 Open access peer-reviewed chapter
Open access peer-reviewed chapter
Abstract
Kidney transplantation is considered the best treatment option for end-stage kidney disease, as it provides significant improvements over dialysis in terms of both survival and quality of life. Various developments in the field of immunosuppression have greatly improved graft survival, and the immunosuppressive treatments currently used include molecules inducing T cell depletion and molecules able to control the activation and proliferation of T cells. Combinations of calcineurin inhibitors, antimetabolites, and glucocorticoids remain the gold standard for kidney transplantation. However, novel molecules designed to minimize renal toxicity, through calcineurin inhibitor sparing, are increasingly being identified. These molecules and the existing combination strategies are discussed here, together with novel therapeutic protocols for mitigating the individual risk of graft rejection and adverse events related to long-term immunosuppression.
Keywords
- kidney transplantation
- immunosuppressive drugs
- management of immunosuppression
1. Introduction
Kidney transplantation is now the standard treatment for patients with end-stage renal disease, as it outperforms dialysis in terms of quality of life and survival [1, 2, 3]. In 2019, kidney transplantation was the leading type of solid organ transplantation worldwide, with 107,540 new kidney graft transplantations (rate of 14.01 per million people—data source: GODT—transplant-observatory). Successful kidney transplantation depends as much on the optimal management of immunosuppression as on meticulous clinical care. Historically, graft rejection was the first barrier to successful transplantation. Graft rejection is an immunological response directed against alloantigens present on the graft and mediated by both the cellular and humoral arms of the immune system. Patients undergoing solid organ transplantation require long-term immunosuppression to inhibit T-cell activation and limit the development of antiallograft effector T cells and antibodies, to prevent graft rejection. Various developments in the field of immunosuppression have led to considerable progress in terms of short-term graft survival, with a decrease in the frequency of acute rejection. However, long-term graft survival has remained stable in recent decades. In the United States, the five-year survival for transplanted kidney grafts was 74.4% for cadaveric donor transplants and 85.6% for living donor transplants for the 2008–2015 period (data source: OPTN—optn.transplant.hrsa.gov). The overall graft failure rate after 10 years was 20% [4].
Powerful immunosuppressive drugs can prevent alloimmune responses, but this benefit must be balanced against several adverse effects. Mortality has decreased significantly over the last 20 years, reaching 1.9 per 100 patient-years in 2015–2018, with improvement most marked for the acute post-transplant period (0–3 months after transplantation). However, death rates over time follow a U-shaped curve, reflecting the existence of two periods of particularly high risk—the first 3 months after the death and a progressive increase in the risk of death from 5 years after transplantation. Cardiovascular disease (36.3%), cancers (27.5%), and infections (16.9%) are the main causes of death [5].
2. The alloimmune response
T cells play a key role in establishing the alloimmune response and are, therefore, the main target of immunosuppressive treatment (Figure 1). T-cell activation requires the presence of an antigen within the peptide-binding groove of major histocompatibility complex (MHC) molecules.
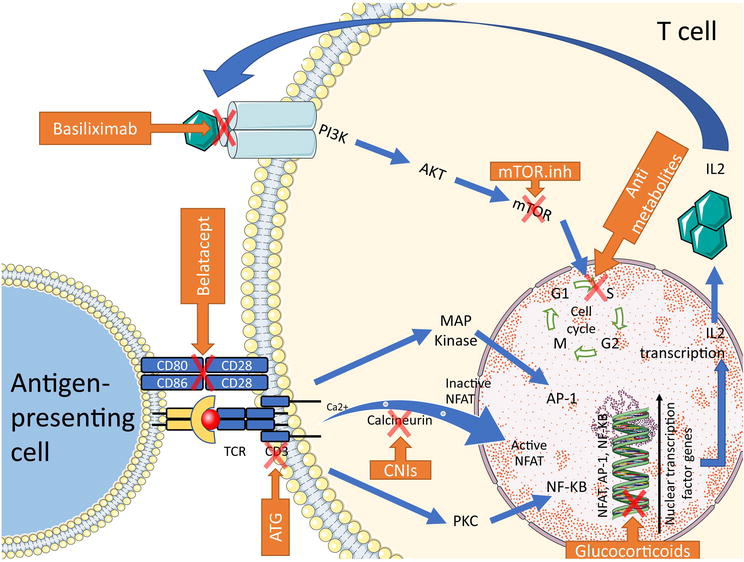
Figure 1.
T-cell activation and sites of action of immunosuppressive agents. ATG: Anti-thymocyte globulins; AP-1: Activator protein 1; CNIs: Calcineurin inhibitors; IL2: Interleukin-2; MAPK: Mitogen-activated protein kinase; mTOR: Mammalian target of rapamycin; mTOR.Inh: Mammalian target of rapamycin inhibitors; NFAT: Nuclear factor of activated T cells, NF-κB: Nuclear factor kappa B; PI3K: Phosphatidylinositol 3 kinases; PKC: Protein kinase C; TCR: T-cell receptor. (Servier medical art – Creative commons attribution 3.0 France license).
Lymphocyte activation requires three signals and results in clonal expansion. The first signal is the interaction between the T cell and an antigen-presenting cell (APC) presenting an allopeptide to the T-cell receptor via its type I or type II MHC. The second costimulatory signal is provided by the interaction between CD28 on the T cell and CD80/CD86 on the APC. These two signals activate the calcium/calcineurin, RAS/MAP-kinase, NFAT, and NFκB pathways, leading to (i) CD40 ligand expression, (ii) the production of various cytokines, including interleukin 2 (IL-2), the key cytokine for T-cell proliferation, and (iii) transcription of the alpha chain of the IL-2 receptor (α-CD25), improving the affinity of IL-2 for its receptor. The third signal involves IL-2 binding to its receptor via an autocrine/paracrine pathway. This binding leads to activation of the Pi3K/AKT/mTOR pathway and the lymphocyte proliferation signal [6].
3. Available immunosuppressants
3.1 Balancing the risk of graft rejection against the risk of infection
Most transplants are allografts. It is almost impossible to obtain a perfect match between donor and recipient, and powerful immunosuppressive drugs are, therefore, needed to limit immunological rejection. In clinical practice, the need to balance the risk of graft rejection against that of infection tends to lead to the personalization of immunosuppression protocols [7].
Calcineurin inhibitors (CNIs) probably provide the best immune protection during the first year of treatment, but in the long term, this advantage is outweighed by chronic renal toxicity and adverse metabolic effects. Novel molecules have been developed to minimize the toxicity of immunosuppressive treatment. However, only a few of these molecules are considered suitable as alternatives to CNIs as their benefits are frequently outweighed by disappointing results in terms of allograft rejection or adverse effects.
3.2 Therapeutic arsenal
Various molecules for eliminating T cells or inhibiting lymphocyte activation and proliferation have been developed (Figure 1).
3.2.1 Anti-thymocyte globulins (ATG)
Anti-thymocyte globulins (ATG) are a mix of polyclonal immunoglobulins G from rabbits or horses directed against human thymocytes and T-cell lines. Its broad antigen targets result in a depletion of T and B cells through complement-mediated or activation-associated death. This approach has been shown to prevent acute rejection in highly sensitized patients [8]. However, its lack of specificity, associated with a need for higher doses, increases the risk of adverse effects [9, 10]. Immune reconstitution takes place over a period of months or years following the initial perfusion, with memory T cells reappearing more rapidly than naive T cells. Prolonged CD4 T-cell lymphopenia has been reported after the administration of ATG. This prolonged lymphopenia was associated with morbidity and mortality [11], with atherosclerosis and cardiovascular events, in particular [12].
3.2.2 Monoclonal anti-CD25 antagonist (anti-IL2 receptor antibody): basiliximab
IL-2 receptor (rIL2) activation leads to T-cell proliferation (Figure 1). Basiliximab (anti-rIL2) is a chimeric antagonist antibody targeting the α-CD25 chain of the IL-2 receptor. This interaction leads to the competitive inhibition of IL-2 binding to the IL-2 receptor, thus inhibiting T-cell proliferation.
3.2.3 Glucocorticoids
Glucocorticoids exert immunosuppressive effects by inhibiting the production of various proinflammatory cytokines, such as IL-1, IL-2, IL-6, and IFNγ. They bind to cytosolic glucocorticoid receptors, resulting in migration toward the nucleus, where they downregulate the expression of genes encoding transcription factors, such as AP1 or NF-κB. At high concentrations, glucocorticoids regulate adaptive immunity by inhibiting lymphocyte activation and promoting lymphocyte apoptosis. They also suppress CD4 T-cell activation indirectly, by modulating dendritic cell function (antigen presentation, costimulation, and cytokine production) [13].
3.2.4 Calcineurin inhibitors (CNIs): cyclosporine-A and tacrolimus
Since their discovery in the 1980s, CNIs have been the cornerstone of immunosuppressive treatment. Two molecules are available—cyclosporine-A (CsA) and tacrolimus. These small molecules interact with cytosolic proteins called immunophilins. CsA binds cyclophilins, whereas tacrolimus binds FKBP12. These interactions lead to the competitive inhibition of calcineurin, a phosphatase responsible for dephosphorylation of the transcription factor NFAT. By inhibiting calcineurin, both drugs block the transcription of NFAT, resulting in the inhibition of the T-cell activation cascade [14].
Tacrolimus has been shown to be superior to CsA. In a seven-year follow-up study, the proportion of patients free from acute rejection was 77% in the tacrolimus arm, but only 59.9% in the CsA arm, with better graft survival in the tacrolimus arm (60.2% in the tacrolimus arm versus 47.0% in the CsA arm; p < 0.0001) [15].
However, some redundancy in this pathway has been observed, and the combination of other molecules with CNIs is required to prevent graft rejection. CNIs have a narrow therapeutic window, requiring constant monitoring to improve their efficacy and reduce their toxicity. Their nephrotoxicity remains a major issue. CNIs cause direct reversible vasoconstriction, which can lead to acute kidney injury. They have also been implicated in chronic vascular lesions, such as arteriole hyalinosis, tubulointerstitial fibrosis, and thrombotic microangiopathy, probably through direct endothelial cell injury. They have also been associated with a higher risk of developing hypertension, dyslipidemia, and de novo diabetes, all of which are associated with cardiovascular risk factors and mortality [16, 17].
3.2.5 Belatacept
Belatacept is a cytotoxic T-lymphocyte-associated protein-4-immunoglobulin fusion protein (CTLA4-Ig). It selectively blocks the CD28-CD80/CD86 pathway, thereby preventing the costimulation required for T-cell activation [18]. The main advantage of this molecule is the absence of nonimmune adverse events, including nephrotoxicity in particular. The phase III BENEFIT study [19, 20] showed significant improvements in the glomerular filtration rate in patients on belatacept. Seven-year follow-up data confirmed these findings, with improvements in allograft kidney function and survival in the longer term. Belatacept also seems to improve the metabolic profile [21]. Interestingly, the frequency of
Persistent viral infections have recently been reported in patients on belatacept. During the initial phase of development, a higher incidence of Epstein–Barr virus (EBV) associated post-transplantation lymphoproliferative disorder (PTLD) related to primary infection with EBV was observed in recipients. The use of belatacept has, therefore, generally been restricted to EBV-seropositive patients. A frequency of opportunistic infections of 12% was recently reported in a retrospective cohort, essentially due to Cytomegalovirus (CMV) reactivation and pneumocystis pneumonia [24]. In CMV-seronegative recipients receiving an organ from a seropositive donor, belatacept use was shown to be associated with a higher incidence of CMV viremia, a higher rate of first-line treatment failure, a longer time to viral clearance, and cases of severe CMV retinitis [25, 26, 27]. Some cases of deadly progressive multifocal leukoencephalopathy related to the JC polyomavirus reactivation have also been described in patients on belatacept [20, 28, 29]. This possible increase in the risk of infection requires confirmation in a larger case–control study, but the available data nevertheless suggest that such events should be monitored very carefully. Belatacept is a CD28-CD80/CD86 costimulation blocker, and, as such, it can induce T-cell anergy. T-cell anergy is defined as a long-term state of T-cell hyporesponsiveness in response to suboptimal activation, including a lack of costimulation due to CD28-CD80/CD86 blockade [30]. It can be very challenging to treat viral infections related to immunological mechanisms of T-cell anergy because we currently have no means of reversing T-cell anergy [31]. Thus, the use of belatacept [32] and future biotherapies capable of inducing T-cell anergy may lead to the emergence of a subset of viral infections that are extremely difficult to manage.
3.2.6 Antimetabolites: azathioprine and mycophenolate mofetil
Azathioprine was the first antimetabolite drug to be developed. It is metabolized to 6-mercaptopurine (6-MP). The thiopurine S-methyltransferase (TPMT) enzyme then catalyzes the metabolism of 6-MP to generate three metabolites—two inactive metabolites and the active 6-thioguanine nucleotide (TGN) metabolite. TGNs are incorporated into the DNA of replicating cells, leading to an inhibition of DNA synthesis, and the impairment of B- and T-cell proliferation. TGNs can also decrease the number of circulating monocytes by inhibiting the cell cycle in bone marrow promyelocytes. Some genetic polymorphisms within the TPMT gene can lead to a decrease in TPMT enzyme activity, or even its total abolition, resulting in higher levels of TGNs and a higher risk of severe bone marrow suppression [33, 34].
The use of azathioprine has decreased considerably since the turn of the century, and this molecule has been largely replaced by mycophenolate mofetil (MMF). MMF is a reversible inhibitor of inosine monophosphate dehydrogenase (IMDPH) isoform 2, an enzyme required for the
3.2.7 Mammalian target of rapamycin inhibitors (mTOR.Inh): sirolimus and everolimus
Mammalian target of rapamycin inhibitors (mTOR.inh) blocks T-cell progression through the cell cycle by inhibiting the mTOR kinase. mTOR.inh bind FKBP-12 and inhibit the mTOR kinase, resulting in cell-cycle arrest in the G1-S phase. This effect leads to the inhibition of the T-cell proliferation signal [39].
Two molecules are currently available—sirolimus and everolimus. Despite the antiviral properties of these molecules and their potential value compared to CNI, the generalized use of mTor.inh has been limited by their adverse effects and the higher incidence of acute rejection in pivotal clinical trials. In the SYMPHONY study, the rate of acute rejection in patients treated with rapamycin was 60% higher than that in patients treated with low doses of cyclosporine with the same associated immunosuppressive therapies, and twice that in patients treated with low doses of tacrolimus [40]. In addition, mTOR.inh may cause renal lesions via various mechanisms, including proteinuria and microangiopathy. The key benefits of mTOR.inh treatment may be the antiproliferative effects of these drugs, which are beneficial in the long term for preventing chronic graft dysfunction and reducing the risk of cancers [41, 42]. Several studies have highlighted the effect of these drugs in reducing tumor development or preventing cancer recurrence.
4. Conventional protocols
Immunosuppression protocols are based on the rational combination of multiple immunosuppressive drugs with different mechanisms of action on T cells. The choice of immunosuppression protocol should be made after an individual assessment of the risks of rejection and opportunistic infection and based on the adverse effects of the drugs concerned. The variables usually considered are the immunological risk and the age of the recipient, the type of donor (living or deceased), HLA matching, and the presence of comorbid conditions and infections.
Current protocols usually combine (i) induction therapy and (ii) maintenance therapy based on corticosteroids, CNIs, and antimetabolites. Traditionally, the immunological risk of the recipient has dictated the intensity of immunosuppression. Patients are therefore classified as described below:
Recipients with low immunological risk: Antibody induction may not be necessary for recipients of organs from HLA-identical donors. Moreover, protocols without continuous exposure to CNIs or steroids can also be used in such patients [43, 44, 45]. Nevertheless, physicians should be aware that less intensive treatment, particularly with a secondary minimization of immunosuppression, may result in acute rejection, highlighting the need for meticulousness when trying to minimize immunosuppression [43].
Recipients with a standard risk: This category includes recipients without HLA sensitization, and with a first isolated kidney transplant from a non-identical HLA donor. In these patients, anti-rIL2 antibodies have been shown to prevent acute rejection effectively, and generally have a better tolerability profile than that reported for anti-thymocyte globulin [46, 47]. Moreover, anti-rIL2 induction makes it possible to reduce the CNI dose. In the SYMPHONY trial [40], induction with an anti-rIL2 antibody in association with low doses of CsA gave similar acute rejection rates to a standard dose of CsA without rIL2 induction. Nevertheless, as basiliximab is not a depleting antibody and induces milder immunosuppression than ATG, it is considered to be associated with a higher immunological risk and is not advised in highly sensitized patients [48].
Recipients with high immunological risk: This category includes recipients with high levels of PRA (panel reactive antibodies—usually ≥85%), DSA, or who have undergone re-transplantation or simultaneous (kidney and another organ) transplantation, and have delayed graft function. In these patients, more intense immunosuppression is required, through the use of anti-thymocyte globulin induction and a combination of potent immunosuppressive drugs [49, 50].
Recipients with a very high immunological risk: This category includes ABO-incompatible transplant recipients, patients with positive cross-matching, and patients undergoing HLA desensitization. These patients require very complex management due to the high risk of severe hyperacute rejection. Powerful immunosuppression protocols are available, including the use of polyclonal immunoglobulins, plasmapheresis, rituximab, anti-thymocyte globulin induction, and maintenance immunosuppression with tacrolimus, mycophenolate mofetil, and corticosteroids [51, 52].
4.1 Induction therapy
The aim of induction therapy is to prevent the activation of the immune system provoked by new alloantigens. ATG and anti-rIL2 antibodies are typically used. High doses of corticosteroids are also part of the induction treatment, as they decrease the initial phase of inflammation by remodeling the innate and adaptive immune responses.
In addition to increasing the risk of neoplasia and infection, the use of ATG has been associated with higher cardiovascular mortality [53]. Based on these limitations, it seems reasonable to reserve this treatment, as far as possible, for patients with a high immunological risk, and to prefer basiliximab for more general use, particularly given the increasing evidence of its safety.
4.2 Maintenance therapy
Long-term maintenance therapy is crucial to maintain control over the immune system so as to avoid allograft rejection. Maintenance therapy usually consists of a combination of CNIs, typically tacrolimus, an antimetabolite molecule, as mycophenolate mofetil and corticosteroids. The combination of these different molecules blocks T-cell activation via different pathways simultaneously, making it possible to decrease the dose of each treatment, thereby limiting the toxicity of individual molecules.
4.3 Treatment of allograft rejection episodes
Three main types of allograft rejection have been described based on the time course of their development:
Hyperacute rejection – resulting from pre-existing DSA reacting with the endothelium. This type of rejection can occur within minutes of transplantation, following massive complement activation. Remarkable advances in our understanding of HLA and correct management of immunosuppression have greatly decreased the risk of hyperacute rejection.
Acute rejection – acute rejection is an immunological response mediated primarily by T cells. Treatments based on corticosteroids and ATG are, therefore, usually effective for the treatment of acute cellular rejection.
Chronic rejection – chronic rejection may occur over a period of months or years after transplantation. It involves several immunological mechanisms, including, in particular, humoral immunity, with activation of the complement pathway. The success of current immunosuppression regimes has resulted in the survival of more than 90% of kidney allografts 1 year after transplantation. However, chronic rejection remains a major issue and its treatment is still a matter of debate. Plasma exchange, polyclonal immunoglobulins, corticosteroids, and rituximab are the most common treatments, but the standard of care practices have not yet been clearly defined [54].
5. New developments
5.1 CNI minimization or sparing with mTOR inhibitors
An alternative protocol based on a combination of mTOR.inh, MMF, and steroids was initially developed for CNI sparing. Unfortunately, rejection rates were significantly higher for patients treated with this protocol than for patients receiving CNIs [40, 55]. The risk of
However, mTOR.inh can be used in cases of CNI minimization. In several studies, the authors chose to keep CNI doses as low as possible, whilst adding mTOR.inh treatment. These studies showed this strategy to be effective [56, 57, 58]. Moreover, viral infection rates were 60% lower for CMV infections and 45% lower for BK polyomavirus-associated nephropathy [59]. These results led many teams to review their protocols and to propose similar CNI minimization strategies for patients at low immunological risk. An optimal everolimus concentration (≥3 ng/mL) was found to be associated with significantly lower rates of acute rejection [60]. In addition, mTOR.inh blocks the cell cycle in many different cell types and has been shown to decrease the incidence of skin cancer recurrence, suggesting potential additional benefits of this treatment for preventing tumors in transplant patients.
5.2 CNI sparing by immunotherapy
Belatacept is the first biotherapy to be used as an alternative to CNIs in kidney transplantation. Its lack of nephrotoxicity and better metabolic profile are the main benefits of this molecule. Furthermore, belatacept is administered as intravenous perfusion, which makes it easier to ensure that it is administered effectively, thereby decreasing the risk of occult nonadherence.
Since 2010, belatacept has been tested in combination with MMF and steroids, in association with anti-rIL2 induction therapy. Two studies, BENEFIT (for donors with standard criteria [19]) and BENEFIT EXT (for donors with extended criteria [61]), have compared the
Surprisingly, higher rates of acute cellular rejection have been reported in patients on belatacept, and various effector memory T-cell phenotypes have been observed [62, 63]. Several studies have identified “risky” memory T-cell subsets associated with belatacept-resistant rejection, including CD4 T cells with a CD28+ effector-memory phenotype or a CD57+ PD1− profile. TIGIT, an immune checkpoint receptor, is expressed on these memory CD4 T-cell subsets. The use of a neutralizing anti-TIGIT antibody significantly increased apoptotic cell death rates for these cells [64]. Recently, it has also been shown that the proliferation of belatacept-resistant T cells requires early IFNα pathway activation. The inhibition of type I IFN or IL-6 reduces the proliferation of belatacept-resistant T cells [65]. These new developments will pave the way for the identification of new therapeutic targets for preventing belatacept-resistant rejection after kidney transplantation.
5.3 Viral infection and the management of immunosuppression
Viral infections remain a challenging issue after transplantation. Usually, treatment includes antiviral agents if available and/or a decrease in the intensity of immunosuppression. The principal goal is the restoration of effective antiviral immunity without increasing the risk of rejection. In solid organ transplantation, the individual risk of viral infection is determined largely by the “net state of immune deficiency”. Many parameters, including immunosuppression regimen, contribute to this state, and other individual predisposing factors, such as immune-aging, concomitant viral infection, and antiviral T-cell efficiency, are also involved [66].
CD8 T cells play a key role in controlling viral infection. Clinicians, therefore, decrease the intensity of immunosuppression by initially decreasing or withdrawing antimetabolites and/or CNIs, but this may increase the risk of graft rejection. A change in the “standard” regimen to another protocol for controlling or preventing viral infection has been proposed. As the mTOR pathway is largely involved in viral replication [67], there are several lines of evidence suggesting that mTOR.inh have antiviral properties, with a lower risk of CMV, polyomavirus, and Human herpes virus-8 (HHV8) infection than for standard protocols [68, 69, 70].
Various acute and recurrent viral infections may occur after kidney transplantation, but two viruses predominate CMV and BK polyomavirus (BKv). Despite recent advances, CMV infection is still associated with progressive disease, graft loss, morbidity and mortality, and antiviral drug resistance. CMV is usually efficiently controlled by specific antiviral therapy. Immunosuppression is not, therefore, systematically reduced, but such reductions may be proposed in cases of severe CMV disease, antiviral drug resistance, or inadequate clinical response [71].
In the last 20 years, BKv infection has emerged as a major complication of kidney transplantation, causing BKv-associated nephropathy, which has a prevalence of 10% and leads to graft loss in >50% of cases. The worldwide seroprevalence for BKv is more than 90%. Nevertheless, BKv disease is particularly frequent in the context of kidney transplantation but rare in non-kidney solid organ transplantation. In the absence of a specific antiviral treatment, a reduction of immunosuppression intensity is recommended for recipients with sustained plasma BKv reactivation or in cases of proven BKv-associated nephropathy. However, in the absence of randomized clinical trials, there are no standardized protocols for immunosuppressant minimization. A decrease in CNI exposure (CNI dose decrease of 25–50%), associated with or followed by a decrease in the dose (by 50%) of the antiproliferative drug, or its complete withdrawal, is proposed. This strong reduction of immunosuppression intensity may expose the patient to a higher risk of graft rejection. Some authors have also suggested switching patients from tacrolimus to low-dose CsA, and/or from mycophenolate mofetil to mTOR.inh. No recommendations have been made concerning the addition of intravenous immunoglobulins, leflunomide, or cidofovir [72].
5.4 Immunovirological monitoring to mitigate the risk of viral infection
An innovative approach to mitigating the viral risk, without increasing the risk of rejection, involves assessing the ability of the antiviral cellular immune response to control viral reactivation [73]. Functional specific antiviral T cells can control viral replication in cases of CMV infection [74, 75]. For example, CMV-specific T-cell monitoring has been shown to predict CMV control after solid organ transplantation [76]. Accordingly, CMV immune monitoring can be added to standard follow-up to improve CMV management. Similarly, the BKv-specific cellular immune response is crucial for the control and clearance of BKv. BKv-specific T-cell dysfunction increases the risk of uncontrolled BKv infection, whereas an increase in the number of BKv-specific CD8 T cells is associated with a better prognosis for BKv-associated nephropathy [77]. Various tools have been developed used to assess the quality of antiviral T-cell responses, but few are currently available in clinical practice. Additional studies are required to determine the potential utility of these tools for predicting infection risk after transplantation.
5.5 Transplantation in the context of cancer
Neoplastic complications are, unfortunately, among the most frequent and severe adverse events occurring after kidney transplantation, constituting the second leading cause of post-transplantation death [5]. Skin cancers are the most frequent post-transplantation tumors, with non-melanoma skin cancer particularly prevalent [78].
CNIs are directly involved in cancer pathogenesis, and their ability to promote the development of cancer cells has been clearly demonstrated in a mouse model [79]. It is, therefore, tempting to stop CNI treatment in the context of cancer.
mTOR.inh inhibit the mTOR pathway, which is involved in cancer cell growth. These molecules are, therefore, the treatment of choice in cases of cancer, as they can decrease the frequency of non-melanoma skin cancer without increasing the risk of acute rejection [80]. Similarly, mTOR.inh-based immunosuppression has been shown to be associated with lower rates of cancer and non-melanoma skin cancer, particularly in cases of late conversion from a standard immunosuppression regimen [81, 82]. In these initial reports, mTOR.inh was used in place of CNIs. However, recent observations of a higher risk of
5.6 Transplantation in the context of pregnancy
End-stage kidney disease is often associated with infertility and high-risk pregnancies. In women of reproductive age, kidney transplantation generally restores fertility, and more than 14,000 pregnancies in the context of kidney transplantation have already been reported worldwide [84]. However, complications, such as pre-eclampsia, hypertension, low-birth weight, preterm birth, and gestational diabetes, are more common in kidney transplant recipients than in women who have not undergone transplantation. Furthermore, acute rejection can occur during pregnancy, at rates similar to those in the non-pregnant transplant recipient population. Pregnancies in the context of kidney transplantation require careful monitoring, with joint management by an obstetrician with experience in managing high-risk pregnancies [85]. It is generally recommended to wait at least 1 year after transplantation before trying to conceive, due to the risk of graft failure. Conception can be considered safe if graft function is stable, with no recent episodes of graft rejection, a serum creatinine concentration < 1.5 mg/dL, proteinuria <500 mg a day, and normal blood pressure or well-controlled hypertension [84].
Immunosuppressive treatment and antihypertensive medication must be carefully managed. Immunosuppressive treatment modification should occur about 3 months before planned conception, to minimize the risk of graft rejection and adverse events due to treatment modification. CNIs are usually considered safe and should be continued throughout pregnancy. However, the levels of these drugs in the blood may fluctuate due to the physiological increase in blood volume. Careful monitoring is, therefore, required to ensure that the patient is not under-treated and exposed to a risk of rejection. By contrast, MMF is teratogenic and should be replaced by azathioprine at least a few weeks before any attempt at conception. Limited evidence is available concerning the safety of belatacept during pregnancy. mTOR.inh should be stopped as soon as possible, due to the involvement of the mTOR pathway in embryonic development and growth [86]. Finally, prednisone may be used safely in pregnant women. The preferred regimen during pregnancy is, therefore, a combination of CNIs, azathioprine, and prednisolone. For patients with hypertension, the optimal management of blood pressure is crucial to ensure a safe pregnancy. Nevertheless, renin-angiotensin-aldosterone system inhibitors should be stopped during pregnancy, due to the risk of renal agenesis.
5.7 Management of immunosuppressive drugs in the context of a loss of graft function and a return to dialysis
Returning to dialysis after transplantation is a complex transition, associated with an increase in morbidity and mortality [87]. Large numbers of transplant recipients eventually need a new graft. There may, therefore, be an indication for maintaining low doses of immunosuppression in this population to minimize the risk of alloimmunization. The benefits of pursuing immunosuppressive treatment after a return to dialysis must be weighed up against the risks of long-term immunosuppression. Several parameters must be considered, including the prospect of retransplantation, the prevention of graft intolerance, and immunosuppression-related adverse events. When managing immunosuppressive drugs, physicians should initially consider withdrawing antimetabolites. A schedule of CNI tapering can then be proposed and personalized on the basis of the benefit–risk ratio of pursuing such treatments. Slow tapering of CNIs over a period of 6 months following the recommencement of dialysis is recommended. If a new transplant is expected, pursuing low-dose CNI treatment is probably a better option than the complete withdrawal of these drugs. mTOR.inh can be tapered slowly, much like CNIs. There are no recommendations concerning the decreasing of prednisone dose [88].
6. Conclusion
Kidney transplantation is now the standard treatment for patients with end-stage renal disease. Current protocols usually combine (i) induction therapy and (ii) maintenance therapy based on corticosteroids, CNIs, and antimetabolite drugs. Nevertheless, new combinations of therapies are emerging as potential alternatives to the standard of care, with the hope of decreasing CNI toxicity, the occurrence of infections or tumors, and the occurrence of antibody-mediated rejection. In this context, mTOR.inh and belatacept are promising emerging candidates for inclusion in new combinations of molecules. New trials will be required to confirm the encouraging results reported and to adapt treatments to the diversity of patients undergoing kidney transplantation.
Abbreviations
| 6-MP | 6-mercaptopurine |
| α-CD25 | Alpha chain of the IL-2 receptor |
| APC | Antigen-presenting cell |
| ATG | Anti-thymocyte globulins |
| AUC | Area under the curve |
| BKv | BK polyomavirus |
| CMV | Cytomegalovirus |
| CNI | Calcineurin inhibitors |
| CsA | Cyclosporine-A |
| CTLA4-Ig | Cytotoxic T-lymphocyte associated protein-4-immunoglobulin |
| DSA | Donor-specific antibody |
| EBV | Epstein–Barr virus |
| HHV | Human herpesvirus |
| HLA | Human leukocyte antigen |
| IFNγ | Interferon γ |
| IL | Interleukin |
| IMDPH | Inosine monophosphate dehydrogenase |
| MHC | Major histocompatibility complex |
| MMF | Mycophenolate mofetil |
| mTOR | Mammalian target of rapamycin |
| mTOR.inh | Mammalian target of rapamycin inhibitors |
| PTLD | Post-transplantation lymphoproliferative disorder |
| rIL2 | Interleukin-2 receptor |
| TGN | Thioguanine nucleotide |
| TPMT | Thiopurine S-methyltransferase |
References
- 1.
Wolfe RA, Ashby VB, Milford EL, Ojo AO, Ettenger RE, Agodoa LYC, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. The New England Journal of Medicine. 1999; 341 :1725-1730. DOI: 10.1056/NEJM199912023412303 - 2.
Chapman JR. The KDIGO clinical practice guidelines for the care of kidney transplant recipients. Transplantation. 2010; 89 :644-645. DOI: 10.1097/TP.0b013e3181d62f1b - 3.
Tonelli M, Wiebe N, Knoll G, Bello A, Browne S, Jadhav D, et al. Systematic review: Kidney transplantation compared with dialysis in clinically relevant outcomes. American Journal of Transplantation. 2011; 11 :2093-2109. DOI: 10.1111/j.1600-6143.2011.03686.x - 4.
Hart A, Smith JM, Skeans MA, Gustafson SK, Wilk AR, Robinson A, et al. OPTN/SRTR 2016 annual data report: Kidney. American Journal of Transplantation. 2018; 18 :18-113. DOI: 10.1111/ajt.14557 - 5.
Ying T, Shi B, Kelly PJ, Pilmore H, Clayton PA, Chadban SJ. Death after kidney transplantation: An analysis by era and time post-transplant. Journal of the American Society of Nephrology. 2020; 31 :2887-2899. DOI: 10.1681/ASN.2020050566 - 6.
Kapsenberg ML. Dendritic-cell control of pathogen-driven T-cell polarization. Nature Reviews. Immunology. 2003; 3 :984-993. DOI: 10.1038/nri1246 - 7.
Halloran PF. Immunosuppressive drugs for kidney transplantation. The New England Journal of Medicine. 2004; 351 :2715-2729. DOI: 10.1056/NEJMra033540 - 8.
Mourad G, Morelon E, Noël C, Glotz D, Lebranchu Y. The role of thymoglobulin induction in kidney transplantation: An update. Clinical Transplantation. 2012; 26 :E450-E464. DOI: 10.1111/ctr.12021 - 9.
Cherikh WS, Kauffman HM, McBride MA, Maghirang J, Swinnen LJ, Hanto DW. Association of the type of induction immunosuppression with posttransplant lymphoproliferative disorder, graft survival, and patient survival after primary kidney transplantation. Transplantation. 2003; 76 :1289-1293. DOI: 10.1097/01.TP.0000100826.58738.2B - 10.
Lim WH, Turner RM, Chapman JR, Ma MKM, Webster AC, Craig JC, et al. Acute rejection, T-cell–depleting antibodies, and cancer after transplantation. Transplantation. 2014; 97 :817-825. DOI: 10.1097/01.TP.0000442773.38510.32 - 11.
Ducloux D, Courivaud C, Bamoulid J, Vivet B, Chabroux A, Deschamps M, et al. Prolonged CD4 T cell lymphopenia increases morbidity and mortality after renal transplantation. Journal of the American Society of Nephrology. 2010; 21 :868-875. DOI: 10.1681/ASN.2009090976 - 12.
Ducloux D, Challier B, Saas P, Tiberghien P, Chalopin J-M. CD4 cell lymphopenia and atherosclerosis in renal transplant recipients. Journal of the American Society of Nephrology. 2003; 14 :767-772. DOI: 10.1097/01.asn.0000048718.43419.44 - 13.
Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nature Reviews. Immunology. 2017; 17 :233-247. DOI: 10.1038/nri.2017.1 - 14.
Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunology Today. 1992; 13 :136-142. DOI: 10.1016/0167-5699(92)90111-J - 15.
Krämer BK, Montagnino G, Krüger B, Margreiter R, Olbricht CJ, Marcen R, et al. Efficacy and safety of tacrolimus compared with ciclosporin-A in renal transplantation: 7-year observational results. Transplant International. 2016; 29 :307-314. DOI: 10.1111/tri.12716 - 16.
Hošková L, Málek I, Kopkan L, Kautzner J. Pathophysiological mechanisms of Calcineurin inhibitor-induced nephrotoxicity and arterial hypertension. Physiological Research. 2017; 66 :167-180. DOI: 10.33549/physiolres.933332 - 17.
Rodrigo E, Fernández-Fresnedo G, Valero R, Ruiz JC, Piñera C, Palomar R, et al. New-onset diabetes after kidney transplantation: Risk factors. Journal of American Society of Nephrology. 2006; 17 :S291-S295. DOI: 10.1681/ASN.2006080929 - 18.
van der Zwan M, Hesselink DA, van den Hoogen MWF, Baan CC. Costimulation blockade in kidney transplant recipients. Drugs. 2020; 80 :33-46. DOI: 10.1007/s40265-019-01226-6 - 19.
Vincenti F, Charpentier B, Vanrenterghem Y, Rostaing L, Bresnahan B, Darji P, et al. A phase III study of Belatacept-based immunosuppression regimens versus cyclosporine in renal transplant recipients (BENEFIT study). American Journal of Transplantation. 2010; 10 :535-546. DOI: 10.1111/j.1600-6143.2009.03005.x - 20.
Durrbach A, Pestana JM, Pearson T, Vincenti F, Garcia VD, Campistol J, et al. A phase III study of belatacept versus cyclosporine in kidney transplants from extended criteria donors (BENEFIT-EXT study). American Journal of Transplantation. 2010; 10 :547-557. DOI: 10.1111/j.1600-6143.2010.03016.x - 21.
Vincenti F, Rostaing L, Grinyo J, Rice K, Steinberg S, Gaite L, et al. Belatacept and long-term outcomes in kidney transplantation. The New England Journal of Medicine. 2016; 374 :333-343. DOI: 10.1056/NEJMoa1506027 - 22.
Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity. 2014; 41 :1026-1039. DOI: 10.1016/j.immuni.2014.12.005 - 23.
Kim EJ, Kwun J, Gibby AC, Hong JJ, Farris AB, Iwakoshi NN, et al. Costimulation blockade alters germinal center responses and prevents antibody-mediated rejection. American Journal of Transplantation. 2014; 14 :59-69. DOI: 10.1111/ajt.12526 - 24.
Bertrand D, Chavarot N, Gatault P, Garrouste C, Bouvier N, Grall-Jezequel A, et al. Opportunistic infections after conversion to belatacept in kidney transplantation. Nephrology, Dialysis, Transplantation. 2020; 35 :336-345. DOI: 10.1093/ndt/gfz255 - 25.
Karadkhele G, Hogan J, Magua W, Zhang W, Badell IR, Mehta A, et al. CMV high-risk status and posttransplant outcomes in kidney transplant recipients treated with belatacept. American Journal of Transplantation. 2021; 21 :208-221. DOI: 10.1111/ajt.16132 - 26.
Deliège P-G, Bastien J, Mokri L, Guyot-Colosio C, Arndt C, Rieu P. Belatacept associated - cytomegalovirus retinitis in a kidney transplant recipient: A case report and review of the literature. BMC Ophthalmology. 2020; 20 :468. DOI: 10.1186/s12886-020-01741-1 - 27.
Fan J, Gong D, Truong C, Miko B, Horowitz J, Chen RWS. Cytomegalovirus retinitis with belatacept immunosuppression. Retinal Cases and Brief Reports. 2019; 16 (2):199-203. DOI: 10.1097/ICB.0000000000000928 - 28.
Dekeyser M, de Goër de Herve M-G, Hendel-Chavez H, Labeyrie C, Adams D, Nasser GA, et al. Refractory T-cell Anergy and rapidly fatal progressive multifocal leukoencephalopathy after prolonged CTLA4 therapy. Open Forum Infectious Diseases. 2017; 4 :ofx100. DOI: 10.1093/ofid/ofx100 - 29.
Klintmalm GB, Feng S, Lake JR, Vargas HE, Wekerle T, Agnes S, et al. Belatacept-based immunosuppression in de novo liver transplant recipients: 1-year experience from a phase II randomized study. American Journal of Transplantation. 2014; 14 :1817-1827. DOI: 10.1111/ajt.12810 - 30.
Fathman CG, Lineberry NB. Molecular mechanisms of CD4+ T-cell anergy. Nature Reviews. Immunology. 2007; 7 :599-609. DOI: 10.1038/nri2131 - 31.
Valdor R, Macian F. Induction and stability of the anergic phenotype in T cells. Seminars in Immunology. 2013; 25 :313-320. DOI: 10.1016/j.smim.2013.10.010 - 32.
Bristol-Myers Squibb Company. Risk Evaluation and Mitigation Strategy (REMS). NULOJIX™ (belatacept). 2011. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/125288Orig1s000remsNulojix.pdf [Accessed: 8 August 2011] - 33.
Dean L. Azathioprine therapy and TPMT and NUDT15 genotype. In: Pratt VM, Scott SA, Pirmohamed M, Esquivel B, Kane MS, Kattman BL, Malheiro AJ, editors. Medical Genetics Summaries. Bethesda (MD): National Center for Biotechnology Information (US); 2012 Bookshelf ID: NBK100661 - 34.
Zaza G, Cheok M, Krynetskaia N, Thorn C, Stocco G, Hebert JM, et al. Thiopurine pathway. Pharmacogenetics and Genomics. 2010; 20 :573-574. DOI: 10.1097/FPC.0b013e328334338f - 35.
Allison AC. Mechanisms of action of mycophenolate mofetil. Lupus. 2005; 14 (Suppl 1):s2-s8. DOI: 10.1191/0961203305lu2109oa - 36.
Allison AC, Eugui EM. Mechanisms of action of mycophenolate mofetil in preventing acute and chronic allograft rejection. Transplantation. 2005; 80 :S181-S190. DOI: 10.1097/01.tp.0000186390.10150.66 - 37.
Sollinger HW. Mycophenolates in transplantation. Clinical Transplantation. 2004; 18 :485-492. DOI: 10.1111/j.1399-0012.2004.00203.x - 38.
Tornatore KM, Sudchada P, Dole K, DiFrancesco R, Leca N, Gundroo AC, et al. Mycophenolic acid pharmacokinetics during maintenance immunosuppression in African American and Caucasian renal transplant recipients. Journal of Clinical Pharmacology. 2011; 51 :1213-1222. DOI: 10.1177/0091270010382909 - 39.
Kirken RA, Wang YL. Molecular actions of sirolimus: Sirolimus and mTor. Transplantation Proceedings. 2003; 35 :227S-230S. DOI: 10.1016/s0041-1345(03)00230-6 - 40.
Ekberg H, Tedesco-Silva H, Demirbas A, Vítko Š, Nashan B, Gürkan A, et al. Reduced exposure to Calcineurin inhibitors in renal transplantation. The New England Journal of Medicine. 2007; 357 :2562-2575. DOI: 10.1056/NEJMoa067411 - 41.
Oberbauer R, Segoloni G, Campistol JM, Kreis H, Mota A, Lawen J, et al. Early cyclosporine withdrawal from a sirolimus-based regimen results in better renal allograft survival and renal function at 48 months after transplantation. Transplant International. 2005; 18 :22-28. DOI: 10.1111/j.1432-2277.2004.00052.x - 42.
Campistol JM, Eris J, Oberbauer R, Friend P, Hutchison B, Morales JM, et al. Sirolimus therapy after early cyclosporine withdrawal reduces the risk for cancer in adult renal transplantation. Journal of American Society of Nephrology. 2006; 17 :581-589. DOI: 10.1681/ASN.2005090993 - 43.
Ossman R, Jamme M, Moulin B, Legendre C, Morelon E, Frimat L, et al. Immunosuppression and graft rejection in living-related HLA-identical renal transplantation: The RADOVFULL study. Transplantation. 2020; 104 :1256-1262. DOI: 10.1097/TP.0000000000002937 - 44.
Verghese PS, Dunn TB, Chinnakotla S, Gillingham KJ, Matas AJ, Mauer MS. Calcineurin inhibitors in HLA-identical living related donor kidney transplantation. Nephrology, Dialysis, Transplantation. 2014; 29 :209-218. DOI: 10.1093/ndt/gft447 - 45.
Woodle ES, Peddi VR, Tomlanovich S, Mulgaonkar S, Kuo PC. A prospective, randomized, multicenter study evaluating early corticosteroid withdrawal with thymoglobulin® in living-donor kidney transplantation. Clinical Transplantation. 2010; 24 :73-83. DOI: 10.1111/j.1399-0012.2009.01127.x - 46.
Mourad G, Rostaing L, Legendre C, Garrigue V, Thervet E, Durand D. Sequential protocols using Basiliximab versus anti-Thymocyte globulins in renal-transplant patients receiving mycophenolate Mofetil and steroids. Transplantation. 2004; 78 :584-590. DOI: 10.1097/01.TP.0000129812.68794.CC - 47.
Lebranchu Y, Bridoux F, Büchler M, Meur YL, Etienne I, Toupance O, et al. Immunoprophylaxis with Basiliximab compared with antithymocyte globulin in renal transplant patients receiving MMF-containing triple therapy. American Journal of Transplantation. 2002; 2 :48-56. DOI: 10.1034/j.1600-6143.2002.020109.x - 48.
Brennan DC, Daller JA, Lake KD, Cibrik D, Del Castillo D. Thymoglobulin induction study group. Rabbit antithymocyte globulin versus basiliximab in renal transplantation. The New England Journal of Medicine. 2006; 355 :1967-1977. DOI: 10.1056/NEJMoa060068 - 49.
Thiyagarajan UM, Ponnuswamy A, Bagul A. Thymoglobulin and its use in renal transplantation: A review. American Journal of Nephrology. 2013; 37 :586-601. DOI: 10.1159/000351643 - 50.
Hardinger KL, Brennan DC, Klein CL. Selection of induction therapy in kidney transplantation. Transplant International. 2013; 26 :662-672. DOI: 10.1111/tri.12043 - 51.
Sethi S, Choi J, Toyoda M, Vo A, Peng A, Jordan SC. Desensitization: Overcoming the immunologic barriers to transplantation. Journal of Immunology Research. 2017; 2017 :6804678. DOI: 10.1155/2017/6804678 - 52.
Salvadori M, Tsalouchos A. Current protocols and outcomes of ABO-incompatible kidney transplantation. World Journal of Transplantation. 2020; 10 :191-205. DOI: 10.5500/wjt.v10.i7.191 - 53.
Meier-Kriesche H-U, Arndorfer JA, Kaplan B. Association of Antibody Induction with short- and long-term cause-specific mortality in renal transplant recipients. Journal of the American Society of Nephrology. 2002; 13 :769-772. DOI: 10.1681/ASN.V133769 - 54.
Sood P, Cherikh WS, Toll AE, Mehta RB, Hariharan S. Kidney allograft rejection: Diagnosis and treatment practices in USA-A UNOS survey. Clinical Transplantation. 2021; 35 :e14225. DOI: 10.1111/ctr.14225 - 55.
Flechner SM, Glyda M, Cockfield S, Grinyó J, Legendre C, Russ G, et al. The ORION study: Comparison of two Sirolimus-based regimens versus tacrolimus and mycophenolate Mofetil in renal allograft recipients. American Journal of Transplantation. 2011; 11 :1633-1644. DOI: 10.1111/j.1600-6143.2011.03573.x - 56.
Pascual J, Berger SP, Witzke O, Tedesco H, Mulgaonkar S, Qazi Y, et al. Everolimus with reduced Calcineurin inhibitor exposure in renal transplantation. Journal of the American Society of Nephrology. 2018; 29 :1979-1991. DOI: 10.1681/ASN.2018010009 - 57.
Tedesco Silva H, Cibrik D, Johnston T, Lackova E, Mange K, Panis C, et al. Everolimus plus reduced-exposure CsA versus mycophenolic acid plus standard-exposure CsA in renal-transplant recipients. American Journal of Transplantation. 2010; 10 :1401-1413. DOI: 10.1111/j.1600-6143.2010.03129.x - 58.
Chan L, Greenstein S, Hardy MA, Hartmann E, Bunnapradist S, Cibrik D, et al. Multicenter, randomized study of the use of everolimus with tacrolimus after renal transplantation demonstrates its effectiveness. Transplantation. 2008; 85 :821-826. DOI: 10.1097/TP.0b013e318166927b - 59.
Berger SP, Sommerer C, Witzke O, Tedesco H, Chadban S, Mulgaonkar S, et al. Two-year outcomes in de novo renal transplant recipients receiving everolimus-facilitated calcineurin inhibitor reduction regimen from the TRANSFORM study. American Journal of Transplantation. 2019; 19 :3018-3034. DOI: 10.1111/ajt.15480 - 60.
Chan L, Hartmann E, Cibrik D, Cooper M, Shaw LM. Optimal everolimus concentration is associated with risk reduction for acute rejection in de novo renal transplant recipients. Transplantation. 2010; 90 :31-37. DOI: 10.1097/TP.0b013e3181de1d67 - 61.
Durrbach A, Pestana JM, Florman S, Del Carmen RM, Rostaing L, Kuypers D, et al. Long-term outcomes in Belatacept- versus cyclosporine-treated recipients of extended criteria donor kidneys: Final results from BENEFIT-EXT, a phase III randomized study. American Journal of Transplantation. 2016; 16 :3192-3201. DOI: 10.1111/ajt.13830 - 62.
Kumar J, Reccia I, Virdis F, Podda M, Sharma AK, Halawa A. Belatacept in renal transplantation in comparison to tacrolimus and molecular understanding of resistance pattern: Meta-analysis and systematic review. World Journal of Transplantation. 2021; 11 :70-86. DOI: 10.5500/wjt.v11.i3.70 - 63.
Espinosa JR, Samy KP, Kirk AD. Memory T cells in organ transplantation: Progress and challenges. Nature Reviews. Nephrology. 2016; 12 :339-347. DOI: 10.1038/nrneph.2016.9 - 64.
Sun H, Hartigan CR, Chen C-W, Sun Y, Tariq M, Robertson JM, et al. TIGIT regulates apoptosis of risky memory T cell subsets implicated in belatacept-resistant rejection. American Journal of Transplantation. 2021; 21 :3256-3267. DOI: 10.1111/ajt.16571 - 65.
Herr F, Desterke C, Bargiel K, Vernochet A, Vanhove B, Vadanici R, et al. The proliferation of belatacept-resistant T cells requires early IFNα pathway activation. American Journal of Transplantation; 22 (2):489-503. DOI: 10.1111/ajt.16811 - 66.
Roberts MB, Fishman JA. Immunosuppressive agents and infectious risk in transplantation: Managing the “net state of immunosuppression”. Clinical Infectious Diseases. 2021; 73 :e1302-e1317. DOI: 10.1093/cid/ciaa1189 - 67.
Pant A, Dsouza L, Yang Z. Alteration in cellular signaling and metabolic reprogramming during viral infection. MBio. 2021; 12 :e0063521. DOI: 10.1128/mBio.00635-21 - 68.
Montero N, Quero M, Melilli E, Pérez-Sáez MJ, Redondo-Pachón D, Bestard O, et al. Mammalian target of rapamycin inhibitors combined with Calcineurin inhibitors as initial immunosuppression in renal transplantation: A meta-analysis. Transplantation. 2019; 103 :2031-2056. DOI: 10.1097/TP.0000000000002769 - 69.
Bowman LJ, Brueckner AJ, Doligalski CT. The role of mTOR inhibitors in the management of viral infections: A review of current literature. Transplantation. 2018; 102 :S50-S59. DOI: 10.1097/TP.0000000000001777 - 70.
Pascual J, Royuela A, Fernández AM, Herrero I, Delgado JF, Solé A, et al. Role of mTOR inhibitors for the control of viral infection in solid organ transplant recipients. Transplant Infectious Disease. 2016; 18 :819-831. DOI: 10.1111/tid.12601 - 71.
Kotton CN, Kumar D, Caliendo AM, Huprikar S, Chou S, Danziger-Isakov L, et al. The third international consensus guidelines on the management of cytomegalovirus in solid-organ transplantation. Transplantation. 2018; 102 :900-931. DOI: 10.1097/TP.0000000000002191 - 72.
Hirsch HH, Randhawa PS. AST infectious diseases Community of Practice. BK polyomavirus in solid organ transplantation-guidelines from the American Society of Transplantation infectious diseases Community of Practice. Clinical Transplantation. 2019; 33 :e13528. DOI: 10.1111/ctr.13528 - 73.
Sester M, Leboeuf C, Schmidt T, Hirsch HH. The “ABC” of virus-specific T cell immunity in solid organ transplantation. American Journal of Transplantation. 2016; 16 :1697-1706. DOI: 10.1111/ajt.13684 - 74.
Yong MK, Lewin SR, Manuel O. Immune monitoring for CMV in transplantation. Current Infectious Disease Reports. 2018; 20 :4. DOI: 10.1007/s11908-018-0610-4 - 75.
Dekeyser M, Ladrière M, Audonnet S, Frimat L, Bittencourt MDC. An early immediate early protein IE-1–specific T-cell Polyfunctionality is associated with a better control of cytomegalovirus reactivation in kidney transplantation. Kidney International Reports. 2017; 2 :486-492. DOI: 10.1016/j.ekir.2017.02.016 - 76.
Rezahosseini O, Møller DL, Knudsen AD, Sørensen SS, Perch M, Gustafsson F, et al. Use of T cell mediated immune functional assays for adjustment of immunosuppressive or anti-infective agents in solid organ transplant recipients: A systematic review. Frontiers in Immunology. 2020; 11 :567715. DOI: 10.3389/fimmu.2020.567715 - 77.
Dekeyser M, François H, Beaudreuil S, Durrbach A. Polyomavirus-specific cellular immunity: From BK-virus-specific cellular immunity to BK-virus-associated nephropathy? Frontiers in Immunology. 2015; 6 :307. DOI: 10.3389/fimmu.2015.00307 - 78.
Webb MC, Compton F, Andrews PA, Koffman CG. Skin tumours posttransplantation: A retrospective analysis of 28 years’ experience at a single Centre. Transplantation Proceedings. 1997; 29 :828-830. DOI: 10.1016/S0041-1345(96)00152-2 - 79.
Ohsawa I, Murakami T, Uemoto S, Kobayashi E. In vivo luminescent imaging of cyclosporin A-mediated cancer progression in rats. Transplantation. 2006; 81 :1558-1567. DOI: 10.1097/01.tp.0000209448.50238.de - 80.
Campbell SB, Walker R, Tai SS, Jiang Q , Russ GR. Randomized controlled trial of sirolimus for renal transplant recipients at high risk for nonmelanoma skin cancer. American Journal of Transplantation. 2012; 12 :1146-1156. DOI: 10.1111/j.1600-6143.2012.04004.x - 81.
Knoll GA, Kokolo MB, Mallick R, Beck A, Buenaventura CD, Ducharme R, et al. Effect of sirolimus on malignancy and survival after kidney transplantation: Systematic review and meta-analysis of individual patient data. BMJ. 2014; 349 :g6679. DOI: 10.1136/bmj.g6679 - 82.
Alberú J, Pascoe MD, Campistol JM, Schena FP, Rial MDC, Polinsky M, et al. Lower malignancy rates in renal allograft recipients converted to sirolimus-based, calcineurin inhibitor-free immunotherapy: 24-month results from the CONVERT trial. Transplantation. 2011; 92 :303-310. DOI: 10.1097/TP.0b013e3182247ae2 - 83.
Sommerer C, Suwelack B, Dragun D, Schenker P, Hauser IA, Witzke O, et al. An open-label, randomized trial indicates that everolimus with tacrolimus or cyclosporine is comparable to standard immunosuppression in de novo kidney transplant patients. Kidney International. 2019; 96 :231-244. DOI: 10.1016/j.kint.2019.01.041 - 84.
McKay DB, Josephson MA, Armenti VT, August P, Coscia LA, Davis CL, et al. Reproduction and transplantation: Report on the AST consensus conference on reproductive issues and transplantation. American Journal of Transplantation. 2005; 5 :1592-1599. DOI: 10.1111/j.1600-6143.2005.00969.x - 85.
Agarwal KA, Pavlakis M. Sexuality, contraception, and pregnancy in kidney transplantation. Kidney Medicine. 2021; 3 :837-847. DOI: 10.1016/j.xkme.2021.05.009 - 86.
Gangloff Y-G, Mueller M, Dann SG, Svoboda P, Sticker M, Spetz J-F, et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Molecular and Cellular Biology. 2004; 24 :9508-9516. DOI: 10.1128/MCB.24.21.9508-9516.2004 - 87.
Rao PS, Schaubel DE, Jia X, Li S, Port FK, Saran R. Survival on dialysis post-kidney transplant failure: Results from the scientific registry of transplant recipients. American Journal of Kidney Diseases. 2007; 49 :294-300. DOI: 10.1053/j.ajkd.2006.11.022 - 88.
Lubetzky M, Tantisattamo E, Molnar MZ, Lentine KL, Basu A, Parsons RF, et al. The failing kidney allograft: A review and recommendations for the care and management of a complex group of patients. American Journal of Transplantation. 2021; 21 :2937-2949. DOI: 10.1111/ajt.16717