Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
Quinazoline derivatives have shown promising pharmacological activities against various diseases, including cancer, inflammation, and cardiovascular disorders. Computational studies have become an important tool in the discovery and optimization of new quinazoline derivatives. In this chapter, the importance and application of computational studies in finding new active quinazoline derivatives were discussed. The various computational techniques, such as molecular docking, molecular dynamics simulations, quantum mechanics calculations, and machine learning algorithms, which have been used to predict the biological activities and optimize the structures of quinazoline derivatives, were described. Examples of successful applications of computational studies in the discovery of new quinazoline derivatives with improved pharmacological activities were added. Overall, computational studies have proven to be valuable in the development of new quinazoline derivatives and have the potential to accelerate the drug discovery process.
Department of Medicinal Chemistry, Faculty of Pharmacy, Sana’a University, Sana’a, Yemen
Department of Pharmacy, Faculty of Medical Sciences, Al-Nasser University, Sana’a, Yemen
*Address all correspondence to: wafalmadhaji2020@gmail.com
1. Introduction
Computer-aided drug design (CADD) is a computational method used in drug discovery. Nowadays, the discovery of new and novel therapeutic compounds for the treatment of different diseases needs to pass through the clinical trials. The examples of clinical trials used for this method are:
inhibitor of carbonic anhydrase dorzolamide, accepted in 1995 [1].
the inhibitor of angiotensin-converting enzyme (ACE) captopril, accepted in 1981 as an antihypertensive drug [2]
three therapeutics for the treatment of human immunodeficiency virus [3]: saquinavir, accepted in 1995.
ritonavir and indinavir, both approved in 1996 [4].
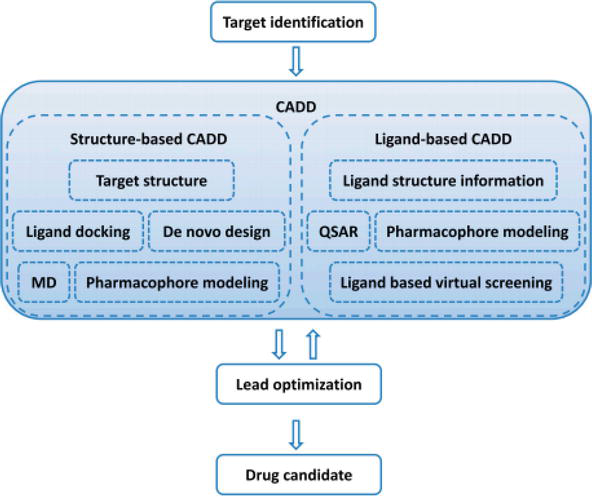
Two main categories of computer-aided drug design have been classified as structure-based and ligand-based. Structure-based CADD depends on the understanding of the target protein structure to calculate the binding energies for all tested compounds, whereas ligand-based CADD relies on the data of chemical similarity finding or building of predictive, quantitative structure-activity relation (QSAR) models for known active and inactive compounds [5]. The place of CADD in drug discovery is summarized in Figure 1, where the target site for a drug to be designed and developed is determined therapeutically, accounting on the knowledge availability of structure information, a structure-based approach, or a ligand-based approach. The data obtained from successful CADD will permit the identification of multiple lead compounds that is often followed by numerous cycles of lead optimization and successive lead identification [6].
Figure 1.
Computer-aided drug design in drug design and discovery [6].
Structure-based CADD is mostly favored compared to other methods because of the high-resolution structural data of the target protein that can be obtained, that is, soluble proteins that can readily be crystallized. On other hand, ligand-based CADD is more commonly preferred when there is either no or slight structural information in existence, regularly if the process involves membrane protein targets. The fundamental aim of structure-based CADD is to design compounds that bind firmly to the target with lower binding energy, enhancing drug metabolism and pharmacokinetics/absorption, distribution, metabolism, elimination, and toxicity (DMPK/ADME/T), that is, would show less off-target properties [7].
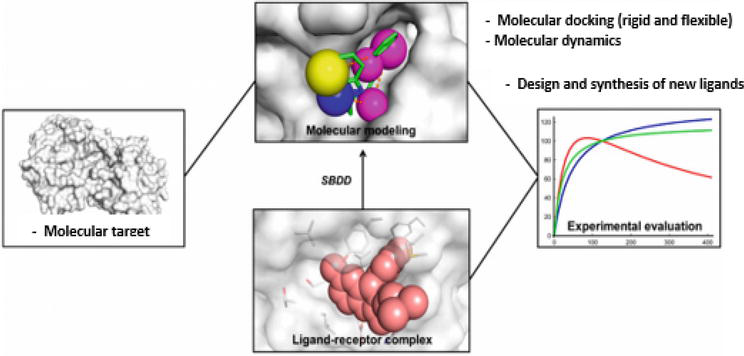
Structure-based drug design (SBDD) depends on study 3D structures of biological molecules with the aid of a computer. The main concept of this approach accounts on the ability of the compound to bind to the binding site to utilize the desired biological activity. Hence, the molecules that share similar favorable binding will give similar biological activities, as showed in Figure 2.
Figure 2.
Structure-based drug design outlines [8].
The new and novel compounds can be further explained through the careful study of a protein’s binding site. SBDD analysis needs structural information for target protein because the SBDD will assist in discovering a large number of compound drug targets [9]. Large 3D database of human and pathogenic proteins structure depending on the biophysical results of X-ray crystallography and NMR spectroscopy are also accessible in the database. The case of example that relates to this is the record of over 81,000 protein structures that could be found in RCSB Protein Data Bank, with 5671 PDBbind database and 129 co-crystal of ligand-protein [10]. Therefore, it is safe to conclude that computational approaches in drug discovery permit rapid screening of a large compound library and allow for the evaluation of binder potential during modeling/simulation and visualization methods [11].
Molecular docking is a computational technique that explores the probable binding pose of protein for a given ligand and calculates the binding affinity. Thus, research of a computational tool to find out new objectives is required. The adverse outcome can be eliminated or reduced if the unfavorable and unexpected binding of drugs or lead molecules to the targets happen. In addition, the drug’s indication can also be increased through reposition of drugs. As a result, reverse docking approaches have many more potential for further exploration and are currently receiving an increase in interest from researchers [12].
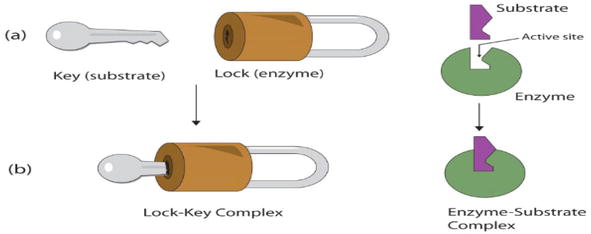
Molecular docking is a computational technique that gives prediction of a macromolecule (receptor) binding efficiency to a small molecule (ligand). In addition, it gives illumination to the fundamental biochemical practices [13]. Docking can be obtained through two correlated ways; the first being ligand conformation in the protein active sites, followed by a ranking of these conformations by scoring function [14]. The mechanism of ligand-receptor binding is formerly elucidated according to a lock-and-key theory proposed by Fischer [15] where the ligand fits into the receptor according to lock-and-key approach as illustrated in Figure 3 in which the ligand and receptor are rigid.
Figure 3.
Lock-and-key theory such as enzyme mechanism of action [15].
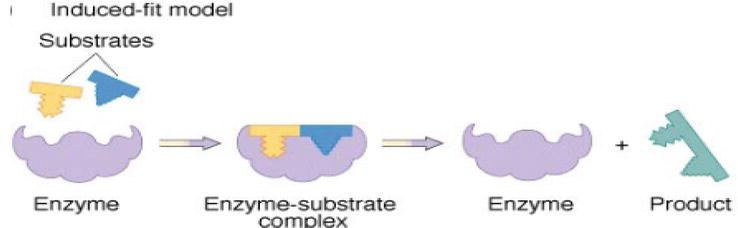
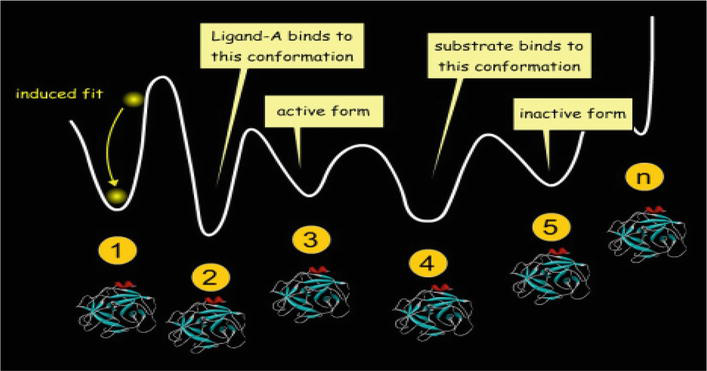
Then, Koshland improves the lock-and-key theory into “induced-fit theory,” by arguing that the protein active site is reshaped by binding to a ligand; thus, the ligand and receptor should be flexible during docking (Figure 4) [16]. This theory covers more accurate binding events in comparison to the rigid run. Nowadays, the most popular method is to perform docking with flexible ligand and a rigid receptor [17, 18].
Figure 4.
Induced fit model [16].
In addition, there is a theory that has been recently discovered that focuses on how the conformation ensemble model describes the proteins as preexisting ensemble of conformational states and that the protein’s flexibility permits large changes in conformation (Figure 5).
Figure 5.
Conformation ensemble model.
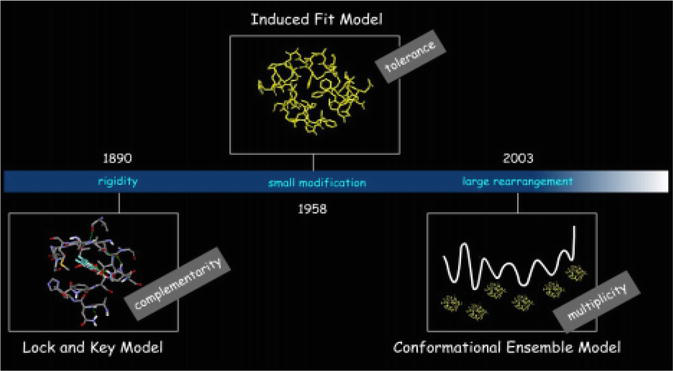
The three different models, the lock-and-key, induced-fit, and the conformation ensemble model all focus on the process of recognition. To be simply put, the lock-and-key module presents the 3D complementarity concepts, the induced-fit model clarifies how complementarity is done, and the ensemble model explains the proteins conformational complexity (Figure 6).
Figure 6.
Different ligand-receptor binding models.
3.1 Docking approach
There are two docking approaches that are well recognized in the molecular docking community. The first approach employs the technique of matching, which defines the protein and the ligand as complementary planes [19, 20, 21]. The second approach simulates the actual docking practice in which the interactions of ligand-protein energies are calculated [22].
3.2 Binding sites
Binding sites are known sites of protein where ligands/compounds are bound actively. The different site of binding is commonly labeled as binding modes. They also predict the binding strength, the complex energy, the types of the signal produced, and the binding affinity’s calculation between two molecules by using scoring functions. The most awaited situation is the protein–ligand interaction type, which has prominent functions in medicine.
3.3 Types of interactions
The interactions between particles are classified into four groups:
Electrostatic forces: Forces of electrostatic origin due to the charges residing in the matter. The most common interactions are charge-charge, charge-dipole, and dipole-dipole.
Electrodynamics forces: The most common force is the Van der Waals interaction, which is intermolecular forces between macromolecules and ligand such as charge-charge, charge-dipole, and dipole-dipole.
Steric forces: Steric forces produced by entropy. Such as, in cases where entropy is limited, there may be forces to minimize the free energy of the system, which are due to entropy.
Solvent-related forces: Solvent-related forces are formed because of the solvent structural changes. These structural changes are produced when ions, colloids, proteins, and so forth are added to the solvent structure like hydrogen bond and hydrophobic interactions.
Other physical factors: The conformational changes in the protein and the ligand are often required for a successful docking process.
3.4 Reverse docking
Reverse docking is a computational study and is commonly used to identify receptors and explore new targets for ligand used (drugs or natural compounds) over a large number of receptors. In addition, it gives the necessary explanation for polypharmacology, molecular mechanism, alternative usage through the repositioning of drugs, and adverse effect and toxicity determination [12]. The reverse docking is classified as structure-based approach that uses 2D fingerprint-based reverse virtual screening, 3D similarity search, and pharmacophore (reverse pharmacophore mapping) [23, 24]. Reverse pharmacophore mapping, for example, pharmacophore model of proteins in the list of targets, is the starting point for the comparison of the pharmacophore models with the studied ligand [23]. A freely accessible web server known as PharmMapper is considered as a valuable resource of pharmacophore-based reverse screening [25]. It attempts to find the potential target applicants for small-molecule drugs, natural compounds, or novel compounds whose targets are still unidentified [26, 27]. As of now, PharmMapper produces the “best mapping poses of the query” against all pharmacophore models in the core database.
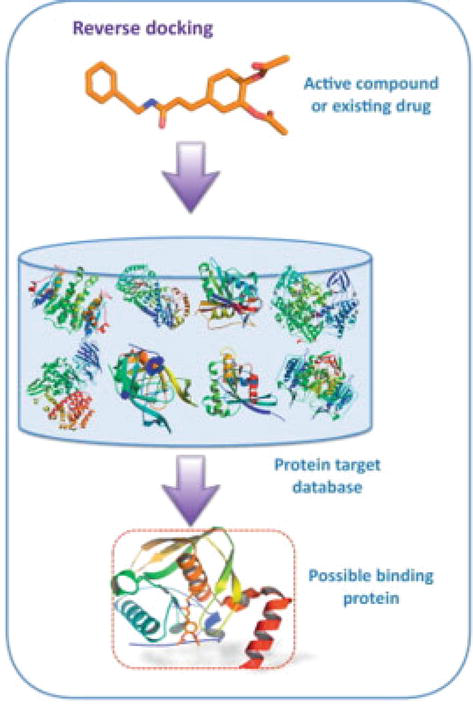
Generally, the ligand-based methods may offer extra benefits, some of which are the immediate computations and ability to support a large variety of data. Direct or indirect use of target structural information is compulsory, depending if one is concerned in exploring potential targets for a set of ligands. Comparable to traditional virtual screening, the collective use of structure and ligand-based tools in reversed virtual screening can give rise to higher enhancement of the hits [26]; the overview of reverse docking was summarized in Figure 7 [12].
Figure 7.
Overview of reverse docking [28].
The computational docking of a specific small molecule to receptor structure library is firstly explained by Chen and Zhi [29] using the term “inverse docking technique”. This technique is beneficial for the identification of the new potential biological targets for known compounds [30, 31, 32], as it specifies compounds targets over a family of related receptors [33]. The approach gives rise to the achievement in identifying the receptors homology models [33]. The approach may also be useful for generating a predicted pharmacological profile for compound [34] or to create a virtual selectivity profile that illustrates the promiscuity of the inhibitors [35]. In addition, inverse docking is also able to give multifaceted nature to pharmacologically active compound and can produce new hypotheses for the mechanism of action [28].
Thus, it has been proven that reverse docking is a vital tool in the computational study, particularly in identifying novel macromolecular targets for a drug or ligand related to its mechanism of action and/or the side effects [28, 36]. Moreover, it is the tool that involves docking of small molecules or drugs in potential binding sites of clinically relevant macromolecular targets [26].
Target structure collection with details of potential binding sites is important for computational efficiency, applicability, and accuracy of docking results. Receptor binding sites or binding pockets are the precise areas of the receptors where ligands bind to form interactions. Known binding pockets are supportive for reverse docking because the time needed to examine proper docking site between ligands and receptors [12] would be decreased. Mostly, there are two ways that are used to get the binding site; one is formed by protein data bank (PDB) complex structures, and the other meaning is to use pocket-searching programs such as SiteMap or F pocket. An automatic procedure to define binding sites is more desirable and favorable because a large number of targets should be examined in reverse docking process [12].
There are different databases used for a target structure, as summarized in Table 1, for example, sc-PDB, a library for 3D ligand binding site that has entries for protein-ligand complex structures in PDB [37]. The sc-PDB keeps three databases for structure information: (i) the target protein coordinate data, (ii) the ligand coordinate data, and (iii) the active site coordinate data. ScPDB’s database explains the active sites of the reference proteins using information in PDB, UniProt [38], and the information are presented on the web (http://bioinfo-pharma.u-strasbg.fr/scPDB/). Currently, it has 9283 entries of binding site from 3678 proteins and 5608 ligands. In addition to that, potential drug target database (PDTD) is an alternative web with accessible target database for small molecules target identification [39]. The target proteins of PDTD are collected from both scientific studies and other databases containing therapeutic target database (TTD), Drug Bank, and Thomson Pharma. However, it is important to note that only protein ligand of complex structures are collected. PDTD covered relevant information about related illnesses, biological purpose, and associated signaling pathways. In addition to determination of the target protein and active sites. One of the benefits of PDTD is that users can alter the target list as they wish. The PDTD’s active site definition seems to match with sc-PDB. Note that PDTD also allows redundant records, so the proteins flexibility could be evaluated even with rigid receptor docking. PDTD is available for use on the web server (http://www.dddc.ac.cn/pdtd/), and at the moment, it contains 1207 entries covering 841 target proteins.
Information type
Database name
Content
Coverage
Protein
PDB
Structural of proteins
107,667 proteins structures
UniProt
Cross references of proteins
550,552 proteins for Swiss-Prot60, 971,489 proteins for TrEMBL
Target protein
TTD
Disease, pathway, drug-related to therapeutic targets
2025 targets, 17,816 drugs
sc-PDB
Coordinate data of proteins, ligands, and active sites
9283 binding sites 3678 proteins, 5608 ligands
PDTD
Binding structures, related diseases, biological function, associated pathways
1207 binding structures 841 target proteins
Target DB
Integrated databases of TTD, PDTD, Drug Bank, and PDB
6920 protein entries
DART
Target associated with drug adverse reaction
618 proteins, 529 ligands
DITOP
Targets associated with drug-induced toxicity
236 proteins, 1327 ligands
CSD
Cambridge structural database
>800,000 protein entries
Drug
Drug Bank
Drugs and drug targets
7677 drugs
PubChem
Structures and activities of chemicals
52 million chemicals
ChEMBL
Information on drug-like bioactive compounds
2.4 million bioassays
ZINC
Chemical compounds
>35 million compounds
Protein-ligand complex
Astex diverse set
Apo and holo form of protein complexes with drug-like ligand
85 protein-ligand complexes
Table 1.
Different docking-based target identification databases.
4.1 Drug-like properties (Lipinski’s “rule of five”)
For oral delivery and according to the world drug index (WDI), [40] Lipinski “rule of five’’ are required to be considered. These properties are:
This rule of five needs to be visible as a qualitative absorption/permeability predictor [41], in preference to a quantitative predictor [42]. The assets distribution in drug-related chemical databases has been studied as any other method to apprehend “drug-likeness” [43, 44]. These aforementioned analyses all point to a crucial aggregate of physicochemical and structural properties [45], which to a massive extent may be manipulated by the medicinal chemist (Figure 8) [46]. Physicochemical properties as well as pharmacokinetic and toxic properties had been ignored for a long time by most medicinal chemists, who in lots of cases best had the quest for most powerful receptor binding as the ultimate goal.
Figure 8.
Property-based design [46].
Based on the Lipinski’s rule of five, if two parameters of the compound were out of the range, a poor absorption or permeability may be arisen. Unless, few oral active drugs were found out to follow the Lipinski rule of 5 such as antibiotics, antifungals, vitamins, and cardiac glycosides. The main reason for activity of such therapeutics groups was structural features that lead compounds to act as a substrate for naturally occurring transporters [40].
4.2 ADME/T studies
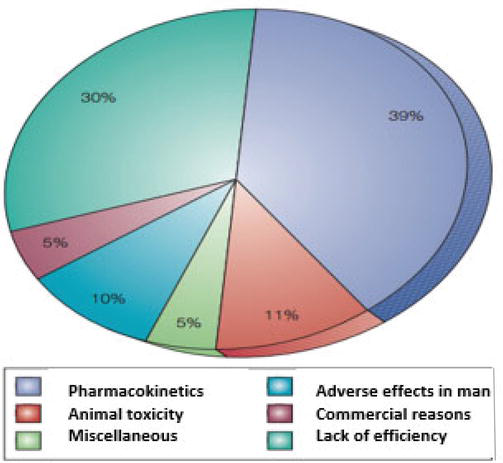
Because the capability for organic screening and chemical synthesis has dramatically expanded, the needs for large portions of early records on absorption, distribution, metabolism, excretion (ADME), and toxicity data (collectively called ADME/T information) have been increased. In addition, the study of pharmacokinetics was very critical in drug development as the failure of drug development could be recognized because of poor pharmacokinetics (39%) and animal toxicity (11%) as summarized in Figure 9.
Figure 9.
The main reasons for attrition in drug development [47].
Those early predictions of ADME/T residences help researchers to pick out the excellent applicants for drug development, in addition to rejecting those with a low chance of success. The evaluation of ADME/T can be in one of three approaches; first, selection of different in vitro assays that further automated through the use of robotics and miniaturization. Second, in silico models have been used to help in the selection of both appropriate analyses as well as in the selection of compounds to go through these screens. Third, predictive approaches have been established that might ultimately emerge as sophisticated enough to replace in vitro assays and/or in vivo assays [47]. The combination data obtained from ADME/T properties prediction and computational (in silico) had been significantly satisfying [48, 49]. Hou et al. [50] have achieved broad study on in silico modeling of various ADME/T-related properties, among them are the blood-brain barrier, human intestinal absorption (HIA), Caco-2 permeability, oral absorption, oral bioavailability, and P-glycoprotein inhibition [50]. Combined information based PKKB [Pharmaco Kinetics Knowledge Base] [51], collecting structures, pharmacological data, significant experimental or predicted physiochemical properties and experimental ADME/T data for 1685 drugs. This information plays an important role as valuable resources for pharmacokinetic studies and for endorsing the accuracy of current ADME/T predicative models reliable.
4.3 Computational studies for the quinazoline derivatives
Molecular properties refer to the physical and chemical characteristics of a molecule that determine its behavior and interactions with other molecules. These properties are important in understanding the biological activity and pharmacological effects of a compound [52]. Some of the key molecular properties that are often studied in computational chemistry include:
Molecular weight: the molecular weight of a compound is the sum of the atomic weights of all the atoms in the molecule. Molecular weight can influence the pharmacokinetic properties of a compound, such as its absorption, distribution, metabolism, and excretion.
Lipophilicity: lipophilicity refers to the ability of a molecule to dissolve in lipids or nonpolar solvents. Lipophilic compounds tend to have better membrane permeability and can more easily cross cell membranes to reach their targets.
Polarizability: polarizability is a measure of the ability of a molecule to be distorted by an electric field. Polarizable molecules tend to have more interactions with other molecules, such as protein-ligand interactions.
Hydrogen bonding: hydrogen bonding is a type of intermolecular interaction in which a hydrogen atom is shared between two electronegative atoms such as oxygen or nitrogen. Hydrogen bonding can affect the solubility, stability, and activity of a compound.
Electrostatic potential: electrostatic potential is a measure of the electrostatic forces between the charged particles in a molecule. It can provide insight into the interactions between a molecule and its target protein.
Computational methods such as molecular modeling, molecular dynamics simulations, and quantum mechanics calculations can be used to predict and analyze these molecular properties, which can help in the design and optimization of new drug candidates
Surface area: the surface area of a molecule is an important factor in determining its solubility, as well as its interactions with other molecules and surfaces.
Charge distribution: the distribution of charges within a molecule can affect its interactions with other charged species, including proteins and other biomolecules.
Dipole moment: the dipole moment of a molecule is a measure of its polarity and can influence its interactions with other polar molecules and surfaces.
Molecular shape: the shape of a molecule is important in determining its interactions with other molecules, particularly in the case of protein-ligand interactions where the shape of the ligand must complement the shape of the protein binding site.
Conformational flexibility: the ability of a molecule to adopt different conformations can affect its interactions with other molecules, as well as its pharmacological properties.
Computational methods can be used to predict and analyze these molecular properties, which can provide insight into the potential biological activity and pharmacological effects of a compound. By understanding these properties, researchers can design and optimize new drug candidates with improved efficacy and safety profiles.
Quinazoline derivatives have been the subject of numerous computational studies due to their diverse biological activities, including anticancer, antitumor, antiviral, and anti-inflammatory properties. Computational studies have been used to predict the molecular properties of these compounds, investigate their biological activities, and design new derivatives with improved activity [53].
One common approach in computational studies of quinazoline derivatives is molecular docking, which involves the prediction of the binding affinity of a ligand to a target protein. This technique has been used to investigate the binding of quinazoline derivatives to various protein targets, including tyrosine kinases and tubulin. In addition to molecular docking, other computational techniques such as molecular dynamics simulations, quantum mechanics are used.
Quinazoline derivatives have been found to exhibit a wide range of biological activities, making them potentially useful in the development of therapeutic agents for various diseases. Some of the most commonly studied biological activities of quinazoline derivatives include:
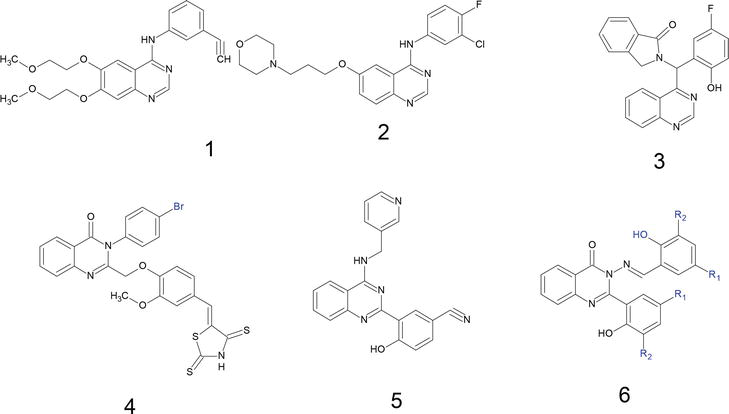
Anticancer and antitumor activity: quinazoline derivatives have been shown to exhibit potent anticancer and antitumor activity by inhibiting various signaling pathways involved in cancer cell growth and proliferation. For example, some quinazoline derivatives have been found to inhibit the activity of tyrosine kinases such as EGFR (epidermal growth factor receptor) and HER2 (human epidermal growth factor receptor 2), which are commonly overexpressed in cancer cells [54], and there are numerous quinazoline scaffold-based compounds present in the market such as erlotinib, gefitinib (structures 1 and 2), and structures 3–6, with high cytotoxic activity toward different cancerous cell lines (Figure 10) [55]
Antiviral activity: quinazoline derivatives have also been found to exhibit antiviral activity against various viruses, including HIV, herpes simplex virus (HSV), and hepatitis C virus (HCV). Some of these compounds act by inhibiting the viral enzymes responsible for viral replication, while others inhibit viral entry into host cells.
Anti-inflammatory activity: quinazoline derivatives have been shown to exhibit anti-inflammatory activity by inhibiting the activity of enzymes such as cyclooxygenase (COX) and lipoxygenase (LOX), which are involved in the production of inflammatory mediators such as prostaglandins and leukotrienes.
Antioxidant activity: some quinazoline derivatives have been found to exhibit antioxidant activity by scavenging free radicals and preventing oxidative damage to cells and tissues.
Figure 10.
Examples of anticancer agents with quinazoline scaffold.
Here are a few examples of computational studies on quinazoline derivatives:
“Design, synthesis, and biological evaluation of novel quinoline derivatives as small molecule mutant EGFR inhibitors targeting resistance in NSCLC: In vitro screening and ADME predictions’’ by [56]. This study used computational methods such as molecular docking and molecular dynamics simulations to design and evaluate novel quinazoline derivatives as inhibitors of the EGFR kinase [54].
“Molecular docking, molecular dynamics simulation and QM/MM calculations study of quinazoline derivatives as tubulin inhibitors” by Mir et al. [57]. This study used a combination of computational methods to investigate the binding of quinazoline derivatives to tubulin, a protein involved in cell division. The study also included quantum mechanics/molecular mechanics (QM/MM) calculations to investigate the reaction mechanism of these compounds [57].
“3D-QSAR and molecular docking studies on a series of 2-phenylquinazoline-4(3H)-ones as antitumor agents” by Liu et al. [27]. This study used 3D-QSAR modeling and molecular docking to investigate the structure-activity relationships of a series of quinazoline derivatives with antitumor activity [58].
“Computational investigation of quinazoline derivatives as potential inhibitors of hepatitis C virus NS5B polymerase” by Nain et al. [59]. This study used molecular docking and molecular dynamics simulations to investigate the binding of quinazoline derivatives to the NS5B polymerase of the hepatitis C virus, with the aim of identifying potential inhibitors of viral replication [59].
Some additional examples of computational studies that have been used to investigate quinazoline derivatives:
Structure-based drug design: structure-based drug design involves using the three-dimensional structure of a target protein to design small molecules that bind to it with high affinity and specificity. This approach has been used to design quinazoline derivatives that target specific tyrosine kinases involved in cancer cell growth and proliferation.
Pharmacophore modeling: pharmacophore modeling is a computational method used to identify the essential structural and chemical features of a ligand that are required for binding to a protein target. This approach has been used to identify the pharmacophoric features of quinazoline derivatives that are responsible for their binding to specific protein targets.
ADMET prediction: ADMET (absorption, distribution, metabolism, excretion, and toxicity) prediction is a computational method used to predict the pharmacokinetic and toxicological properties of compounds. This approach has been used to predict the ADMET properties of quinazoline derivatives and identify potential drug candidates with improved pharmacokinetic profiles and reduced toxicity.
Molecular modeling of drug resistance: molecular modeling techniques have been used to investigate the mechanisms of drug resistance in cancer cells, particularly in the case of tyrosine kinase inhibitors. These studies have provided insights into the structural changes that occur in target proteins that lead to drug resistance and have helped in the design of new quinazoline derivatives that overcome drug resistance.
Structure-activity relationship (SAR) studies: SAR studies involve investigating the relationship between the chemical structure of a compound and its biological activity. SAR studies have been used extensively to optimize the biological activity of quinazoline derivatives, particularly in the case of anticancer activity.
Overall, computational studies have been instrumental in the discovery and optimization of quinazoline derivatives with diverse biological activities and have provided valuable insights into the molecular mechanisms of action and structure-activity relationships of these compounds. Here are some ways in which computational studies can help in the design of quinazoline derivatives:
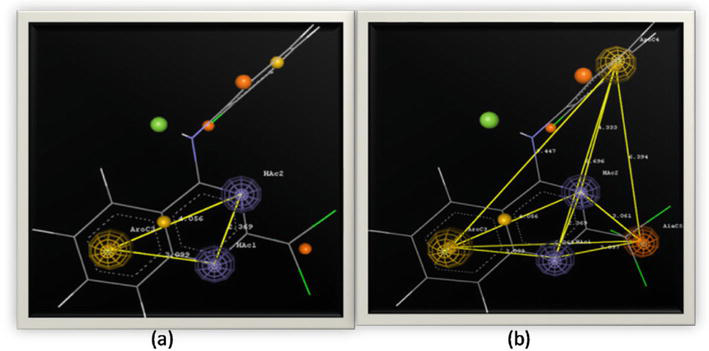
Identification of pharmacophoric features: computational methods such as pharmacophore modeling and molecular docking can help identify the essential structural and chemical features of quinazoline derivatives that are required for binding to specific protein targets. This information can be used to design new derivatives with improved binding affinity and selectivity; Figure 11 shows an example of pharmacophoric features to on a set of 133 compounds of quinazoline derivatives to identify the pharmacophoric feature [60].
Optimization of molecular properties: computational methods such as molecular dynamics simulations and ADMET prediction can be used to optimize the molecular properties of quinazoline derivatives, such as their solubility, stability, and pharmacokinetic properties. This can help improve the efficacy and safety profiles of these compounds.
Exploration of chemical space: computational methods such as virtual screening and machine learning can be used to explore large chemical space and identify potential drug candidates with desired biological activities. This can help speedup the drug discovery process and reduce the cost of drug development.
Prediction of drug resistance: computational methods can be used to predict the potential for drug resistance in cancer cells, particularly in the case of tyrosine kinase inhibitors. This information can be used to design new quinazoline derivatives that overcome drug resistance.
Structural optimization: computational methods such as structure-based drug design and SAR studies can be used to optimize the chemical structure of quinazoline derivatives for specific biological activities. This can help improve the potency and selectivity of these compounds.
Figure 11.
Pharmacophoric features (a) three point biophore, and (b) five point biophore [60].
Overall, computational studies provide a powerful tool for the design and optimization of quinazoline derivatives with diverse biological activities. By combining computational approaches with experimental methods, researchers can accelerate the drug discovery process and develop more effective and safer drugs for the treatment of various diseases. Here is an example of a quinazoline derivative that was optimized using computational methods:
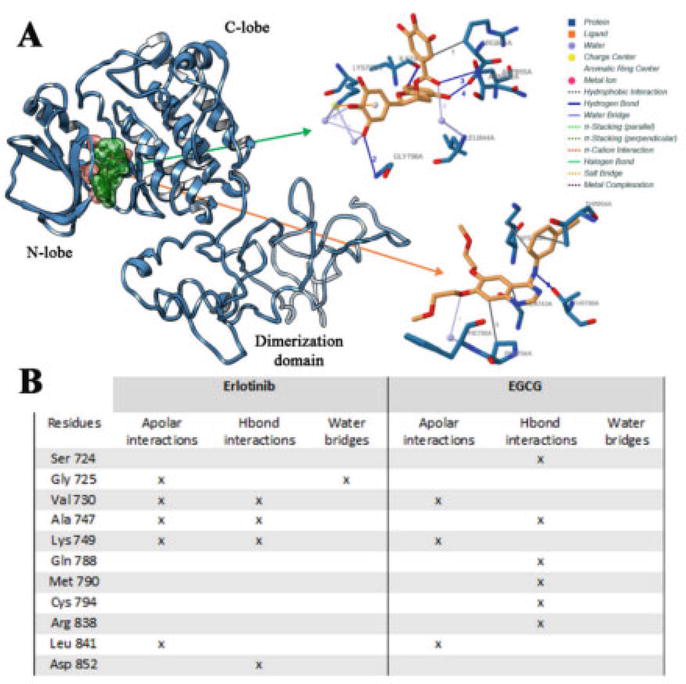
Erlotinib (trade name Tarceva) is a quinazoline derivative that acts as an inhibitor of the epidermal growth factor receptor (EGFR) tyrosine kinase, which is overexpressed in many types of cancer. Erlotinib was designed using a combination of computational and experimental methods. Computational studies were used to optimize the chemical structure of erlotinib for binding to the EGFR kinase domain. Molecular docking studies were used to predict the binding mode and affinity of erlotinib to the EGFR kinase (Figure 12), and molecular dynamics simulations were used to investigate the stability and flexibility of the erlotinib-EGFR complex [61]. Based on the results of these computational studies, a series of erlotinib analogs were synthesized and tested for their ability to inhibit EGFR kinase activity. The most promising analogs were then subjected to further optimization using a combination of computational and experimental methods. This approach led to the development of erlotinib, which was approved by the FDA in 2004 for the treatment of non-small cell lung cancer and later for pancreatic cancer. Erlotinib has since been used in the treatment of various types of cancer and has been shown to provide significant clinical benefits in some patients.
Figure 12.
Binding mode of erlotinib and EGCG in association with wild-type EGFR domain (A), and interactions list of erlotinib and EGCG with wild-type EGFR (B). The 3D binding mode of erlotinib (orange) and EGCG (green) has been reported [61].
Overall, the successful development of erlotinib highlights the importance of using computational methods in the design and optimization of quinazoline derivatives for the treatment of cancer.
Quinazoline derivatives have been studied extensively for their potential as anticancer agents and have been investigated for the treatment of various types of cancer [62]. Here are some examples:
Non-small cell lung cancer (NSCLC): quinazoline derivatives such as erlotinib and gefitinib have been approved for the treatment of NSCLC, which is the most common type of lung cancer [63].
Colorectal cancer: quinazoline derivatives such as lapatinib and afatinib have been investigated for the treatment of colorectal cancer, which is the third most common cancer worldwide [62].
Breast cancer: quinazoline derivatives such as lapatinib and afatinib have also been investigated for the treatment of breast cancer, particularly in the case of HER2-positive breast cancer [62].
Head and neck cancer: quinazoline derivatives such as gefitinib and erlotinib have been investigated for the treatment of head and neck cancer, which is the sixth most common cancer worldwide.
Glioblastoma: quinazoline derivatives such as lapatinib and erlotinib have been investigated for the treatment of glioblastoma, which is a highly aggressive form of brain cancer.
Prostate cancer: quinazoline derivatives such as zalutumumab and lapatinib have been investigated for the treatment of prostate cancer, which is the second most common cancer in men worldwide.
Overall, quinazoline derivatives have shown promise as anticancer agents in the treatment of various types of cancer. The development of new quinazoline derivatives with improved potency, selectivity, and pharmacokinetic properties is an active area of research in medicinal chemistry.
4.4 Other types of biological quinazoline derivatives
Quinazoline derivatives have been investigated for the treatment of various diseases beyond cancer. Here are some examples:
Neurological disorders: quinazoline derivatives have been investigated for the treatment of neurological disorders such as Alzheimer’s disease, Parkinson’s disease, and epilepsy. For example, quinazoline derivatives such as donepezil and tacrine have been used as acetylcholinesterase inhibitors for the treatment of Alzheimer’s disease [64].
Inflammation: quinazoline derivatives have been investigated for their anti-inflammatory activity. For example, quinazoline derivatives such as quinazoline-4-carboxylic acid and its derivatives have shown anti-inflammatory activity by inhibiting the production of pro-inflammatory cytokines [65].
Cardiovascular diseases: quinazoline derivatives have been investigated for the treatment of cardiovascular diseases such as hypertension and heart failure. For example, quinazoline derivatives such as prazosin and terazosin have been used as alpha-1 adrenergic receptor antagonists for the treatment of hypertension [66].
Diabetes: quinazoline derivatives have been investigated for the treatment of diabetes. For example, quinazoline derivatives such as imatinib have been shown to improve insulin sensitivity and glucose metabolism in animal models of diabetes.
Immune system: quinazoline derivatives have been investigated for their immunomodulatory activity and potential applications in the treatment of autoimmune diseases and cancer immunotherapy. For example, quinazoline derivatives such as imatinib have been shown to have immunomodulatory activity by inhibiting the c-kit receptor, which is expressed on mast cells and plays a role in the immune response.
Infectious diseases: quinazoline derivatives have been investigated for their activity against infectious diseases such as malaria, tuberculosis, and viral infections. For example, quinazoline derivatives such as mefloquine and hydroxyquinoline have been used as antimalarial agents, and quinazoline derivatives have shown activity against viruses such as HIV and hepatitis C.
Metabolic disorders: quinazoline derivatives have been investigated for the treatment of metabolic disorders such as diabetes and obesity. For example, quinazoline derivatives such as imatinib have been shown to improve insulin sensitivity and glucose metabolism in animal models of diabetes.
Overall, quinazoline derivatives have a wide range of biological activities and potential therapeutic applications beyond cancer. Ongoing research in this area is focused on developing new quinazoline derivatives with improved potency and selectivity for the treatment of various diseases.
4.5 Determination of the selectivity of quinazoline derivatives for specific targets
Researchers determine the selectivity of quinazoline derivatives for specific targets using a variety of experimental and computational methods. Here are some examples [67]:
Biochemical assays: biochemical assays are commonly used to measure the inhibitory activity of quinazoline derivatives against specific targets, such as tyrosine kinases. These assays can provide information on the potency and selectivity of quinazoline derivatives for specific targets.
Cell-based assays: cell-based assays are used to measure the activity of quinazoline derivatives in living cells and can provide information on the selectivity of these compounds in a more physiological context. For example, cell-based assays can be used to measure the activity of quinazoline derivatives against cancer cells that overexpress specific tyrosine kinases.
Proteomics: proteomics is a large-scale analysis of proteins and can be used to identify the targets of quinazoline derivatives in a complex biological system. For example, mass spectrometry-based proteomics can be used to identify the proteins that are bound by quinazoline derivatives in cancer cells.
Computational modeling: computational methods such as molecular docking and molecular dynamics simulations can be used to predict the binding affinity and selectivity of quinazoline derivatives for specific targets. These methods can provide valuable information on the structural and energetic factors that contribute to the binding of quinazoline derivatives to specific targets.
Kinome profiling: kinome profiling is a high-throughput method used to measure the activity of quinazoline derivatives against a panel of kinases. Kinome profiling can provide information on the selectivity of quinazoline derivatives against a large number of kinases and can help identify them.
There are several examples of quinazoline derivatives that have been found to be selective for specific targets. Here are some examples:
Lapatinib: lapatinib is a dual tyrosine kinase inhibitor that selectively targets both the epidermal growth factor receptor (EGFR) and HER2, which are overexpressed in many types of cancer. Lapatinib has been approved for the treatment of HER2-positive breast cancer.
Vandetanib: vandetanib is a tyrosine kinase inhibitor that selectively targets the vascular endothelial growth factor receptor (VEGFR), epidermal growth factor receptor (EGFR), and RET kinase, which are involved in the development and progression of various types of cancer. Vandetanib has been investigated for the treatment of thyroid cancer and non-small cell lung cancer.
Gefitinib: gefitinib is a tyrosine kinase inhibitor that selectively targets the EGFR kinase, which is overexpressed in many types of cancer. Gefitinib has been approved for the treatment of non-small cell lung cancer.
Afatinib: afatinib is a tyrosine kinase inhibitor that selectively targets the EGFR and HER2 kinases, which are overexpressed in many types of cancer. Afatinib has been approved for the treatment of non-small cell lung cancer and HER2-positive breast cancer.
BIBW2992: BIBW2992 is a dual tyrosine kinase inhibitor that selectively targets the EGFR and HER2 kinases. BIBW2992 has been investigated for the treatment of non-small cell lung cancer and HER2-positive breast cancer.
Overall, the development of quinazoline derivatives with increased selectivity for specific targets has been a major focus of research in medicinal chemistry. By developing selective quinazoline derivatives, researchers can minimize the potential for off-target effects and improve the efficacy and safety of these compounds as potential drugs for the treatment of various diseases, including cancer.
5. Challenges of synthesis of new quinazoline derivatives
Developing new quinazoline derivatives as potential drugs can be challenging due to several factors. Here are some of the challenges:
Selectivity: quinazoline derivatives are known to inhibit multiple tyrosine kinases, which can lead to off-target effects and toxicity. Developing derivatives with increased selectivity for specific targets can be challenging.
Resistance: cancer cells can develop resistance to tyrosine kinase inhibitors, including quinazoline derivatives, by acquiring mutations in the target kinase or activating alternative signaling pathways. Developing derivatives that are effective against resistant cancer cells can be challenging.
Pharmacokinetics: quinazoline derivatives can have poor pharmacokinetic properties, such as low solubility, poor bioavailability, and rapid metabolism. Developing derivatives with improved pharmacokinetic properties can be challenging.
Toxicity: quinazoline derivatives can cause various adverse effects, including skin rash, diarrhea, and liver toxicity. Developing derivatives with reduced toxicity can be challenging.
Intellectual property: quinazoline derivatives are a well-established class of compounds, and developing new derivatives that are sufficiently different from existing compounds to be patentable can be challenging.
Overall, developing new quinazoline derivatives as potential drugs requires a multidisciplinary approach that includes medicinal chemistry, pharmacology, and computational modeling. Overcoming these challenges is essential for developing new and effective quinazoline derivatives as potential drugs for the treatment of various diseases, including cancer.
References
1.Vijayakrishnan R. Structure-based drug design and modern medicine. Journal of Postgraduate Medicine. 2009;55(4):301. Available from: http://www.jpgmonline.com/article.asp?issn=0022-3859;year=2009;volume=55;issue=4;spage=301;epage=304;aulast=Vijayakrishnan
2.Talele TT, Khedkar SA, Rigby AC. Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Current Topics in Medicinal Chemistry. 2010;10(1):127-141
3.Mohammadzadeh S, Sharriatpanahi M, Hamedi M, Amanzadeh Y, Ebrahimi SES, Ostad SN. Antioxidant power of Iranian propolis extract. Food Chemistry. 2007;103(3):729-733. Available from: http://ac.els-cdn.com/S0308814606007266/1-s2.0-S0308814606007266-main.pdf?_tid=0cc7a9fa-a185-11e6-aff0-00000aacb361&acdnat=1478150529_7c6b71562fb2dedac61c926e6fe8909b
4.Van Drie JH. Computer-aided drug design: The next 20 years. Journal of Computer-Aided Molecular Design. 2007;21(10-11):591-601
5.Kalyaanamoorthy S, Chen Y-PP. Structure-based drug design to augment hit discovery. Drug Discovery Today. 2011;16(17):831-839
6.Sliwoski G, Kothiwale S, Meiler J, Lowe EW. Computational methods in drug discovery. Pharmacological Reviews. 2014;66(1):334-395
7.Jorgensen WL. Drug discovery: Pulled from a protein's embrace. Nature. 2010;466(7302):42-43
8.Ferreira LG, dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules. 2015;20(7):13384-13421
9.Bambini S, Rappuoli R. The use of genomics in microbial vaccine development. Drug Discovery Today. 2009;14(5):252-260
10.Wang R, Fang X, Lu Y, Wang S. The PDBbind database: Collection of binding affinities for protein−ligand complexes with known three-dimensional structures. Journal of Medicinal Chemistry. 2004;47(12):2977-2980
11.Klebe G. Virtual ligand screening: Strategies, perspectives and limitations. Drug Discovery Today. 2006;11(13):580-594
12.Lee A, Lee K, Kim D. Using reverse docking for target identification and its applications for drug discovery. Expert Opinion on Drug Discovery. 2016;11(7):707-715
13.McConkey BJ, Sobolev V, Edelman M. The performance of current methods in ligand–protein docking. Current Science. 2002;83(7):845-856
14.Meng X, Zhang H, Mezei M, Cui M. Molecular docking: A powerful approach for structure-based drug discovery. Current Computer-Aided Drug Design. 2011;7(2):146-157. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3151162/pdf/nihms-308746.pdf
15.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. Journal of Molecular Biology. 1982;161(2):269-288
16.Hammes GG. Multiple conformational changes in enzyme catalysis. Biochemistry. 2002;41(26):8221-8228
17.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of Medicinal Chemistry. 2004;47(7):1739-1749
18.Moitessier N, Englebienne P, Lee D, Lawandi J, Corbeil CR. Towards the development of universal, fast and highly accurate docking/scoring methods: A long way to go. British Journal of Pharmacology. 2008;153(S1):S7-S26
20.Meng EC, Shoichet BK, Kuntz ID. Automated docking with grid-based energy evaluation. Journal of Computational Chemistry. 1992;13(4):505-524
21.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19(14):1639-1662
22.Feig M, Onufriev A, Lee MS, Im W, Case DA, Brooks CL. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. Journal of Computational Chemistry. 2004;25(2):265-284
23.Bhattacharjee B, Chatterjee J. Identification of proapoptopic, anti-inflammatory, anti-proliferative, anti-invasive and anti-angiogenic targets of essential oils in cardamom by dual reverse virtual screening and binding pose analysis. Asian Pacific Journal of Cancer Prevention. 2013;14(6):3735-3742
24.Kinnings SL, Jackson RM. ReverseScreen3D: A structure-based ligand matching method to identify protein targets. Journal of Chemical Information and Modeling. 2011;51(3):624-634
25.Gurung A, Ali M, Bhattacharjee A, Al-Anazi K, Farah M, Al-Hemaid F, et al. Target fishing of glycopentalone using integrated inverse docking and reverse pharmacophore mapping approach. Genetics and Molecular Research. 2016;15(3):01-13
26.Kharkar PS, Warrier S, Gaud RS. Reverse docking: A powerful tool for drug repositioning and drug rescue. Future Medicinal Chemistry. 2014;6(3):333-342
27.Liu X, Ouyang S, Yu B, Liu Y, Huang K, Gong J, et al. PharmMapper server: A web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Research. 2010;38(suppl 2):W609-W614
28.Grinter SZ, Liang Y, Huang S-Y, Hyder SM, Zou X. An inverse docking approach for identifying new potential anti-cancer targets. Journal of Molecular Graphics and Modelling. 2011;29(6):795-799
29.Chen YZ, Zhi DG. Ligand-protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins: Structure, Function, and Bioinformatics. 2001;43:217-226
30.Do Q-T, Renimel I, Andre P, Lugnier C, Muller CD, Bernard P. Reverse pharmacognosy: Application of Selnergy, a new tool for lead discovery. The example of ε-viniferin. Current Drug Discovery Technologies. 2005;2(3):161-167
31.Muller P, Lena G, Boilard E, Bezzine S, Lambeau G, Guichard G, et al. In silico-guided target identification of a scaffold-focused library: 1, 3, 5-Triazepan-2, 6-diones as novel phospholipase A2 inhibitors. Journal of Medicinal Chemistry. 2006;49(23):6768-6778
32.Zahler S, Tietze S, Totzke F, Kubbutat M, Meijer L, Vollmar AM, et al. Inverse in silico screening for identification of kinase inhibitor targets. Chemistry & Biology. 2007;14(11):1207-1214
33.Schapira M, Abagyan R, Totrov M. Nuclear hormone receptor targeted virtual screening. Journal of Medicinal Chemistry. 2003;46(14):3045-3059
34.Rollinger JM. Accessing target information by virtual parallel screening—The impact on natural product research. Phytochemistry Letters. 2009;2(2):53-58
35.Bissantz C, Logean A, Rognan D. High-throughput modeling of human G-protein coupled receptors: Amino acid sequence alignment, three-dimensional model building, and receptor library screening. Journal of Chemical Information and Computer Sciences. 2004;44(3):1162-1176
36.Chen S, Ren J. Identification of a potential anticancer target of Danshensu by inverse docking. Asian Pacific Journal of Cancer Prevention. 2014;15(1):111-116
37.Desaphy J, Bret G, Rognan D, Kellenberger E. sc-PDB: A 3D-database of ligandable binding sites—10 years on. Nucleic Acids Research. 2014;43(D1):D399-D404
38.Consortium U. The universal protein resource (UniProt) in 2010. Nucleic Acids Research. 2010;38(suppl 1):D142-D148
39.Gao Z, Li H, Zhang H, Liu X, Kang L, Luo X, et al. PDTD: A web-accessible protein database for drug target identification. BMC Bioinformatics. 2008;9(1):104
40.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews. 1997;23(1-3):3-25
41.Pagliara A, Reist M, Geinoz S, Carrupt PA, Testa B. Evaluation and prediction of drug permeation. Journal of Pharmacy and Pharmacology. 1999;51(12):1339-1357
42.Stenberg P, Luthman K, Ellens H, Lee CP, Smith PL, Lago A, et al. Prediction of the intestinal absorption of endothelin receptor antagonists using three theoretical methods of increasing complexity. Pharmaceutical Research. 1999;16(10):1520-1526
43.Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. Journal of Combinatorial Chemistry. 1999;1(1):55-68
44.Oprea TI. Property distribution of drug-related chemical databases. Journal of Computer-Aided Molecular Design. 2000;14(3):251-264
45.Blake JF. Chemoinformatics–predicting the physicochemical properties of ‘drug-like’ molecules. Current Opinion in Biotechnology. 2000;11(1):104-107
46.Van De Waterbeemd H, Smith DA, Beaumont K, Walker DK. Property-based design: Optimization of drug absorption and pharmacokinetics. Journal of Medicinal Chemistry. 2001;44(9):1313-1333
47.Van De Waterbeemd H, Gifford E. ADMET in silico modelling: Towards prediction paradise? Nature Reviews Drug Discovery. 2003;2(3):192-204
48.Butina D, Segall MD, Frankcombe K. Predicting ADME properties in silico: Methods and models. Drug Discovery Today. 2002;7(11):S83-S88
49.Valerio LG. In silico toxicology for the pharmaceutical sciences. Toxicology and Applied Pharmacology. 2009;241(3):356-370
50.Hou H, Leung KC-F, Lanari D, Nelson A, Stoddart JF, Grubbs RH. Template-directed one-step synthesis of cyclic trimers by ADMET. Journal of the American Chemical Society. 2006;128(48):15358-15359
51.Chou C-H, Lin F-M, Chou M-T, Hsu S-D, Chang T-H, Weng S-L, et al. A computational approach for identifying microRNA-target interactions using high-throughput CLIP and PAR-CLIP sequencing. BMC Genomics. 2013;14(1):S2
52.Politzer P, Murray JS. The fundamental nature and role of the electrostatic potential in atoms and molecules. Theoretical Chemistry Accounts. 2002;108:134-142
53.Xiang M, Cao Y, Fan W, Chen L, Mo Y. Computer-aided drug design: Lead discovery and optimization. Combinatorial Chemistry & High Throughput Screening. 2012;15(4):328-337
54.El-Azab AS, Al-Omar MA, Alaa A-M, Abdel-Aziz NI, Magda A-A, Aleisa AM, et al. Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: Molecular docking study. European Journal of Medicinal Chemistry. 2010;45(9):4188-4198
55.Emami L, Khabnadideh S, Faghih Z, Farahvasi F, Zonobi F, Gheshlaghi SZ, et al. Synthesis, biological evaluation, and computational studies of some novel quinazoline derivatives as anticancer agents. BMC Chemistry. 2022;16(1):1-14
56.Kardile RA. Sarkate AP, Lokwani DK, Tiwari SV, Azad R, Thopate SR. Design, synthesis, and biological evaluation of novel quinoline derivatives as small molecule mutant EGFR inhibitors targeting resistance in NSCLC: In vitro screening and ADME predictions. European Journal of Medicinal Chemistry. 2023;245:114889
57.Mir SA, Dash GC, Meher RK, Mohanta PP, Chopdar KS, Mohapatra PK, et al. In silico and in vitro evaluations of fluorophoric thiazolo-[2, 3-b] quinazolinones as anti-cancer agents targeting EGFR-TKD. Applied Biochemistry and Biotechnology. 2022;194(10):4292-4318
58.Wan Z, Hu D, Li P, Xie D, Gan X. Synthesis, antiviral bioactivity of novel 4-thioquinazoline derivatives containing chalcone moiety. Molecules. 2015;20(7):11861-11874
59.Nasr T, Aboshanab AM, Mpekoulis G, Drakopoulos A, Vassilaki N, Zoidis G, et al. Novel 6-Aminoquinazolinone derivatives as potential cross GT1-4 HCV NS5B inhibitors. Viruses. 2022;14(12):2767
60.Mishra M, Agarwal S, Dixit A, Mishra VK, Kashaw V, Agrawal RK, et al. Integrated computational investigation to develop molecular design of quinazoline scaffold as promising inhibitors of plasmodium lactate dehydrogenase. Journal of Molecular Structure. 2020;1207:127808
61.Minnelli C, Laudadio E, Mobbili G, Galeazzi R. Conformational insight on WT-and mutated-EGFR receptor activation and inhibition by epigallocatechin-3-gallate: Over a rational basis for the design of selective non-small-cell lung anticancer agents. International Journal of Molecular Sciences. 2020;21(5):1721
62.Ravez S, Castillo-Aguilera O, Depreux P, Goossens L. Quinazoline derivatives as anticancer drugs: A patent review (2011–present). Expert Opinion on Therapeutic Patents. 2015;25(7):789-804
63.Ibrahim MT, Uzairu A, Uba S, Shallangwa GA. Design of more potent quinazoline derivatives as EGFR WT inhibitors for the treatment of NSCLC: A computational approach. Future Journal of Pharmaceutical Sciences. 2021;7:1-11
64.Haghighijoo Z, Zamani L, Moosavi F, Emami S. Therapeutic potential of quinazoline derivatives for Alzheimer's disease: A comprehensive review. European Journal of Medicinal Chemistry. 2022;227:113949
65.Laddha SS, Bhatnagar SP. A new therapeutic approach in Parkinson’s disease: Some novel quinazoline derivatives as dual selective phosphodiesterase 1 inhibitors and anti-inflammatory agents. Bioorganic & Medicinal Chemistry. 2009;17(19):6796-6802
66.Selvam TP, Kumar PV. Quinazoline marketed drugs. Research in Pharmacy. 2015;1(1):1-21
67.Marzaro G, Guiotto A, Chilin A. Quinazoline derivatives as potential anticancer agents: A patent review (2007-2010). Expert Opinion on Therapeutic Patents. 2012;22(3):223-252
Written By
Wafa Mohamed Al Madhagi
Submitted: 28 June 2023Reviewed: 29 June 2023Published: 08 November 2023
 Open access peer-reviewed chapter
Open access peer-reviewed chapter