGF inhibitors. The development of proteinuria often occurs due to disruption of the glomerular filtration barrier. The incidence and degree of proteinuria may vary. In most studies, grading for proteinuria was defined as the following: Grade 1—dipstick 1+ or 0.15 to 1.00 g/24 h; Grade 2—dipstick 2+ to 3+ or 1.0 to 3.5 g/24 h; Grade 3—dipstick 4+ or > 3.5 g/24 h; Grade 4—nephrotic syndrome; and Grade 5—death. The same systematic review by Zhu et al. reported that the incidence of all-grade proteinuria was as high as 63%. Grade 3 proteinuria was significantly lower at 1.8% (even in the high-dose bevacizumab group) [26]. Among patients who are receiving TKIs, the incidence of all-grade and high-grade (Grade 3 or higher) proteinuria was 18.7 and 2.4%, respectively, in another systematic review [50]. As with hypertension, the VEGF inhibitor-induced proteinuria appears to be a dose-dependent effect.
Among patients who develop proteinuria after VEGF inhibition, some may develop endothelial lesions or podocytopathies. Typically, anti-VEGF antibodies (such as Bevacizumab) have been associated with “thrombotic microangiopathy (TMA)-like” endothelial lesions (also known as pseudo-thrombi), while the TKIs have been generally associated with minimal change nephrotic syndrome (MCNS) or focal segmental glomerulosclerosis (FSGS). Although there is a case report of TMA and subsequent development of collapsing FSGS (cFSGS) [78], most patients do not develop both entities concomitantly [30, 78].
Unlike the usual systemic TMA that presents with thrombocytopenia and hemolytic anemia, the bevacizumab-induced injuries are limited to the kidneys only. A unique pattern that is specific to bevacizumab-related TMA is the hyaline occlusive glomerular microangiopathy, which is likely arising from endothelial leakage followed by subendothelial accumulation of serum proteins. The glomeruli undergo duplication of the glomerular basement membrane, loss of fenestration, and detachment of endothelial cells from the basement membrane [18]. Then, over time, the accumulated serum proteins cause the formation of microaneurysms. Solidifying of these leads to the formation of hyalinosis that is also known as “pseudo-thrombi” [79, 80, 81]. Such an entity must be considered in proteinuric cancer patients who are treated with anti-VEGF therapies.
Recent evidence suggests that complement alternative pathway may contribute to endothelial injury in the cases of TMA. Complement factor H (CFH) is considered the most important inhibitor of the alternative pathway and acts to prevent endothelial injury from the complement pathway. In one study, VEGF inhibition decreases local CFH and other complement regulators, thus making the endothelial cells more vulnerable to complement activation [80, 82].
Podocytopathy, mainly minimal change nephrotic syndrome and focal segmental glomerulosclerosis, is another renal structural injury associated with VEGF inhibitors, especially the TKIs. MCNS and FSGS are characterized by podocyte foot effacement. Such structural changes can lead to actin cytoskeleton disorganization and subsequent nephrotic range proteinuria. The podocyte injuries can be the result of changes in podocyte-associated proteins including those that assemble and stabilize the slit diaphragm and those that anchor the foot process to the glomerular basement membrane. Examples of those proteins include nephrin, podocin, synaptopodin, and podoplanin [83].
Nephrin phosphorylation has been found to be essential to maintaining slit diaphragm integrity. Nephrin interacts in vivo with VEGFR2 and activates downstream the Akt/PI3K pathway and regulates actin polymerization and stress fiber formation. Disruption of this crosstalk between VEGF and nephrin signaling has been associated with podocyte injuries [83, 84].
Another mechanism for glomerular injury and proteinuria with TKI use is the role of endothelial-to-mesenchymal transition (EndoMT). EndoMT is a novel source of myofibroblasts and contributes to renal fibrosis and subsequent development of chronic kidney disease. During the EndoMT, endothelial cells tend to change their phenotypes into a mesenchymal phenotype [83, 85]. Several studies already have demonstrated that EndoMT plays a crucial role in glomerular injury and subsequent albuminuria in diabetic nephropathy [86, 87]. The study done by Stavniichuck et al. was able to demonstrate that TKI use (Sorafenib) decreased renal cortical endothelial marker WT-1 mRNA expression while increasing the mesenchymal marker expression by 2–3-fold in a murine study.
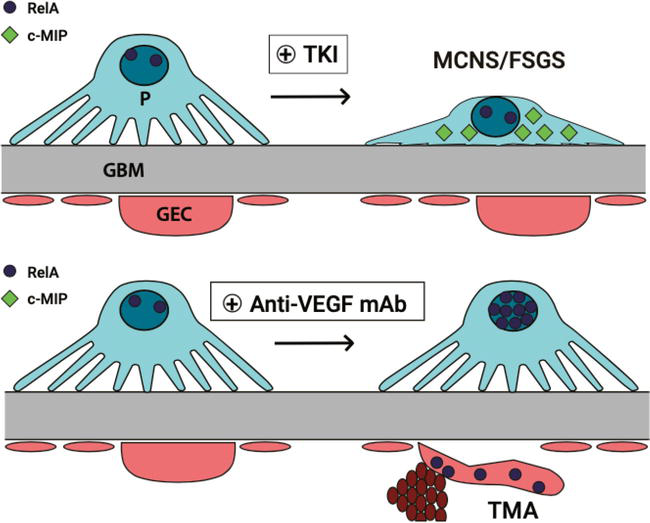
The balance between two intracellular proteins, c-MIP (C-Maf-inducing protein) and RelA (NF-kB transcription factor, also known as p65), has been associated with either the podocytopathy or the endothelial injuries associated with different VEGF inhibitors. TMA due to anti-VEGF therapy has been associated with increased expression of RelA with no detection of c-MIP. This leads to subsequent activation of the pro-inflammatory cascade, leading to endothelial injuries. On the contrary, MCNS/FSGS were only associated with C-MIP overexpression without significant detection of RelA. Overexpression of c-MIP has been associated with MCNS and FSGS, as the c-MIP interferes with the nephrin/Akt signaling pathway and causes subsequent podocyte injuries [43, 81] (Figure 2).
Figure 2.
The effect of TKI vs. anti-VEGF mAb on the balance of intracellular proteins, c-MIP and RelA, and associated structural injuries.
4.3 Other clinical considerations
Due to the frequent comorbidities that are concurrently present (such as hypertension and diabetes), cancer patients are at an increased risk of developing acute kidney injury and chronic kidney disease. Patients who are receiving antiangiogenic therapy may be at increased risk of kidney injury as these agents may induce direct cellular toxicity on endothelial cells and/or podocytes. The effect on kidney function can further be complicated if there is a simultaneous use of other systemic chemotherapies and/or if the patient underwent nephrectomy (which remains the gold standard treatment for non-metastatic renal cell carcinoma (RCC)) [81].
Contrast-induced nephropathy (CIN) appears to be more frequent in cancer patients who are receiving VEGF inhibitors. A small prospective cross-sectional study was able to show that the prevalence of CIN was significantly higher in the anti-VEDGF/R treatment group compared to the control group. No one type or specific VEGF inhibitor seemed to cause more CIN than the others [88].
Patients with previously stable proteinuria and chronic kidney diseases are at risk of developing worsening renal function after initiating VEGF inhibitors. In one study, the authors reported cases of worsening kidney function with biopsy-proven acute interstitial nephritis, diabetic nephropathy, and collapsing FSGS [47].
Unfortunately, research concerning the long-term renal function and chronic kidney disease with chronic VEGF inhibitor use in cancer patients is scarce. Patients who receive these therapies often have advanced cancers with many comorbidities. Long-term follow-up may not be feasible in many cases. It is also more difficult to perform kidney biopsies for histopathological examinations given the stability of these patient populations.
Some of the available currently regarding long-term effects on eGFR after VEGF inhibition show conflicting conclusions. In one study [89], a retrospective review of patients revealed that chronic use of intravitreal VEGF inhibitor (ranibizumab) did not significantly alter the rate of change in eGFR and/or albumin-to-creatinine ratio with an increasing number of treatment injections in patients with and without diabetic kidney disease. All of the participants had diabetic macular edema and received an average of 26.8 intravitreal anti-VEGF injections per patient over 31 months.
In contrast, a different study showed that chronic use of lenvatinib can induce a decline in eGFR, leading to the development of chronic kidney disease, especially with a duration greater than 2 years. This effect was observed regardless of the baseline eGFR. The presence of proteinuria was a risk factor for the decline in eGFR. Of note, patients with better baseline renal function were able to prolong the clinical outcome [90].
Of course, the amount of systemic circulation of the VEGF inhibitors is likely much higher in the study with lenvatinib (used for patients with thyroid cancer compared to intravitreal injections). It can be speculated that the impact on eGFR and subsequent development of chronic kidney disease may be a dose-dependent phenomenon.
4.4 Electrolyte disorders and cell damages
Various electrolyte disorders may occur with VEGF inhibitors as they may have direct nephrotoxic effects on tubular transporters. Of note, hypophosphatemia and hyponatremia are the most reported electrolyte disturbances associated with VEGF inhibitors. In some cases, hypomagnesemia, hypocalcemia, and hypokalemia may also occur [81].
VEGF inhibitors are also suspected to cause cell damage via several mechanisms including disruption of the cytoskeleton [91], tubular cell apoptosis [92], and autophagy [81].
5. Preventive strategies for minimizing nephrotoxicity
Renal toxicities associated with VEGF inhibition are an area of ongoing research as new therapeutic agents are constantly in development. The National Cancer Institute’s Investigational Drug Steering Committee published guidelines in 2010 advising to conduct a formal risk assessment for cardiovascular complications prior to starting treatment. Patients with preexisting hypertension should be identified and addressed before starting therapy, since they are expected to have an exacerbation of hypertension after starting treatment. Blood pressure should be measured throughout the treatment course and managed aggressively to a goal of under 140/90 mmHg. VEGF inhibition may need to be held if BP control is not achieved [93]. Calcium channel blockers (CCBs), potassium-sparing diuretics, and HCTZ are associated with the greatest decrease in BP in patients receiving anti-VEGF TKIs therapy [74, 94].
Renal function should be assessed prior to receiving antiangiogenic therapy. Patients should undergo a screening urinalysis to quantify proteinuria prior to beginning therapy and throughout the treatment course. Patients with preexisting proteinuria should be referred to a nephrologist for possible renal biopsy to determine the histological diagnosis. Inhibition of the RAAS system is first-line therapy for treating patients with proteinuria and hypertension on anti-VEGF agents. Though this remains to be validated in randomized, controlled trials. Of note, the use of ACE-I/ARB should be considered only if the patient has concurrent HTN and proteinuria as the RAAS is downregulated in VEGF inhibitor-induced hypertension [59]. Significant proteinuria, nephrotic syndrome, and TMA are indications for stopping therapy and obtaining a kidney biopsy. Patient education and awareness of expected systemic toxicities and management are important for promoting optimal use and adherence to therapy.
Molecularly targeted therapeutic agents have become a mainstay in the treatment of cancers. Anti-VEGF agents are commonly used in combination with other modalities such as traditional chemotherapy, radiotherapy, and immunotherapy. New antiangiogenic targets and combinations are under investigation. Delta-like ligand 4 (DLL4) is a component of the Notch signaling pathway and is highly expressed in actively growing vessels. It is a key negative regulator downstream of the VEGF signaling pathway and thus a promising target of antiangiogenesis therapy to augment the effects of VEGF inhibitors [95]. Currently, therapies against DLL4 alone and in combination with VEGF pathway inhibitors are being developed. ABT-165 is a bispecific anti-DLLF/VEGF antibody that has shown to be superior to single anti-VEGF treatment in preclinical models of colon cancer, breast cancer, and glioma when used alone and in combination with standard-of-care chemotherapeutics [96].
Currently, there are no set guidelines in the management of renal toxicities from anti-VEGF agents. Cancer patients who require these therapies are often at higher risk for kidney injury due to preexisting kidney disease and concomitant use of nephrotoxic chemotherapeutics. Patients who require intravitreal injection for diabetic macular edema and macular degeneration are also typically older and have some level of baseline renal dysfunction. Further research into the incidence and mechanisms of renal toxicity of antiangiogenic agents would help with the management. The development of early markers of vascular injury can help identify patients at risk for hypertension and proteinuria before they develop. Early recognition may allow for medication dose adjustment rather than premature discontinuation of a potentially beneficial anticancer therapy.
3.Meadows KL, Hurwitz HI. Anti-VEGF therapies in the clinic. Cold Springer Harbour Perspective Medicine. 2012;2(10):1-28
4.Fogli S, Del Re M, Rofi E, Posarelli C, Figus M, Danesi R. Clinical pharmacology of intravitreal anti-VEGF drugs. Eye (London, England). 2018;32(6):1010-1020
5.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. The FASEB Journal. 1999;13(1):9-22
6.Schrijvers BF, Flyvbjerg A, de Vriese AS. The role of vascular endothelial growth factor (VEGF) in renal pathophysiology. Kidney International. 2004;65(6):2003-2017
7.Kretzler M, Schröppel B, Merkle M, Huber S, Mundel P, Horster M, et al. Detection of multiple vascular endothelial growth factor splice isoforms in single glomerular podocytes. Kidney International. 1998;54:S159-S161
8.Simon M, Grone HJ, Johren O, Kullmer J, Plate KH, Risau W, et al. Expression of vascular endothelial growth factor and its receptors in human renal ontogenesis and in adult kidney. American Journal of Physiology-Renal Physiology. 1995;268(2):F240-F250
9.Whittle C, Gillespie K, Harrison R, Mathieson PW, Harper SJ. Heterogeneous vascular endothelial growth factor (VEGF) isoform mRNA and receptor mRNA expression in human glomeruli, and the identification of VEGF148 mRNA, a novel truncated splice variant. Clinical Science. 1999;97(3):303-312
10.Tanabe K, Wada J, Sato Y. Targeting angiogenesis and lymphangiogenesis in kidney disease. Nature Reviews Nephrology. 2020;16(5):289-303
11.Tanabe K, Sato Y, Wada J. Endogenous antiangiogenic factors in chronic kidney disease: Potential biomarkers of progression. International Journal of Molecular Sciences. 2018;19(7):1859
12.Sugimoto H, Hamano Y, Charytan D, Cosgrove D, Kieran M, Sudhakar A, et al. Neutralization of circulating vascular endothelial growth factor (VEGF) by anti-VEGF antibodies and soluble VEGF Receptor 1 (sFlt-1) Induces Proteinuria*. Journal of Biological Chemistry. 2003;278(15):12605-12608
13.Bertuccio C, Veron D, Aggarwal PK, Holzman L, Tufro A. Vascular endothelial growth factor receptor 2 direct interaction with nephrin links VEGF-A signals to actin in kidney podocytes. The Journal of Biological Chemistry. 2011;286(46):39933-39944
14.Veron D, Reidy KJ, Bertuccio C, Teichman J, Villegas G, Jimenez J, et al. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney International. 2010;77(11):989-999
15.Sison K, Eremina V, Baelde H, Min W, Hirashima M, Fantus IG, et al. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. Journal of the American Society of Nephrology: JASN. 2010;21(10):1691-1701
16.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117-1134
17.Dimke H, Maezawa Y, Quaggin SE. Crosstalk in glomerular injury and repair. Current Opinion in Nephrology and Hypertension. 2015;24(3):231-238
18.Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, et al. VEGF inhibition and renal thrombotic microangiopathy. The New England Journal of Medicine. 2008;358(11):1129-1136
19.Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. The Journal of Clinical Investigation. 2003;111(5):707-716
20.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380(6573):435-439
21.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380(6573):439-442
22.Tanabe K, Maeshima Y, Sato Y, Wada J. Antiangiogenic therapy for diabetic nephropathy. BioMed Research International. 2017;2017:5724069
23.Lee YT, Tan YJ, Oon CE. Molecular targeted therapy: Treating cancer with specificity. European Journal of Pharmacology. 2018;834:188-196
24.Zirlik K, Duyster J. Anti-Angiogenics: Current situation and future perspectives. Oncology Res earchTreatment. 2018;41(4):166-171
25.Ranpura V, Pulipati B, Chu D, Zhu X, Wu S. Increased risk of high-grade hypertension with bevacizumab in cancer patients: A meta-analysis. American Journal of Hypertension. 2010;23(5):460-468
26.Zhu X, Wu S, Dahut WL, Parikh CR. Risks of proteinuria and hypertension with bevacizumab, an antibody against vascular endothelial growth factor: Systematic review and meta-analysis. American Journal of Kidney Diseases. 2007;49(2):186-193
27.Wu S, Kim C, Baer L, Zhu X. Bevacizumab increases risk for severe proteinuria in cancer patients. Journal of the American Society of Nephrology. 2010;21(8):1381-1389. DOI: 10.1681/asn.2010020167
28.Nikkhah H, Karimi S, Ahmadieh H, Azarmina M, Abrishami M, Ahoor H, et al. Intravitreal injection of anti-vascular endothelial growth factor agents for ocular vascular diseases: Clinical practice guideline. Journal of Ophthalmic and Vision Research. Apr-Jun 2018;13(2):158-169. DOI: 10.4103/jovr.jovr_50_18
29.Rasier R, Artunay O, Yuzbasioglu E, Sengul A, Bahcecioglu H. The effect of intravitreal bevacizumab (avastin) administration on systemic hypertension. Eye. 2009;23(8):1714-1718
30.Izzedine H, Escudier B, Lhomme C, Pautier P, Rouvier P, Gueutin V, et al. Kidney diseases associated with anti-vascular endothelial growth factor (VEGF): An 8-year observational study at a single center. Medicine (Baltimore). 2014a;93(24):333-339
31.Chen HX, Cleck JN. Adverse effects of anticancer agents that target the Vegf pathway. Nature Reviews Clinical Oncology. Aug 2009;6(8):465-477. DOI: 10.1038/nrclinonc.2009.94
32.Drevs J, Siegert P, Medinger M, Mross K, Strecker R, Zirrgiebel U, et al. Phase I clinical study of Azd2171, an oral vascular endothelial growth factor signaling inhibitor, in patients with advanced solid tumors. Journal of Clinical Oncology. 20 Jul 2007;25(21):3045-3054. DOI: 10.1200/jco.2006.07.2066
33.Teuma, C., Muzet, C., & Pelletier, S. (2015). New insights in renal toxicity of BRAF inhibitor vemurafenib in patients with metastatic melanoma. Cancer and Kidney International Network (CKIN). Brussels, Belgium
34.Launay-Vacher V, Zimner-Rapuch S, Poulalhon N, Fraisse T, Garrigue V, Gosselin M, et al. Acute renal failure associated with the new BRAF inhibitor vemurafenib: A case series of 8 patients. Cancer. 2014;120(14):2158-2163
35.FDA U. Highlights of Prescribing Information. 2014. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/202806s002lbl.pdf
36.Wanchoo R, Jhaveri KD, Deray G, Launay-Vacher V. Renal effects of BRAF inhibitors: A systematic review by the Cancer and the Kidney International Network. Clinical Kidney Journal. 2016;9(2):245-251
37.Neves KB, Montezano AC, Lang NN, Touyz RM. Vascular toxicity associated with anti-angiogenic drugs. Clinical Science (London). 2020;134(18):2503-2520
38.Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: Involvement of vascular endothelial growth factor. Nature Medicine. 2002;8(2):128-135
39.Sartelet H, Toupance O, Lorenzato M, Fadel F, Noel LH, Lagonotte E, et al. Sirolimus-induced thrombotic microangiopathy is associated with decreased expression of vascular endothelial growth factor in kidneys. American Journal of Transplantation. 2005;5(10):2441-2447
40.Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Frontiers in oncology. 2014;4:64
41.Kaplan B, Qazi Y, Wellen JR. Strategies for the management of adverse events associated with mTOR inhibitors. Transplant Rev (Orlando). 2014;28(3):126-133
42.Cinà DP, Onay T, Paltoo A, Li C, Maezawa Y, De Arteaga J, et al. Inhibition of MTOR disrupts autophagic flux in podocytes. Journal of American Society Nephrology. 2012;23(3):412-420
43.Izzedine H, Mangier M, Ory V, Zhang S-Y, Sendeyo K, Bouachi K, et al. Expression patterns of RelA and c-mip are associated with different glomerular diseases following anti-VEGF therapy. Kidney International. 2014b;85(2):457-470
44.Pellé G, Shweke N, Duong Van Huyen JP, Tricot L, Hessaïne S, Frémeaux-Bacchi V, et al. Systemic and kidney toxicity of intraocular administration of vascular endothelial growth factor inhibitors. American Journal of Kidney Diseases. 2011;57(5):756-759
45.Cheungpasitporn W, Chebib FT, Cornell LD, Brodin ML, Nasr SH, Schinstock CA, et al. Intravitreal antivascular endothelial growth factor therapy may induce proteinuria and antibody mediated injury in renal allografts. Transplantation. 2015;99(11):2382-2386
46.Hanna RM, Barsoum M, Arman F, Selamet U, Hasnain H, Kurtz I. Nephrotoxicity induced by intravitreal vascular endothelial growth factor inhibitors: Emerging evidence. Kidney International. 2019;96(3):572-580
47.Shye M, Hanna RM, Patel SS, Tram-Tran N, Hou J, Mccannel C, et al. Worsening proteinuria and renal function after intravitreal vascular endothelial growth factor blockade for diabetic proliferative retinopathy. Clinical Kidney Journal. 2020;13(6):969-980
48.Qi WX, Shen Z, Tang LN, Yao Y. Risk of hypertension in cancer patients treated with aflibercept: A systematic review and meta-analysis. Clinical Drug Investigation. 2014;34(4):231-240
49.Liu B, Ding F, Liu Y, Xiong G, Lin T, He D, et al. Incidence and risk of hypertension associated with vascular endothelial growth factor receptor tyrosine kinase inhibitors in cancer patients: A comprehensive network meta-analysis of 72 randomized controlled trials involving 30013 patients. Oncotarget. 2016;7(41):67661-67673
50.Zhang Z-F, Wang T, Liu L-H, Guo H-Q. Risks of proteinuria associated with vascular endothelial growth factor receptor tyrosine kinase inhibitors in cancer patients: A systematic review and meta-analysis. PLOS ONE. 2014;9(3):e90135
51.Zhang W, Feng L-J, Teng F, Li Y-H, Zhang X, Ran Y-G. Incidence and risk of proteinuria associated with newly approved vascular endothelial growth factor receptor tyrosine kinase inhibitors in cancer patients: An up-to-date meta-analysis of randomized controlled trials. Expert Review of Clinical Pharmacology. 2020;13(3):311-320
52.Terashima T, Yamashita T, Takata N, Takeda Y, Kido H, Iida N, et al. Safety and efficacy of sorafenib followed by regorafenib or lenvatinib in patients with hepatocellular carcinoma. Hepatology Research. 2021;51(2):190-200
53.Yamazaki H, Iwasaki H, Takasaki H, Suganuma N, Sakai R, Masudo K, et al. Efficacy and tolerability of initial low-dose lenvatinib to treat differentiated thyroid cancer. Medicine (Baltimore). 2019;98(10):e14774
54.Okamoto I, Miyazaki M, Takeda M, Terashima M, Azuma K, Hayashi H, et al. Tolerability of nintedanib (BIBF 1120) in combination with docetaxel: A phase 1 study in Japanese patients with previously treated non-small-cell lung cancer. Journal of Thoracic Oncology. 2015;10(2):346-352
55.Ruebner RL, Copelovitch L, Evageliou NF, Denburg MR, Belasco JB, Kaplan BS. Nephrotic syndrome associated with tyrosine kinase inhibitors for pediatric malignancy: Case series and review of the literature. ogy. 2014;29(5):863-869
56.Tao L, Zhang H, An G, Lan H, Xu Y, Ge Y, et al. Balancing the risk–benefit ratio of immune checkpoint inhibitor and Anti-VEGF combination therapy in renal cell carcinoma: A systematic review and meta-analysis. Frontiers in Oncology. 2021;11:739263
57.Zhao J, He M, Li J, Li D, Zhao Y, Li X, et al. Apatinib combined with paclitaxel and cisplatin neoadjuvant chemotherapy for locally advanced esophageal squamous cell carcinoma. Cancer Biotherapy and Radiopharmaceuticals. May 2022;37(4):324-331
58.Agarwal M, Thareja N, Benjamin M, Akhondi A, Mitchell GD. Tyrosine kinase inhibitor-induced hypertension. Current Oncology Reports. 2018;20(8):65
60.Jin Z-G, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circulation Research. 2003;93(4):354-363
61.Neagoe P-E, Lemieux C, Sirois MG. Vascular endothelial growth factor (VEGF)-A165-induced prostacyclin synthesis requires the activation of VEGF Receptor-1 and -2 Heterodimer *. Journal of Biological Chemistry. 2005;280(11):9904-9912
62.Hamnvik O-PR, Choueiri TK, Turchin A, McKay RR, Goyal L, Davis M, et al. Clinical risk factors for the development of hypertension in patients treated with inhibitors of the VEGF signaling pathway. Cancer. 2015;121(2):311-319
63.Henry TD, Annex BH, McKendall GR, Azrin MA, Lopez JJ, Giordano FJ, et al. The VIVA Trial. Circulation. 2003;107(10):1359-1365
64.Hayman SR, Leung N, Grande JP, Garovic VD. VEGF inhibition, hypertension, and renal toxicity. Current Oncology Reports. 2012;14(4):285-294
65.Zou A-P, Cowley AW. Role of nitric oxide in the control of renal function and salt sensitivity. Current Hypertension Reports. 1999;1(2):178-186
66.Jesus-Gonzalez de N, Robinson E, Penchev R, Mehren M. von Heinrich MC, Tap W, Wang Q, Demetri G, George S, & Humphreys BD. Regorafenib induces rapid and reversible changes in plasma nitric oxide and endothelin-1. American Journal of Hypertension. 2012;25(10):1118-1123
67.Kappers MHW, de Beer VJ, Zhou Z, Danser AHJ, Sleijfer S, Duncker DJ, et al. Sunitinib-induced systemic vasoconstriction in swine is endothelin mediated and does not involve nitric oxide or oxidative stress. Hypertension. 2012;59(1):151-157
68.Kappers MHW, van Esch JHM, Sluiter W, Sleijfer S, Danser AHJ, van den Meiracker AH. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension. 2010;56(4):675-681
69.Mourad J-J, des Guetz G, Debbabi H, Levy BI. Blood pressure rise following angiogenesis inhibition by bevacizumab. A crucial role for microcirculation. Annals of Oncology. 2008;19(5):927-934
70.Steeghs N, Gelderblom H, Roodt J, Christensen O, Rajagopalan P, Hovens M, et al. Hypertension and rarefaction during treatment with telatinib, a small molecule angiogenesis inhibitor. Clinical Cancer Research. 2008;14(11):3470-3476
71.van der Veldt AAM, de Boer MP, Boven E, Eringa EC, van den Eertwegh AJM, van Hinsbergh VW, et al. Reduction in skin microvascular density and changes in vessel morphology in patients treated with sunitinib. Anti-Cancer Drugs. 2010;21(4):439-446
72.Kanellis J, Fraser S, Katerelos M, Power DA. Vascular endothelial growth factor is a survival factor for renal tubular epithelial cells. American Journal of Physiology-Renal Physiology. 2000;278(6):F905-F915
73.Grisk O, Koenen A, Meissner T, Donner A, Braun D, Steinbach A, et al. Rho kinase inhibition mitigates sunitinib-induced rise in arterial pressure and renal vascular resistance but not increased renal sodium reabsorption. Journal of Hypertension. 2014;32(11):2199-2210
74.Witte J, Lampe J, Koenen A, Urbaneck I, Steinbach A, Rettig R, et al. The role of distal tubule and collecting duct sodium reabsorption in sunitinib-induced hypertension. Journal of Hypertension. 2018;36(4):892-903
75.Lv B, Chen J, Liu X-L. Anlotinib-induced hypertension: Current concepts and future prospects. Current Pharmaceutical Design. 2021;2021:27
76.Rini BI, Cohen DP, Lu DR, Chen I, Hariharan S, Gore ME, et al. Hypertension as a biomarker of efficacy in patients with metastatic renal cell carcinoma treated with sunitinib. JNCI: Journal of the National Cancer Institute. 2011;103(9):763-773
77.Xie L, Xu J, Sun X, Tang X, Yan T, Yang R, et al. Anorexia, hypertension, pneumothorax, and hypothyroidism: Potential signs of improved clinical outcome following apatinib in advanced osteosarcoma. Cancer Management and Research. 2020;12:91-102
78.Phadke G, Hanna RM, Ferrey A, Torres EA, Singla A, Kaushal A, et al. Review of intravitreal VEGF inhibitor toxicity and report of collapsing FSGS with TMA in a patient with age-related macular degeneration. Clinical Kidney Journal. 2021;14(10):2158-2165
79.Ozawa M, Ohtani H, Komatsuda A, Wakui H, Takahashi N. VEGF-VEGFR2 inhibitor-associated hyaline occlusive glomerular microangiopathy: A Japanese single-center experience. Clinical and Experimental Nephrology. 2021;25(11):1193-1202
80.Tonooka A, Ohashi R. Current trends of anti-cancer molecular targeted therapies: A narrative review focusing on renal complications and their histological features. Journal of Nippon Medical School. 2022;89(2):128-138
81.van Wynsberghe M, Flejeo J, Sakhi H, Ollero M, Sahali D, Izzedine H, et al. Nephrotoxicity of Anti-Angiogenic Therapies. Diagnostics (Basel, Switzerland). 2021;11(4). DOI: 10.3390/diagnostics11040640. Available online: https://www.ncbi.nlm.nih.gov/pubmed/33916159
82.Keir LS, Firth R, Aponik L, Feitelberg D, Sakimoto S, Aguilar E, et al. VEGF regulates local inhibitory complement proteins in the eye and kidney. The Journal of Clinical Investigation. 2017;127(1):199-214
83.Stavniichuk A, Savchuk O, Khan AH, Jankiewicz WK, Imig JD. A sorafenib induced model of glomerular kidney disease. Visnyk Kyivs’koho Natsional’noho Universytetu Imeni Tarasa Shevchenka. Biolohiia. 2020;81(2):25-31
84.New LA, Martin CE, Scott RP, Platt MJ, Keyvani Chahi A, Stringer CD, et al. Nephrin tyrosine phosphorylation is required to stabilize and restore podocyte foot process architecture. Journal of the American Society of Nephrology: JASN. 2016;27(8):2422-2435
85.Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. The American Journal of Pathology. 2011;179(3):1074-1080
86.Liu F, Zhang S, Xu R, Gao S, Yin J. Melatonin attenuates endothelial-to-mesenchymal transition of glomerular endothelial cells via regulating miR-497/ROCK in diabetic nephropathy. Kidney and Blood Pressure Research. 2018;43(5):1425-1436
87.Ma Z, Zhu L, Liu Y, Wang Z, Yang Y, Chen L, et al. Lovastatin alleviates endothelial-to-mesenchymal transition in glomeruli via suppression of oxidative stress and TGF-β1 signaling. Frontiers in Pharmacology. 2017;8:473
88.Gökyer A, Küçükarda A, Köstek O, Hacıoğlu MB, Uzunoğlu S, Kula O, et al. Contrast nephropathy in cancer patients receiving anti-VEGF therapy: A prospective study. International Journal of Clinical Oncology. 2020;25(10):1757-1762
89.O’Neill RA, Gallagher P, Douglas T, Little J-A, Maxwell AP, Silvestri G, et al. Evaluation of long-term intravitreal anti-vascular endothelial growth factor injections on renal function in patients with and without diabetic kidney disease. BMC Nephrology. 2019;20(1):478
90.Masaki C, Sugino K, Kobayashi S, Hosoi Y, Ono R, Yamazaki H, et al. Impact of lenvatinib on renal function: Long-term analysis of differentiated thyroid cancer patients. BMC Cancer. 2021;21(1):894
91.Calizo RC, Bhattacharya S, van Hasselt JGC, Wei C, Wong JS, Wiener RJ, et al. Disruption of podocyte cytoskeletal biomechanics by dasatinib leads to nephrotoxicity. Nature Communications. 2019;10(1):2061
92.Xiao J, Wang J, Yuan L, Hao L, Wang D. Study on the mechanism and intervention strategy of sunitinib induced nephrotoxicity. European Journal of Pharmacology. 2019;864:172709
93.Maitland ML, Bakris GL, Black HR, Chen HX, Durand JB, Elliott WJ, et al. Initial assessment, surveillance, and management of blood pressure in patients receiving vascular endothelial growth factor signaling pathway inhibitors. Journal of the National Cancer Institute. 2010;102(9):596-604
94.Waliany S, Sainani KL, Park LS, Zhang CA, Srinivas S, Witteles RM. Increase in blood pressure associated with tyrosine kinase inhibitors targeting vascular endothelial growth factor. JACC: CardioOncology. Sep 2019;1(1):24-36. DOI: 10.1016/j.jaccao.2019.08.012
95.Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, et al. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proceedings of the National Academy Science USA. 2007;104(9):3219-3224
96.Li Y, Hickson JA, Ambrosi DJ, Haasch DL, Foster-Duke KD, Eaton LJ, et al. ABT-165, a dual variable domain immunoglobulin (DVD-Ig) targeting DLL4 and VEGF, demonstrates superior efficacy and favorable safety profiles in preclinical models. Molecular Cancer Therapeutics. 2018;17(5):1039-1050
Written By
Hoon Chang, Shanshan Gustafson and Tulsi Mehta
Submitted: 31 March 2022Reviewed: 16 April 2022Published: 08 September 2022
 Open access peer-reviewed chapter
Open access peer-reviewed chapter