Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
We report that mitochondrial encephalomyopathy, lactic acidosis, and stroke-like eposodes (MELAS) was also present in a patient with a preemptive living renal transplantation who had autosomal dominant polycystic kidney disease (ADPKD) as a primary disease. A 20-years-old female was referred to our hospital from a nearby hospital 4 years ago for preemptive living renal transplantation (Donor: her mother blood type incompatible A → O type) for chronic renal failure due to ADPKD. She was hospitalized X-3 years for renal transplantation. Since the rate of GFR decline was 18 ml/min/1.73 mm2/year and the progression of renal function was fast for ADPKD, detailed examination of other diseases was performed. The sensory deafness, impaired glucose tolerance and dilated heart were recognized, and the presence of MELAS was confirmed, and renal transplantation was postponed and hemodialysis was introduced. The donor was also diagnosed with mitochondrial abnormality, but it was judged that abnormality was slight and donation was possible. Recipients are expected to improve ADL by renal transplantation, after approval by the ethics committee, living renal transplantation was performed 2 years ago. At present, the general condition is stable and the renal function is stable at Cr 0.67 mg/dl. There are few reports of renal transplantation in patients with MELAS and we report a rare case with some review of the literature.
Kinuyama Clinic, Matsuyama City, Ehime Prefecture, Japan
*Address all correspondence to: shirodesu2010@yahoo.co.jp
1. Introduction
Preemptive kidney transplantation (PEKT) is on the rise, with 42% of living donor kidney transplants performed in Japan in 2018 [1]. A large portion of patients with chronic renal failure due to autosomal dominant polycystic kidney disease (ADPKD) as a primary disease are reportedly eligible for PEKT because of the slow progression of renal function [2]. ADPKD is a mutation of the PKD gene that causes tubular cells to dilate, resulting in cysts. With age, renal function progressively declines, and about half of patients develop end-stage renal failure by the age of 60 [3, 4]. Glomerular filtration rate (GFR) begins to decline at around 40 years of age, with an average decline rate of 4.4 to 5.9 ml/min/year, and genetic factors, renal volume, and hypertension may affect the rate of decline in renal function [5]. In this case, the total renal volume capacity was 541 ml and less than 750 ml, the age was less than 30 years, and ADPKD classification was 1D. Despite the rate of decline in GFR predicted to be 3.48 ml/min/1.73 mm2/year [6], the rate of decline in GFR in the past year was as fast as 18 ml/min/1.73 mm2/year. A close examination of concomitant diseases as a cause of decreased renal function revealed sensorineural hearing loss, short stature, hypertension, glucose intolerance, and cardiac enlargement. Further examination revealed Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes (MELAS). MELAS causes Central nervous system symptoms and muscle diseases due to mitochondrial dysfunction caused by mitochondrial gene mutations, and also decreased renal function in some cases [7, 8]. We report a case of a patient who underwent living donor kidney transplantation after hemodialysis with a good outcome.
Complaint General malaise, weakness attacks, headache, vomiting.
Family medical history Father, ADPKD, living donor kidney transplant.
Past medical history 19 years old, chronic glomerulonephritis, 19 years old, ADPKD.
History of present illness The patient was diagnosed as ADPKD by a nearby doctor after proteinuria was detected in her school medical examination, and was referred to our hospital in X-4 for a prior kidney transplant with her mother as the donor (blood type incompatible donor type A → recipient type O) for chronic renal failure due to ADPKD. After outpatient follow-up, the patient was admitted for renal transplantation in X-3. The rate of decline in GFR was fast for ADPKD, 18 ml/min/1.73 mm2/year, and general malaise and weakness attacks appeared just before admission. A close examination of concomitant diseases as a cause of decreased renal function revealed sensorineural hearing loss, short stature, hypertension, glucose intolerance, and cardiac enlargement. Further examination revealed MELAS. Renal transplantation was postponed and hemodialysis was started.
Illness on admission Height 135 cm, Weight 29.8 kg, Physical examination Moderate sensorineural hearing loss, Intermittent weakness attacks in both upper and lower limbs.
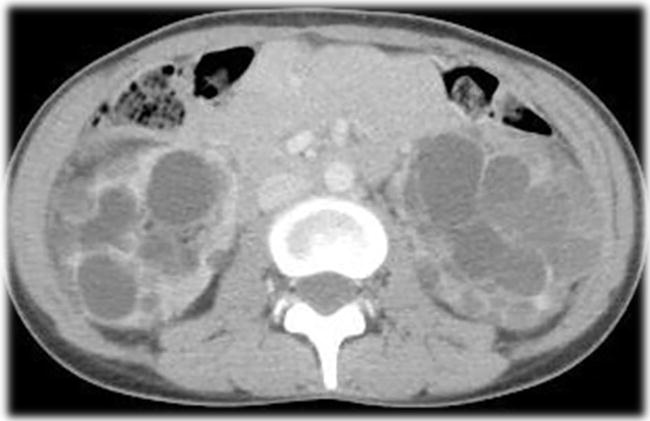
Finding on admission CT scan showed multiple cysts in both kidneys. Renal volume was 228 ml in the right kidney and 313 ml in the left kidney (Figure 1). Blood tests (Table 1) showed mild anemia, decreased renal function, and high levels of lactate 29.8 mg/dl (reference value 4.2–17), pyruvate 1.6 mg/dl (0.3–0.9), and myoglobin 327.4 ng/ml (below 60). CSF findings showed a significantly high lactate level of 29.8 mg/dl. General chest imaging showed significant cardiac enlargement and bilateral pleural effusions. Cardiac ultrasonography showed cardiac ejection fraction of 33%, wall thickening and granular sparkling, and a diagnosis of cardiomyopathy. MRI showed cerebellar atrophy and EEG showed phase reversal of a spike in the right occipital lobe. Audiological examination revealed sensorineural hearing loss (average hearing level of 53.8 dB on the right and 56.3 dB on the left according to the quadrant method).
Muscle biopsy showed ragged-red-fibers (RRF) with red-purple staining of enlarged mitochondria by Gomori-Trichrome staining, and strong SDH-reactive blood vessels (SSV) by Succinate dehydrogenase (SDH) staining, consistent with MELAS.
With the suspected MELAS based on the clinical symptoms, blood samples, spinal fluid examination, and imaging studies, we performed genetic testing after obtaining written consent from the patient and guardian. The mitochondrial gene 3243 A > G mutation was found (mutation rate 50%), and together with the clinical symptoms, MELAS was diagnosed. Since the disease is maternally inherited, the mother, as the donor, also underwent genetic testing and was found to have a 3243 A > G mutation in the mitochondrial gene. However, the mutation rate was low at 6%, and the clinical symptoms were only mild deafness and glucose intolerance, which were not diagnosed as mitochondrial disease (Table 2), but as mitochondrial dyscrasia. Renal transplantation was postponed, and hemodialysis was started. After 6 months of hemodialysis, we confirmed the recipient’s stable general condition. We reconfirmed her intention to undergo renal transplantation, and both the patient and her mother were very eager to undergo renal transplantation. For the problems of the recipients, the prognosis of MELAS is grim, with a mean age of death of 40 years in the adult form, and there is a risk of malignant hyperthermia, prolonged action of muscle relaxants and anesthetics, and hyperlactatemia during general anesthesia management [9, 10]. A joint conference was held with neurology, anesthesiology, and cardiology, and confirmed that although the patient already had cerebral atrophy and cardiac disease, ADL could be improved by renal transplantation, and that perioperative management could be addressed by devising anesthetics and infusions (using bicarbonate Ringer’s solution). No other donor candidates were found, and although the mother had mitochondrial abnormalities, she did not meet the diagnostic criteria for mitochondrial disease. Taken together, the risk of renal function progression was judged to be low. After obtaining approval from the ethics committee of our hospital, we explained the risks to the patient and her family again, confirmed their willingness to undergo transplantation, and decided to proceed with the kidney transplantation.
Main symptoms
① Progressive muscle weakness, deafness, diabetes mellitus or external ophthalmoplegia
② One or more of the following Central nervous system symptoms: intellectual regression, memory impairment, seizures, psychiatric symptoms, aphasia/phasia, severe vision loss, transient paralysis, hemianopsia/cortical blindness, myoclonus, dystonia, cerebellar ataxia
③ One or more of the following: cardiac symptoms such as cardiac transmission disorder and cardiomyopathy, renal symptoms such as glomerulosclerosis and renal tubular dysfunction, hematologic symptoms such as severe anemia, and hepatic symptoms of moderate severity or greater.
Examination and imaging findings
① Repeatedly high lactate levels in serum or CSF at rest or lying down, or a clear lactate peak in the lesion area on MR spectroscopy
② Brain CT/MRI showing infarct-like lesions, cerebral or cerebellar atrophy, or bilateral symmetrical lesions in the basal ganglia or brainstem
③ Muscle biopsy or abnormal mitochondrial morphology in the symptomatic organ
④ Deficiency of mitochondria-related enzymes or deficiency of intermediate metabolites such as coenzyme Q10
⑤ Qualitative or quantitative abnormalities of mitochondrial DNA or mitochondria-related nuclear gene mutations.
Diagnosis of mitochondrial disease
Definite cases: at least one of (1) ① to ③ and at least two of (2) ① to ⑤
Suspicious cases: at least one of (1) ① to ③ and at least one of (2) ① to ⑤
Table 2.
Diagnostic criteria for mitochondrial disease.
3.1 Perioperative course
The blood group antibody titer was 64-fold in an incompatible blood type living donor kidney transplant (donor type A and recipient type O). Anti-donor specific antibodies were negative. Preoperatively, the patient underwent one double-membrane filtration plasma exchange and one simple plasma exchange, and rituximab 200 mg/body was administered preoperatively. Immunosuppressive drugs such as tacrolimus 0.15 mg/kg, Cellcept 500 mg/body, and methylprednisolone 10 mg/body were started 1 week before surgery, and the patient underwent living donor kidney transplantation in X-2 years. For anesthesia, intravenous anesthetics (propofol and remifentanil) were used instead of inhalation anesthetics and muscle relaxants, and lactate Ringer’s solution was used instead of starting solution and bicarbonate Ringer’s solution to prevent lactate level from rising. The operation time was 368 minutes, the anesthesia time was 501 minutes, the total blood loss was 73 minutes and 43 seconds, and the blood loss was 322 ml. First urination was immediately observed, and there was no delay in postoperative awakening or prolonged respiratory depression. Although a weakness seizure appeared on postoperative day 4, the symptoms resolved with conservative treatment. Her renal function stabilized at 0.77 mg/dl of Cr without rejection, and she was discharged on postoperative day 35. Two years after the surgery, no rejection was observed, her renal function was 0.67 mg/dl and no proteinuria was observed, and her MELAS was stable with no exacerbation of stroke symptoms such as convulsions and paralysis, although she complained of intermittent headaches.
ADPKD reportedly occurs in 1 in 1000 people, and is a good indication for renal transplantation because of the better survival rate of renal transplantation than that of non-ADPKD patients, and is also a good indication for PEKT because of the slow progression of renal function [11]. In this case, mitochondrial disease (MELAS) was found during the examination of concomitant diseases due to rapid progression of renal function. Mitochondria exist in all cells in the body except red blood cells, while mitochondrial disease is a systemic disease caused by mitochondrial dysfunction mainly due to mitochondrial gene mutations [12].
Mitochondrial dysfunction causes abnormalities in energy production and apoptosis, resulting in symptoms in various organs throughout the body, especially in the brain, heart, and skeletal muscles, requiring energy, and is often associated with diabetes and nephropathy (Table 2) [13]. In addition to MELAS, myoclonic epilepsy with ragged red fibers (MERRF), chronic progressive external ophthalmoplegia (Kearns-Sayre syndrome), and myoclonic epilepsy with ragged red fibers (MELAS) are classified according to the combination of characteristic symptoms. In addition to MELAS, there are chronic progressive external ophthalmoplegia/Kearns-Sayre syndrome (CPEO/KSS) and Leigh’s encephalopathy, and MELAS is the most common, accounting for 30% [14].
The diagnostic criteria for mitochondrial disease are shown in Table 2 [15]. In this case, the main symptoms were muscle weakness, cardiomyopathy, hearing loss, glucose intolerance, and renal symptoms, and the examination and imaging findings were high blood and spinal fluid lactate levels, cerebellar atrophy on MRI, abnormal mitochondrial morphology on muscle biopsy, and mitochondria-related gene mutations. The patient was diagnosed as mitochondrial disease, meeting the three criteria for mitochondrial disease and the four criteria for laboratory and imaging findings. In addition, according to the diagnostic criteria for MELAS (Table 3), the patient showed symptoms of headache, vomiting, and cerebellar atrophy, as well as high lactate levels, abnormal mitochondrial morphology, and mitochondrial gene mutation (A3243G mutation) on muscle biopsy. Taken together, she was diagnosed as having MELAS, fulfilling two items in A and three items in B [16]. ATP deficiency due to mitochondrial disorders reportedly causes tubular damage, and furthermore, glomerulosclerosis due to podocyte dysfunction and microvascular damage, which can lead to Fanconi syndrome, tubulointerstitial nephritis, and secondary focal glomerulosclerosis [8, 17]. The possibility of mitochondrial disease, including MELAS, should be actively suspected in patients with progressive renal dysfunction refractory to treatment, renal biopsy findings of tubular damage and secondary focal glomerulosclerosis, multiple organ abnormalities other than renal, growth abnormalities and growth retardation, and suspected maternal inheritance. Since the symptoms of headache and vomiting may overlap with those of uremia, it is often difficult to differentiate between the two, as there have been case reports of renal failure preceding multi-organ symptoms [18].
Clinical findings of stroke
Headache/vomiting 2.Convulsion 3.Hemiplegia
homonymous hemianopsia or cortical blindness
Abnormal brain localization on brain imaging (CT, MRI, etc.)
Evidence of mitochondrial abnormality
Repeatedly high lactate levels in blood or CSF, or deficiency of mitochondria-related enzymes
Abnormal mitochondrial morphology (e.g., ragged-red fibers) on muscle biopsy
Known genetic abnormality (MELAS-related) (A3243G abnormality)
Certification criteria/Definite cases
Two items in A and two items in B above (at least 4 items in total)
Certification criteria/Suspicious cases
Two items in A and one item in B above (at least three items in total)
Table 3.
Diagnostic criteria for MELAS.
In this case, the patient’s father also had chronic renal failure due to ADPKD and had received a kidney transplant. The patient was also diagnosed with chronic renal failure due to ADPKD, and the delay in examining other diseases may have contributed to the delay in diagnosis. For MELAS, without fundamental treatment to improve mitochondrial function, symptomatic treatment of symptoms in each organ has been the mainstay of treatment [12]. Recently, oral L-arginine reportedly improves the unbalanced distribution of blood flow in the brain and prevent stroke-like attacks, and taurine replacement therapy for abnormal taurine modification of tRNA in MELAS with mitochondrial gene mutation (A3243G mutation) prevents recurrence of stroke-like attacks, and has been started in this case [19, 20, 21]. Regarding renal replacement therapy for patients with end-stage renal failure due to mitochondrial disease, it has been reported that patients with cardiomyopathy often have difficulty in continuing hemodialysis due to circulatory instability, and have been shifted to peritoneal dialysis [22]. Regarding the reports of renal transplantation for end-stage renal failure with mitochondrial disease as the primary disease, Paul et al. reported that all 11 observable cases (including 0 cases with MELAS) survived and transplanted kidneys were attached (observation period: 5.6 years). In contrast, two cases of renal transplantation in patients with MELAS-induced renal failure have been reported, although all of them reported only the perioperative course and not the prognosis [23, 24, 25]. Perioperative precautions include the onset of malignant hyperthermia caused by inhalational anesthetics, prolonged action of muscle relaxants and anesthetics, and hyperlactatemia [10, 21]. In this case, we did not use inhalational anesthetics or muscle relaxants, but used a total intravenous anesthetic with propofol [10, 21]. For infusion, lactated Ringer’s solution was not used to prevent lactate level from rising, but mainly starting solution was used. After autonomic urination, bicarbonate Ringer’s solution was also used, and the infusion volume was about 80% of that of normal renal transplant patients (10 ml/kg/hr) to avoid cardiac stress. Awakening from anesthesia was normal, and blood lactate level after extubation was as high as 30.6 mg/dl. However, without exacerbation of MELAS symptoms such as shivering, headache, muscle weakness, and epileptic seizures, Cr was stable at 0.77 mg/dl. Two years have passed since the kidney transplantation, without clinical deterioration of MELAS, and the renal function is stable. We will need to continue to monitor her closely.
We reported that mitochondrial encephalomyopathy, lactic acidosis, and stroke-like eposodes (MELAS) was also present in a patient with a preemptive living renal transplantation who had autosomal dominant polycystic kidney disease (ADPKD) as a primary disease. At present, the patient is taking arginine and taurine, and her general condition is stable, including renal function, without any stroke attacks. We will need to continue to monitor her closely. In cases of unexplained progressive renal hypofunction with multiple organ abnormalities, growth abnormalities, and growth retardation, it is important to consider mitochondrial disease (including MELAS) as a differential when maternal inheritance is suspected.
1.Japanese Society for Clinical Renal Transplantation/The Japan Society for Transplantation. Annual progress report from the Japanese renal transplant registry: Number of renal transplantations in 2017 and a follow-up survey. Japanese Journal of Transplantation. 2018;53(2-3):89-108
2.Ryan AH et al. Identifying barriers to preemptive kidney transplantation in a living donor transplant cohort. Transplant Rirect. 2018;4(4):356
3.Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. The New England Journal of Medicine. 2008;359:1477-1485
4.Higashihara E et al. Prevalence and renal prognosis of diagnosed autosomal dominant polycystic kidney disease in Japan. Nephron. 1998;80:421-427
5.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: The last 3 years. Kidney International. 2009;76(2):149-168
6.Irazabal MV et al. CRISP Investigators. Imaging classification of autosomal dominant polycystic kidney disease: A simple model for selecting patients for clinical trials. J Am Soc Nephrol. 2015;26:160-172
7.Ikawa M, Yoneda M. Pathomechanisms of mitochondrial diseases (Commentary/Special Issue). Medical Science Digest. 2018;44(10):559-562
8.Niaudet P, Rotig A. The Kidney in mitochondrial cytopathies. Kidney International. 1997;51:1000-1007
9.Yatsuga C et al. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochimica et Biophysica Acta. 2012;1820:619-624
10.Hashimoto K et al. Anesthetic management of a patient with mitochondrial encephalomyopathy for renal transplantation. Masui. 2009;58:629-632
11.Jacquet A et al. Outcomes of renal transplantation in patients with autosomal dominant polycystic kidney disease: A nationwide longitudinal study. Transplant International. 2011;24:582-587
12.Ikawa M, Yoneda M. Mitochondrial diseases. Nihon Naika Gakkai Zasshi. 2017;106:1584-1590
13.The Japanese Society of Mitochondrial Research and Medicine. Mitokondoriabyou shindannmanyuaru 2017. Tokyo: sinndann to tiryousha; 2017. pp. 6-7
14.DiMauro S, Scho EA. Mitochondrial respirator-chain diseases. The New England Journal of Medicine. 2003;348:2656-2668
15.Koga Y. Diagnosis and treatment of mitochondrial myopathy: Update review. No to Hattatsu. 2010;42:124-129
16.Koga, Y.: Mitokondoriabyoupanfurextuto, kouseiroudoukagakukennkyuuhishounisixtukanrinnshoukennkyuuzigyoushounikihaxtusyounomitokonndorianoukinnsyounitaisuru L-arugininnoyobizikurorosakusannryouhounokoukahannteitobunnsibyoutaiwohumaetaatarasiitiryouhoukaihatunikannsururinnshoukennkyuu」. pp. 3-8, 2005
17.Kurayama R, Yo K. Mitokondoriaijoushou. jinntotouseki. 2012;72:440-444
18.Motoda A et al. A case of MELAS with G13513A mutation presenting with chronic kidney disease long before stroke-like episodes. Rinshō Shinkeigaku. 2013;53(6):446-451
19.Koga H et al. L-arginine improves the symptoms of stroke-like spisodes in MELAS. Neurology. 2005;64:710-712
20.Sunada Y. Taurine supplemental therapy for MELAS. Igaku no Ayumi. 2017;260:93-97
21.Watanabe M et al. A family demonstrating two cases (mother and daughter) of mitochondrial encephalomyopathy (MELAS) with end-stage renal failure treated by long-term CAPD. Nihon Toseki Igakkai Zasshi. 2004;37:151-156
22.Paul DL et al. Five non-mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes phenotype adult patients with m.3243A>G mutation after kidney transplantation: follow-up and review of the literature. Clinical Kidney Journal. 2019;12(6):840-846
23.Humeidan ML et al. Anesthetic consideration for renal transplant surgery in patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes syndrome: a case report. Journal of Clinical Anesthesia. 2016;34:344-347
24.Lederer SR et al. MELAS: A mitochondrial disorder in an adult patient with a renal transplant. Wien Klin Wochensch. 2010;122(11-12):363-365
25.Veyckemans F et al. More on mitochondrial myopathies. Anesthesia and Analgesia. 2016;122:579-580
Written By
Shiro Fujikata
Submitted: 21 March 2022Reviewed: 25 March 2022Published: 08 September 2022
 Open access peer-reviewed chapter
Open access peer-reviewed chapter