Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
Quinazolines are important stable heterocyclic compounds of great biological activates. Naturally, they are found in many plants that are the source of these quinazolines in addition they are synthesized chemically. Recently quinazolines represent a nucleus of the vast majority of novel compounds that have promising biological activity. They show different activities by acting on different body targets such activities are anticancer, antihypertensive, antimicrobial, antifungal, antibacterial, analgesic, anti-inflammatory, antituberculosis, and antimalarial activity. This chapter highlights the recent advance in the biological activates of quinazolines and quinazolines derivatives on different biological targets.
Faculty of Pharmacy, Medicinal Chemistry Department, Sana’a University, Yemen
Faculty of Medical Sciences, Department of Pharmacy, National University, Yemen
Rana Abdullah Al-Robaidi
Faculty of Pharmacy, Medicinal Chemistry Department, Sana’a University, Yemen
*Address all correspondence to: alialkaf21@gmail.com, alialkaf@national-univ.net
1. Introduction
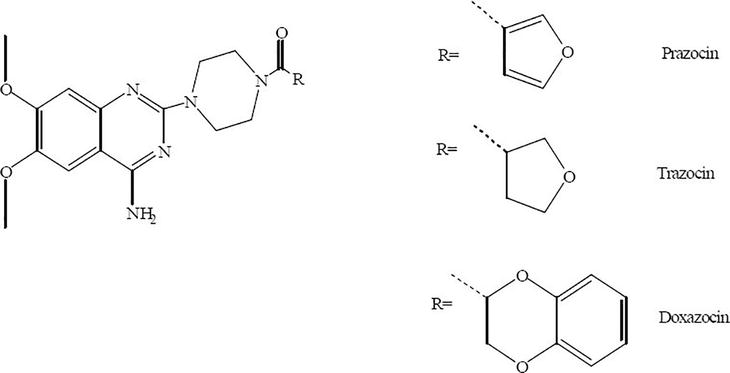
Heterocyclic compounds represent the biggest group in the field of medicinal chemistry for the treatment of diseases and infections; among this group, is quinazoline moiety. Quinazoline is a heterocyclic system having two aromatic six-membered rings. One of them contains two nitrogen atoms named as pyrimidine ring and this ring is fused to the second aromatic benzene ring. Therefore, quinazoline is a phenyl pyrimidine compound. Quinazolines have a wide range of pharmacological activities. They are used as anticancer, antiviral, antibacterial, antitubercular, analgesic, antihypertensive, anti-inflammatory, antidiabetic, sedative-hypnotic, antihistaminic, anticonvulsant, and many other uses [1]. Scientists have been interested in quinazoline alkaloids since 1888, when (+)-peganine, a natural representative of the class, was discovered (vasicine). The vasicine biosynthetic pathway: The amide synthesis that follows the anthranilate nitrogen’s neucleophilic attack on the pyrrolinium cation easily explains the structure of the peganine skeleton. Surprisingly, Justicia adhatoda does not use this pathway; instead, there is a much less predictable sequence involving aspartic acid and acetylanthranilic acid [2]. Due to their extensive spectrum of antibacterial, antifungal, anti-inflammatory, antimalarial, anti-HIV, antiviral, and antituberculosis characteristics, along with their capacity to inhibit thymidylate synthase, poly-(ADP-ribose) polymerase (PARP), and tyrosine kinase, quinazolinones and quinazolines hold significance in the field of medicinal chemistry. Several licensed medications with quinazoline structure are available on the market, including doxazosine mesylate, terazosine hydrochloride, and prazosin hydrochloride (Figure 1) [3].
Figure 1.
Quinazoline-containing approved and marketed medications.
2. Therapeutic potential of quinazoline and quinazolinone compounds
One of the most beneficial heterocyclic substances originating from the perspectives of synthetic and therapeutic chemical analysis are quinazolines and quinazolinones. Because of its wide range of biological efficacy, varied chemical reactivity, and easily accessible nature, the scaffolds’ structural design has attracted a lot of attention. Despite a substantial corpus of literature containing many instances of these themes demonstrating possible biological processes, we have concentrated here on the latest changes in these compounds’ activity profiles [4].
2.1 Anti-inflammatory properties
Two sets of derivatives based on 2-phenyl-4(3H) quinazolinone demonstrated potent anti-inflammatory and analgesic effects in experimental rats, exhibiting an improved gastrointestinal safety profile in comparison to the reference drug. In experimental rats, a few chemical entities showed the strongest inflammation-inhibiting effects when compared to the reference drug indomethacin (Figure 2) [2].
Figure 2.
Derivatives based on 2-phenyl-4(3H) quinazolinone.
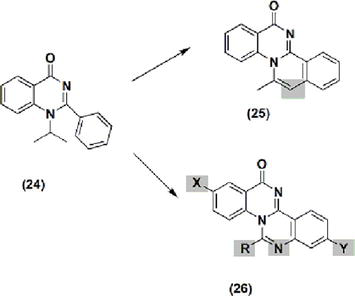
Anti-inflammatory activity [5] of two novel quinazoline derivatives were synthesized, identified as 25 and 26 (Figure 3) and their anti-inflammatory properties were evaluated. Isoquino-quinazolinone 25 and quinazolino-quinazolinone 26 were substituted for these derivatives. However, in contrast to the high potency observed in the substituted 2-phenyl-quinazoline compound 24, neither of them demonstrated a comparable level of activity.
Figure 3.
The quinazolines 24, 25, and 26 have anti-inflammatory properties.
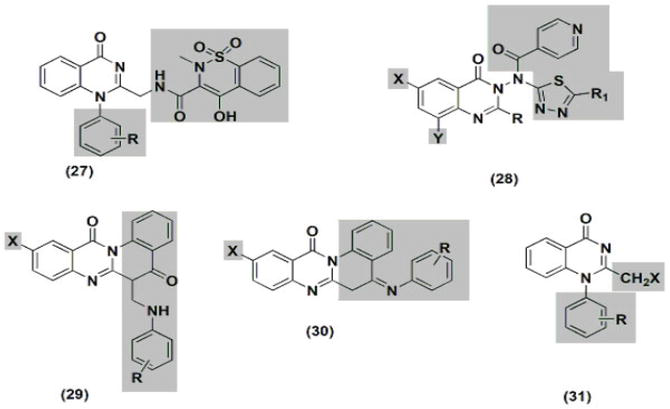
Taking regular piroxicam at 4 mg/kg, the anti-inflammatory activity of additional variations of compound 27 (Figure 4) was evaluated. Compound 27’s derivatives without substitution, those with a 4-methyl substitution, and those with a 3-nitro substitution produced higher activity than regular piroxicam, according to a structure-activity relationship (SAR) study. An administration of three derivatives at a dose of 4 mg/kg exhibited less activity than piroxicam diclofenac. To increase the anti-inflammatory activity, thiadiazole with a nicotinyl moiety was added to the structure at position C-3 of compound 28. The highest activity was produced by substituted 2-phenylquinazoli’s nicotinyl-5-pyridyl thiadiazole, which is similar to regular ibuprofen. The activity range of substituted quinazolinedione derivatives 29 and 30 was 12.7–58.2%. 10-Iodosubstitution significantly increased the anti-inflammatory activity, according to SAR studies. Furthermore, the anti-inflammatory activity was improved by substituting 4-phenylaminomethyl and 6-methoxyphenylaminomethyl.
Figure 4.
The compounds 27, 28, 29, 30, and 31 exhibit anti-inflammatory characteristics.
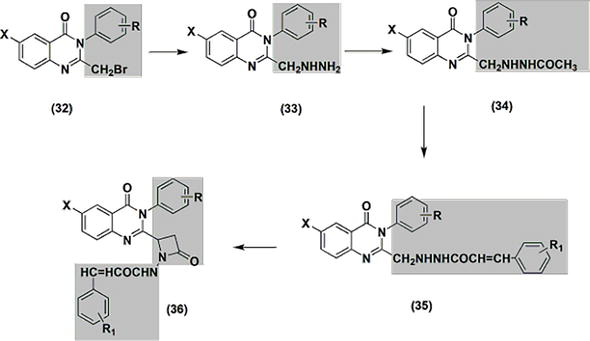
31-Phenyl quinazolinone derivatives were synthesized in order to assess their potential as inflammation-inhibiting drugs. Among these variations, the most potent was the compound with a two-piperidenomethyl substitution., followed by two-methlyl and two-dimethylaminomethyl. The 2-chloromethyl substituents were the least active. The anti-inflammatory potential of the 2,3,6-trisubstituted quinazolinone derivatives 32–36 (depicted in Figure 5) was explored through development, synthesis, and evaluation as potential medications. The variable activity range for these derivatives was 10.28–53.33%. The activity of compounds containing p-dimethylaminophenyl at C-2 and o-methoxyphenyl substituents at C-3 was higher than that of standard phenylbutazone. Furthermore, the ulcerogenic potential of these derivatives was examined. When compared to regular phenylbutazone, the potent anti-inflammatory derivatives showed less ulcerogenic activity.
Figure 5.
The compounds 32, 33, 34, 35, and 36 are agents with anti-inflammatory properties.
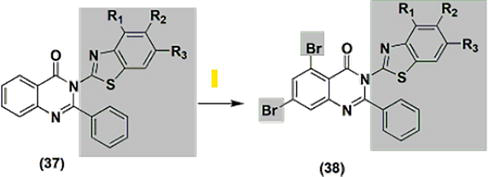
Assessing against indomethacin as the reference, the anti-inflammatory properties of various newly developed derivatives from benzothiazole-substituted 2-phenyl quinazolinones 37, 38 (illustrated in Figure 6) were investigated. Each of the derivatives exhibited reduced efficacy (ranging from 21.3% to 77.5% protection) compared to indomethacin, which provided 80.9% protection. The efficacy of the derivatives with bromine substitution (38) was inferior, with protection ranging from 21.3% to 27%, compared to the unsubstituted derivatives, which showed protection between 30% and 77.5%. A SAR investigation revealed that compounds featuring electron-withdrawing groups, like 4-nitro and halogen, exhibited reduced activity compared to compounds with electron-releasing groups, such as 4-alkyl and alkoxy. Moreover, when assessed for their inhibitory effects on COX-I and COX-II, these derivatives displayed limited activity against COX-I and substantial activity against COX-II. The IC50 values for COX-I ranged from 98 to 100 μM, while for COX-II, they ranged from 0.39 to 1.87 μM. In contrast, COX-I showed 0.22 μM and COX-II showed 2.64 μM with standard indomethacin. Furthermore, at an oral dose of 100 mg/kg/day, the effective derivative exhibited a favorable gastrointestinal safety profile with no ulceration, in stark contrast to the 100% ulceration observed with the equivalent dose of indomethacin.
Figure 6.
Quinazolines 37 and 38 with anti-inflammatory properties.
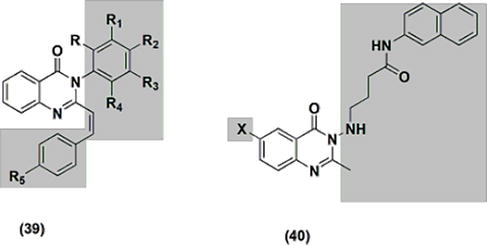
Quinazolinone 39 new derivatives (Figure 7) showed comparable efficacy to ibuprofen at a dose of 200 mg/kg. Four-nitrostyryl-substituted quinazolinone and four-hydroxystyryl-substituted quinazolinone were the most active derivatives. The edema volume was reduced in an activity range of 62.2–80.7%. The anti-inflammatory properties of the quinazolinone derivatives 40, which are substituted with phenylaphtalene (Figure 8), were evaluated at 50 mg/kg. They produced inhibition ranging from 19.69% to 59.61% and demonstrated a favorable gastrointestinal safety profile, with ulcerogenic activity in the range of 30–70%. The most effective derivative among these was 6-bromosubstituted-quinazolinone. Standard phenylbutazone had 50% ulcerogenic activity and 38.9% anti-inflammatory activity.
Figure 7.
The anti-inflammatory quinazolines 39 and 40.
Figure 8.
Quinazoline compound with corresponding chelates shows antifungal activity.
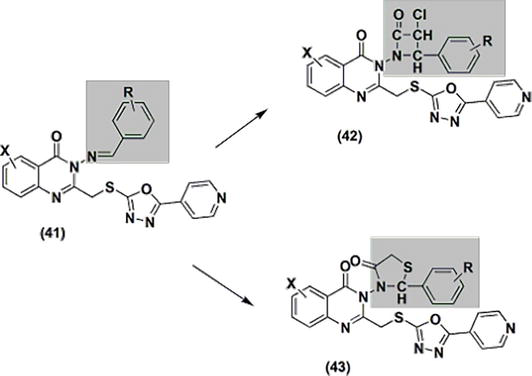
Novel derivatives 41–43 were created when thiazolidindione and azetidinone were added to quinazolinone at C-3 (Figure 9). The anti-inflammatory properties of these derivatives were assessed. At a dosage of 50 mg/kg, they produced activity that was comparable to standard phenylbutazone (16.33–36.3%). Compound 41’s arylidene underwent cyclization, yielding azetidinone derivative 42 and thiazolidinone derivative 43. When compared to the azetidinone derivative series, the thiazolidinone derivative series exhibited greater anti-inflammatory activity.
Figure 9.
Quinazolines 41, 42, and 43 with anti-inflammatory properties.
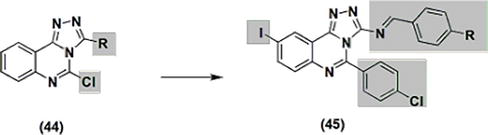
An examination of the anti-inflammatory properties of the triazoloquinazoline derivative with a 5-chloro-2 substitution. Compound 44 (Figure 10) indicated that its activity ranged from more potent to equipotent compared to the reference ketoprofen. The carrageenin-induced paw edema test was employed to formulate, synthesize, and assess the anti-inflammatory activity of an additional set of triazolo-quinazoline derivatives with substitutions, illustrated as compound 45 in Figure 10. The outcomes revealed activity that was similar to that of the reference indomethacin.
Figure 10.
Quinazolines 44 and 45 with anti-inflammatory properties.
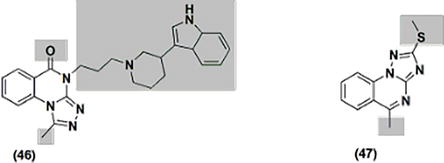
Inflammatory conditions such as rheumatoid arthritis, autoimmune diseases, and ulcerative colitis are addressed using alternative derivatives of piperidinyl-substituted quinazolinone. Compound 46 was assessed for its anti-inflammatory properties. (Figure 11). It proved to have potent anti-inflammatory qualities. Strong anti-inflammatory properties against multiple molecules involved in inflammation, including prostaglandin (PGE-2), nitric oxide (NO), and tumor necrosis factor-alpha (TNF-α), and macrophages stimulated by bacterial endotoxin lipopolysaccharide (LPS), have been reported for 5-chloro-2-methylsulfonyl-triazoloquiazoline 47 (Figure 11).
Figure 11.
Quinazolines 46 and 47 with anti-inflammatory properties.
2.2 Antifungal activity
The antifungal efficacy of the ligand 6-bromo-2[(4-(2,3-dichlorophenyl)) piperazine-1-yl)methyl]-3-[8-hydroxyquinoline-5-yl]-3-quinazolin-4-one and its corresponding transition metal chelates (Figure 8).
By condensing 2-phenyl-3,1-benzoxazine-4-one with 4-substituted phenyl ureas, a collection of innovative compounds, namely 4-oxo-2-phenyl-4H-quinazoline-3-carboxylic acid (4-substituted phenyl amides), was successfully synthesized. The synthesis involved combining N-benzoyl anthranilic acid with acetic anhydride to produce 2-phenyl-3,1-benzoxazine-4-one. Subsequently, sodium cyanide was employed to condense various 4-substituted anilines, leading to the formation of 4-substituted phenyl ureas. The in vitro antifungal activity of all synthesized compounds against four pathogenic fungi was evaluated using the standard agar dilution method. The resulting zone of inhibition was then determined. Clotrimazole was employed as the reference standard. However, none of the compounds exhibited activity against Aspergillus fumigatus (Figure 12) [2].
Figure 12.
4-Oxo-2-phenyl-4H-quinazoline-3-carboxylic acid.
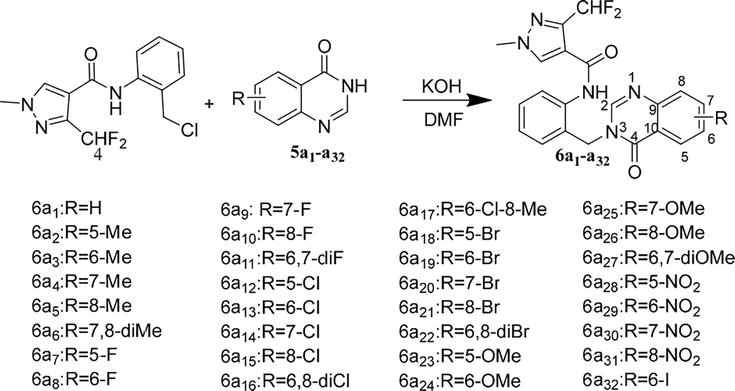
In an effort to find a novel fungicide against Rhizoctonia solani, 32 new pyrazole carbamide derivatives with quinazolinone scaffold were designed and synthesized. 3-(difluoromethyl)-N-(2-((6,7-difluoro-4-oxoquinazolin-3(4H)-yl) methyl)phenyl)-1-methyl-1H-pyrazole-4-carboxamide (6a11) single-crystal X-ray diffraction verified the target compounds’ structure. At 100 μg mL−1, the target compounds’ in vitro antifungal activity against R. solani was assessed. According to the findings of the SAR analysis, antifungal activity peaked at position 6 when the substitution activity was there. Furthermore, the antifungal activity was directly impacted by the location and quantity of chlorine atoms. Following additional in vitro bioassays, it was discovered that 6a16 (EC50 = 9.06 mg/L) exhibited superior antifungal activity against R. solani compared to fluconazole (EC50 = 12.29 mg/L), but less so than bixafen (EC50 = 0.34 mg/L). According to 7.33 (SEM), scanning electron microscopy, N-(2-((6,8-dichloro-4-oxoquinazolin-3(4H)-yl)methyl)phenyl)-3-(difluoromethyl) additionally, -1-methyl-1H-pyrazole-4-carboxamide (6a16) had an impact on the mycelial shape. The results showed that creating antifungal candidates through molecular hybridization was a useful technique. In the meantime, compounds containing a quinazolinone fragment in pyrazolecarbamide showed possible antifungal activity against R. solani (Figure 13) [6].
Figure 13.
Synthesis of the target compounds 6a1–a32 [5].
2.3 Anticancer activity
A class of diseases known as cancer arises when aberrant cells proliferate uncontrollably and have the ability to infiltrate other tissues. Cancer cells have the ability to travel through the blood and lymphatic systems to other areas of the body. Many anti-cancer medications have an incomplete precise mechanism of action, and most of the time it is unknown what causes their marginal anti-tumor selectivity. Promising derivatives’ anti-cancer activity [7, 8, 9, 10, 11] is demonstrated by the following: the percentage of control cells that survive decreases as concentrations increase, indicating the cytotoxic characteristics of the derivatives. Upon analysis, there was a discernible suppression of tumor growth (Figure 14).
After being tested against three human cancer cells for their antiproliferative properties, the following newly synthesized compounds [12, 13, 14] showed a notable inhibition of tumor growth. 8–10. These substances have cytotoxic properties and stop the body from spreading (Figure 15) [15].
Figure 15.
Compounds [15, 16, 17].
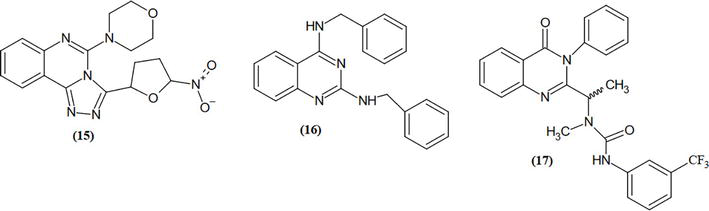
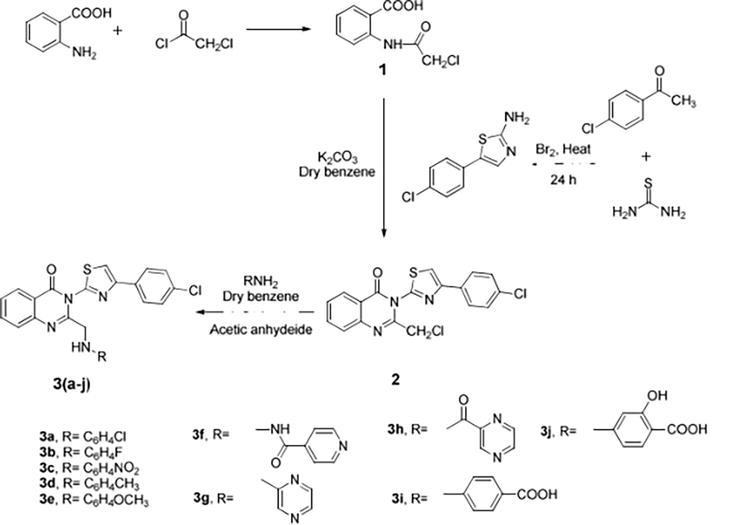
We conceived and synthesized innovative thiazole compounds based on quinazoline as inhibitors of EGFR-mutant kinase, aiming at treating non-small cell lung cancer (NSCLC) along with human breast (MCF-7), liver (HepG2), and lung (A549) cancer cell lines. Figure 16 outlines the pathways for generating novel thiazole derivatives based on quinazoline, while Table 1 presents the structures of the derivatives 4a–j (Table 2) [16].
Figure 16.
The synthetic pathway for preparing the target.
Table 1.
Structures of the derivatives 4a–j.
Compound
IC50 (μM)
MCF-7
HepG2
A549
Vero
4a
6.21 ± 0.33
11.86 ± 0.71
24.73 ± 0.95
>50
4b
6.28 ± 0.58
12.59 ± 1.02
27.04 ± 1.09
>50
4c
7.43 ± 0.91
14.16 ± 0.83
31.16 ± 0.97
>50
4d
9.75 ± 1.03
17.28 ± 0.98
24.97 ± 1.14
>50
4e
8.29 ± 0.44
15.03 ± 0.81
29.61 ± 1.22
>50
4f
3.71 ± 0.47
7.92 ± 0.63
19.02 ± 0.83
>50
4 g
4.14 ± 0.69
9.36 ± 1.15
20.84 ± 1.24
>50
4 h
4.92 ± 0.31
9.85 ± 0.97
22.86 ± 1.06
>50
4i
2.86 ± 0.31
5.91 ± 0.45
14.79 ± 1.03
>50
4j
3.09 ± 0.45
6.87 ± 0.59
17.92 ± 0.95
>50
Erlotinib
3.16 ± 0.22
6.83 ± 0.51
19.42 ± 1.28
>30
Table 2.
Cytotoxicity of compounds 4a–j on cancer cell lines.
2.4 Antihypertensive activity
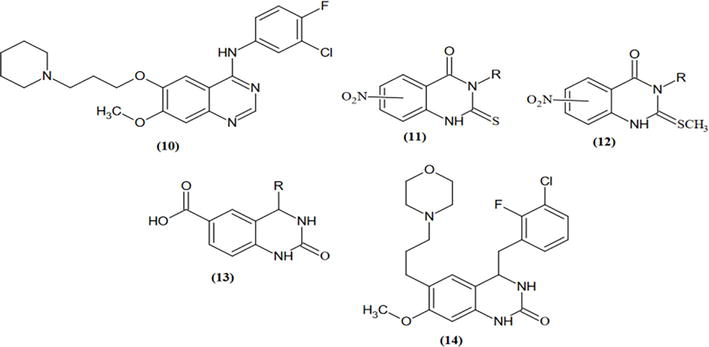
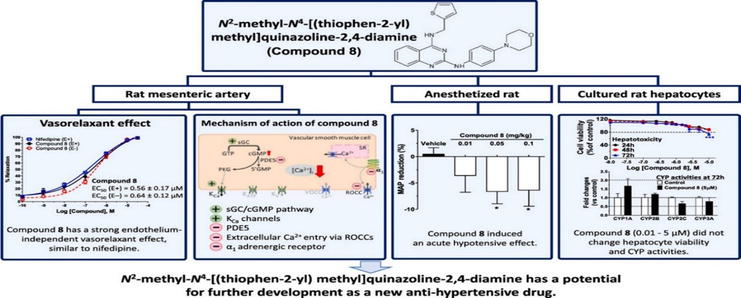
In an attempt to create novel pulmonary hypertension treatments with PDE5 inhibitors, twenty N2, N4-2,4-diamines of disubstituted quinazoline were recently designed based on the sildenafil-like nature of their pharmacophore. Six of them exhibited promising inhibitory effects on rat PDE5 (compounds 4, 5, 8, 9, 10, and 11). One compound (compound 8, N2-methyl-N4-[(thiophen-2-yl)methyl]quinazoline-2,4-diamine) demonstrated when testing their vasorelaxant effect on rat pulmonary arteries in comparison to aortas, they showed five times more selectivity for aortas than pulmonary arteries, highlighting its possible usefulness as a systemic vasodilator. The following were looked at in the current study: (i) in vivo: compound 8’s acute hypotensive effect in normotensive rats, and for the early evaluation of its drugability; (ii) ex vivo: compound 8’s vasorelaxant effect and the mechanisms involved in rat isolated mesenteric artery; (iii) toxicity on isolated hepatocytes; and (iv) its effect on liver cytochrome P450 (CYP) activities. The primary objectives of this study were to determine whether compound 8 can induce vasodilation in resistance arteries within the systemic vasculature and to comprehend its underlying mechanisms. Nifedipine served as the reference comparator (refer to Figure 17) [17].
Figure 17.
A N2-methyl-N4-[quinazoline-2,4-diamine](thiophen-2-yl)methyl) was found in the current study to have a strong vasodilation impact on the resistance vasculature [https://ars.els-cdn.com/content/image/1-s2.0-S0014299923003400-ga1_lrg.jpg].
Vasodilation impact on the resistance vasculature, which was linked to a hypotensive effect. Its relaxing action includes KCa channel opening, α1-adrenergic receptor and transmembrane calcium influx inhibition, and sGC/cGMP Course potentiation. If compound 8 proves to be a promising new drug candidate for the management of high blood pressure, more research needs to be done [17]. The ability of the derivatives of 6,7-di methoxy-2(phenylamino)quinazolin-4(3H)-one (Figure 18) to inhibit alpha-1 adrenergic receptors was tested [7].
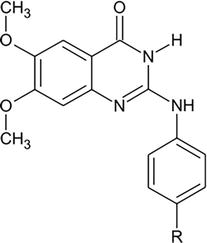
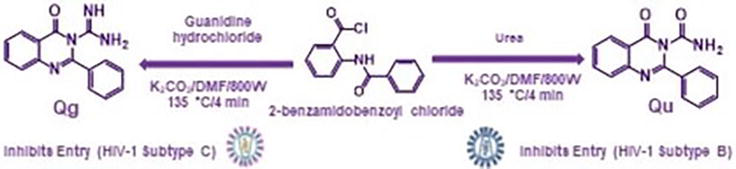
Several dihydrobenzo(h)quinazoline derivatives were synthesized starting from quinazloline derivatives and aryl methylene thiopyrimidine. Based on biological screening, many of these compounds demonstrated significant efficacy against viral infection [7]. Using an industrial-scale microwave-assisted production method, a set of hydrophilic 2,3-substituted quinazolinones, 4-oxo-2-phenylquinazoline-3(4H)-carboximidamide (Qg), and 4-oxo-2-phenylquinazoline-3(4H)-carboxamide (Qu), were produced in promising yields. These agents possess both antiviral and antibacterial properties. They were produced by reacting urea or guanidine hydrochloride with 2-benzamidobenzoyl chloride in DMF’s potassium carbonate solution. Geometrical optimization and vibrational frequency analysis of the compounds were performed using DFT calculations. The DFT calculations of the free energies of solvation (ΔGsol) for Qg and Qu provide evidence in favor of their hydrophilicity. We looked into the anti-HIV-1 properties of the small-molecule quinazolinone derivatives Qu and Qg. In vitro, the synthons have a low cytotoxicity (EC50 = 50.53 nM for Qu and EC50 = 97.92 nM for Qg) and prevent HIV entry in receptor cells. The two benzo-fused pyrimidine derivatives, which were previously reported to exhibit antitubercular activity, are identified in the study in terms of their antiviral characteristics. Our results point to the need for additional synthetic investigations into these quinazolinone scaffolds, which may result in the creation of viable substitutes for use in clinical research, pharmaceutics, and drug design (Figure 19) [8].
Figure 19.
Small-molecule quinazolinone derivatives Qu and Qg.
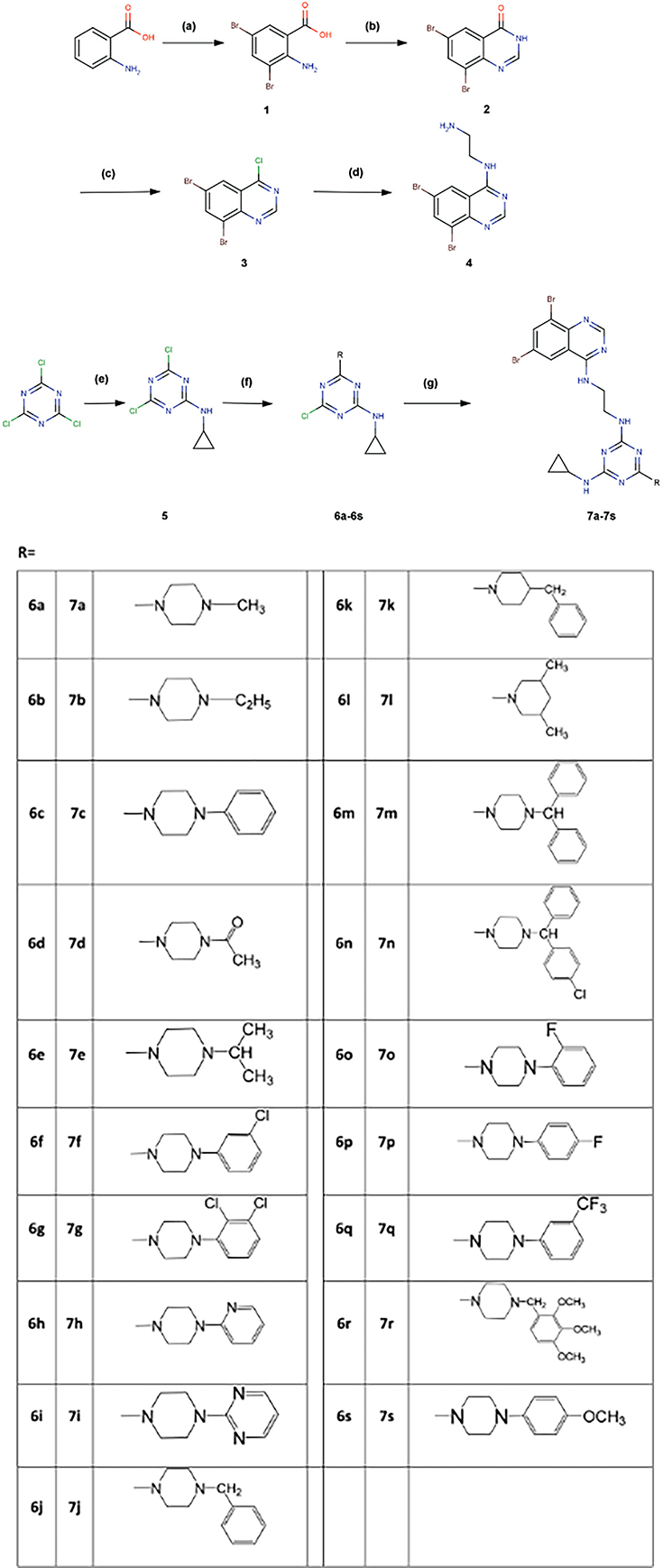
Human T-lymphocyte (MT-4) cells were used to test the novel synthetic compounds 6a–s and 7a–s for their in vitro anti-HIV-1 (strain IIIB) and -HIV-2 (strain ROD) activity. The inhibition of the virus-induced cytopathic effect in MT-4 cells was used to determine the inhibitory concentrations (IC50) of synthetic compounds on HIV-1 and HIV-2 replications (Figure 20) [9].
Figure 20.
Overview of the synthetic strategy for the synthesis of N2-cyclopropyl-N4-(2-(6,8-dibromoquinazolin-4-ylamino)ethyl)-6-R-1,3,5-triazine-2,4-diamine (7a–7s).
2.6 Antioxidant activity
Novel analogs of quinazolines, along with their combined heterocyclic counterparts, have been synthesized. It was observed that certain molecules exhibited a remarkable inhibition of aldehyde oxidase, surpassing 98% [7].
2.7 Antiviral activity
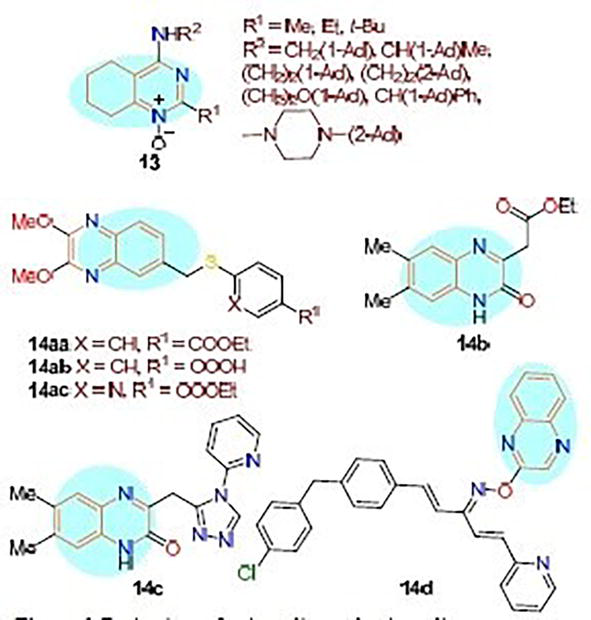
The investigation involved assessing the activity of a representative series of 4-amino-tetrahydroquinazoline derivatives 13 with aliphatic and aromatic substituents, as well as with an adamantyl framework (Figure 21), in porcine embryo kidney (PEK) cells in relation to TBEV reproduction. The antiviral activity was found to be influenced by the presence of a bulky hydrophobic adamantane fragment. Quinoxaline derivatives represent a burgeoning class of heterocyclic compounds that exhibit a broad spectrum of biological activities and find applications in various therapeutic contexts. The synthesis of various quinoxaline derivatives and the assessment of their cytotoxic and antiviral efficacy against diverse RNA and DNA viruses are currently areas of active research. Enteroviruses worldwide distribution of CV-B4 and CV-B5 infections is responsible for numerous serious illnesses. In immunocompromised children, CV-B5 is linked to encephalitis and myocarditis; in older adults, it is linked to central nervous system disease. Three compounds (compounds 14aa–ac) exhibited cytotoxicity against various cell lines (MT-4, MDBK, BHK, and Vero-76) as well as very strong and selective antiviral activity against CV-B5. Furthermore, compound 14aa blocks penetration that may be directed at the viral capsid protein VP1. Several nucleophilic reactions were used to create distinct quinoxalin-2-one derivatives. The synthesized compounds were assessed for their antiviral activity against HCV (Hepatitis C Virus), HBV (Hepatitis B Virus), and HSV-1 (Herpes Simplex Virus type 1).
Figure 21.
Derivatives of quinazoline and quinoxaline as antiviral.
Concurrently, their safety profile and selectivity against various viral strains were studied. Consequently, two of the test compounds, namely quinoxalin-2-ones 14b and 14c, exhibited a low cytotoxicity index concerning cell viability. Moreover, they demonstrated potent activity and selectivity against HCV when compared to ganciclovir. Following synthesis, the antiviral effectiveness of a series of penta-1,4-dien-3-one oxime ethers containing a quinoxaline moiety was evaluated against TMV (Tobacco Mosaic Virus). Compound 14d demonstrated exceptional TMV-specific healing, preventive, and inactivating capabilities. In the control experiment, the commercial agent Ningnanmycin was used to assess the antiviral activity of this compound using the half-leaf method.
This review underscores the increasing interest in the development of heterocyclic compounds and their potential applications in treating viral infections. Antiviral medications with high potency and specificity are currently accessible to treat various viral types. The need for novel chemical scaffolds in targeted therapy stems from the rise in viral resistance. Numerous research teams focus on therapeutic agents that contain heterocycles, as they have the potential to be effective against various virus strains. Future studies will focus on chemically modifying specific heterocyclic moieties to improve their inhibitory efficacy, transforming them into a robust antiviral platform with direct implications for the environment and society [10].
2.8 Antimalarial activity
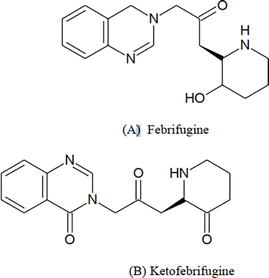
It was possible to create substitute quinazolinoes that structurally resemble febrifugine and ketofebrifugine (Figure 22). The anti-plasmodial activity of the synthesized compounds against Plasmodium berghei in mice was demonstrated at a dose of 5 mg/kg body weight. By comparison, these compounds have a more feasible synthetic scheme than artemisinin and chloroquin [7].
Figure 22.
Febrifugine and ketofebrifugine.
2.9 Antibacterial
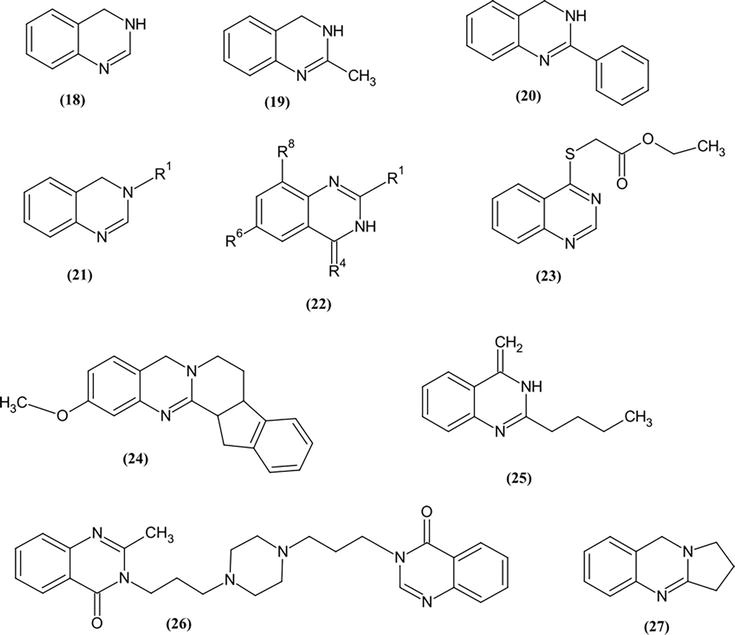
The antimicrobial activity for the following Quinazoline derivatives [18, 19, 20, 21, 22, 23, 24, 25, 26, 27] was carried out by different Techniques. These shows the successful anti-microbial activity against Gram-positive Staphylococcus aureus and Gram-negative Escherichia coli. The present study of Quinazoline derivatives shows the Several successful attempts have been made and recorded in the literature demonstrating promising outcomes (Figure 23) [15].
Figure 23.
Quinazoline derivatives [18, 19, 20, 21, 22, 23, 24, 25, 26, 27] had their antimicrobial activity tested using various methods.
For the recently synthesized compounds, the antibacterial evaluation was achieved were hybrids of quinazolin-2,4-dione with bioactive scaffolds like amide, sulfonamide, hydrazone, thiourea linkage, and/or N-heterocyclic cores like pyrrolidine-2,5-dione, pyrazole, and oxadiazole, utilizing two G +ve (Bacillus subtilis ATCC 6633 and Staphylococcus aureus NRRL B-767) and two G −ve (Escherichia coli ATCC 25955 and Pseudomonas aeruginosa ATCC 10145) bacteria. Compound 3c demonstrated a distinctive antimicrobial efficacy Figure 24 against every tested pathogenic strain at a concentration lower than the tested standard drug, ranging from 2.5 to 10 μg mL−1. The synthesized compounds displayed a range of activities against the tested pathogens.
Figure 24.
Design of the target compounds.
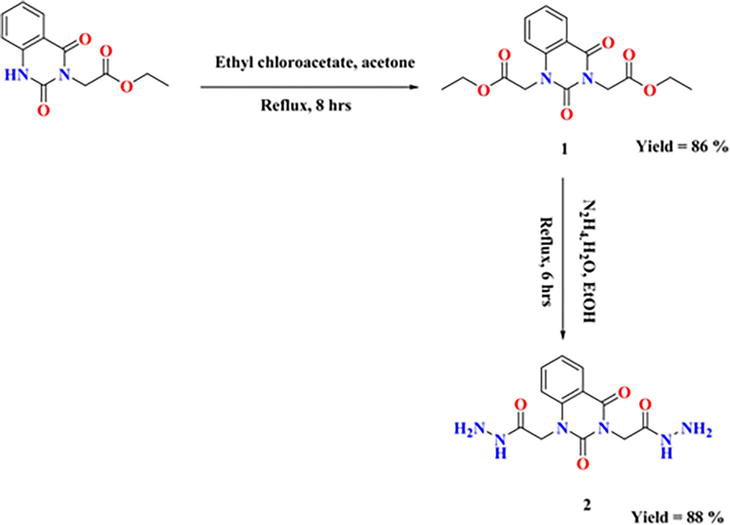
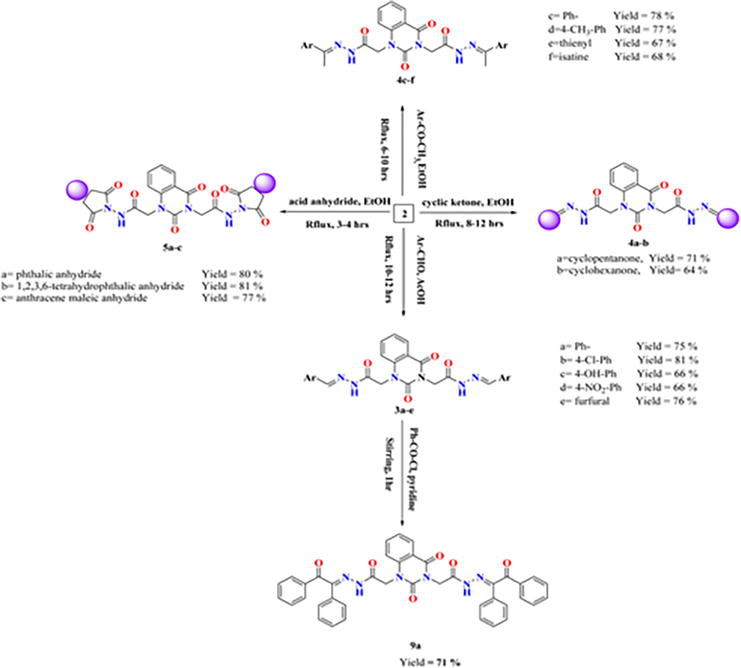
As shown in Figure 25, our starting 2 was created by reacting hydrazine hydrate in absolute ethanol with the synthesized ester diethyl-2,2′-(2,4-dioxoquinazoline-1,3(2H,4H)-diyl)-diacetate 1.
Figure 25.
Synthesis of starting material 2.
By treating 2,2′-(2,4-dioxoquinazoline-1,3(2H,4H)-diyl)di(aceto hydrazide) 2 with different aromatic aldehydes, such as benzaldehyde, 4-hydroxybenzaldehyde, 4-chlorobenzaldehyde, 4-nitrobenzaldehyde, and furfural, respectively, several arylidene hydrazide derivatives (Schiff bases) 3a–e were synthesized in good to excellent yields (Figure 26).
Figure 26.
Compound 3-5 and compound 9a's synthesis.
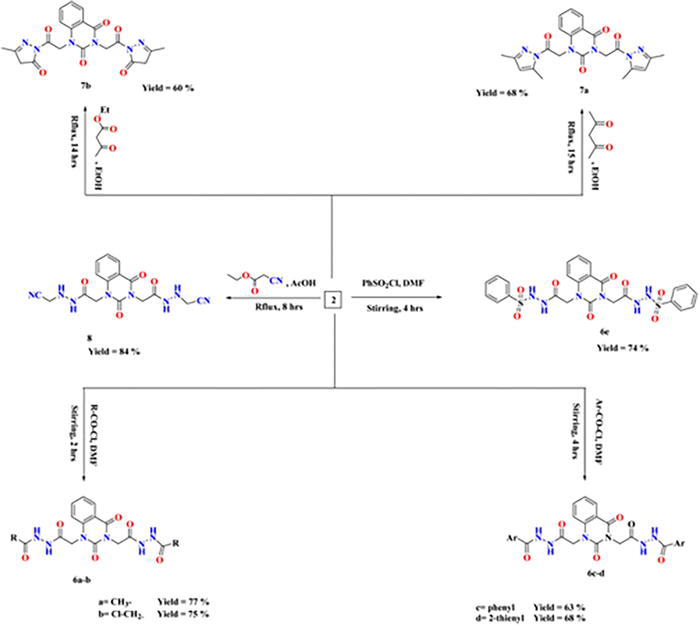
Moreover, quinazolin-2,4-dione containing pyrazole and pyrazolone moieties 7a–b were synthesized based on condensation of 2 with active methylene compounds such as acetylacetone and ethyl acetoacetate via Knorr pyrazole reaction, as declared in Figure 27 [11].
Figure 27.
Synthesis of compounds 6–8.
2.10 Anti-leishmanial hybrids
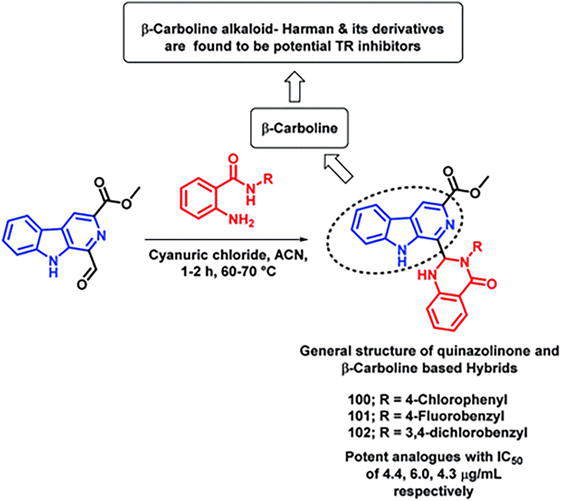
One of the main goals for the development of anti-leishmanial drugs is to demolish the metabolic pathways that are necessary to allow the parasite to remain within the host [12]. One of the vital enzymes in charge of the parasite’s antioxidant defense is trypanothione reductase (TR). In earlier studies, a variety of anti-leishmanial agents were synthesized, demonstrating potent activity across different series, including β-carboline, dihydro-β-carboline, and quinazolinone. Taking these variables into account, Chauhan et al. [12] synthesized hybrids based on quinazolinone and β-carboline and then tested them in vitro for anti-leishmanial activity. After conducting in vitro enzyme inhibition against the LdTR enzyme, numerous compounds exhibited activity in inhibiting the enzyme. It was discovered that compounds on quinazolinone with aromatic groups were just as active as those with aliphatic substituents. Compounds 100, 101, and 102, which are aromatic groups with substituents that withdraw electrons, exhibit greater inhibition than unsubstituted phenyl or phenyl rings with substituents that donate electrons (Figure 28). Competitive inhibition has been demonstrated by all enzyme inhibitors. Additionally, anti-leishmanial activity was assessed, and the anti-leishmanial activity and the outcomes of the enzyme assay were correlated. Compounds 100, 101, and 102 were identified as the most potent in the series, displaying IC50 values of 4.4, 6.0, and 4.3 μg mL−1, respectively, against intracellular amastigotes of Leishmania donovani. The standards employed in this analysis were miltefosine (with an IC50 of 8.1 μg mL−1) and SSG (with an IC50 of 54.4 μg mL−1).
Figure 28.
Formulation for β-carboline-quinazolinone blends.
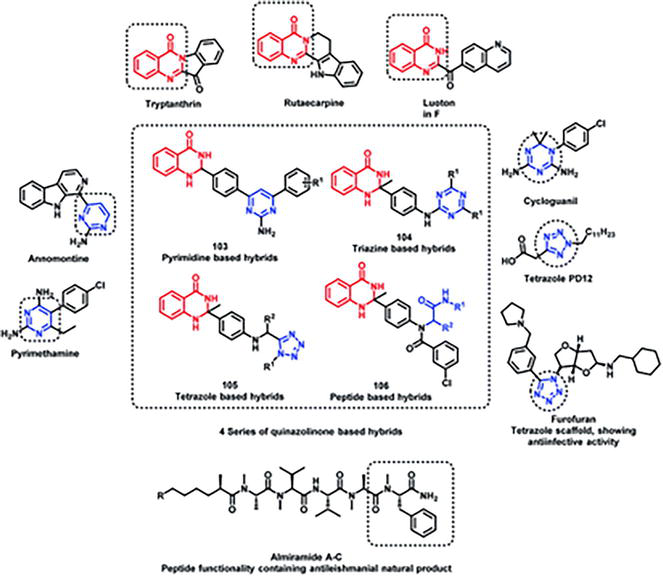
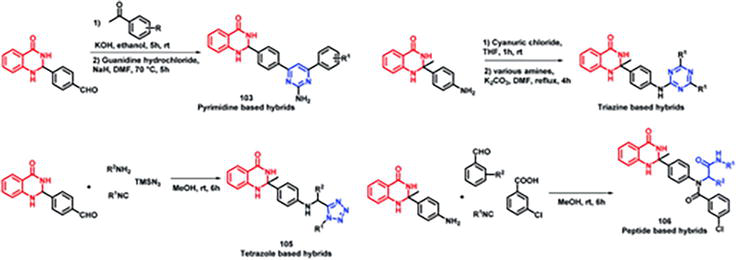
Four series of quinazolinone-based hybrids were created by Sharma et al. [12] (Figures 29 and 30). In these series, there are members such as quinazolinone-pyrimidine hybrids (a), quinazolinone-triazine hybrids (b), quinazolinone-peptide hybrids (c), and quinazolinone-tetrazole hybrids (d). All of the compound series, with the exception of the tetrazole series, were discovered to be active following the viewing process for anti-leishmanial activity.
Figure 29.
Plan the design of quinazoline hybrids containing peptide, tetrazole, triazine, and pyrimidine.
Figure 30.
Quinazoline hybrids: a synthetic scheme involving pyrimidine, triazine, tetrazole, and peptide.
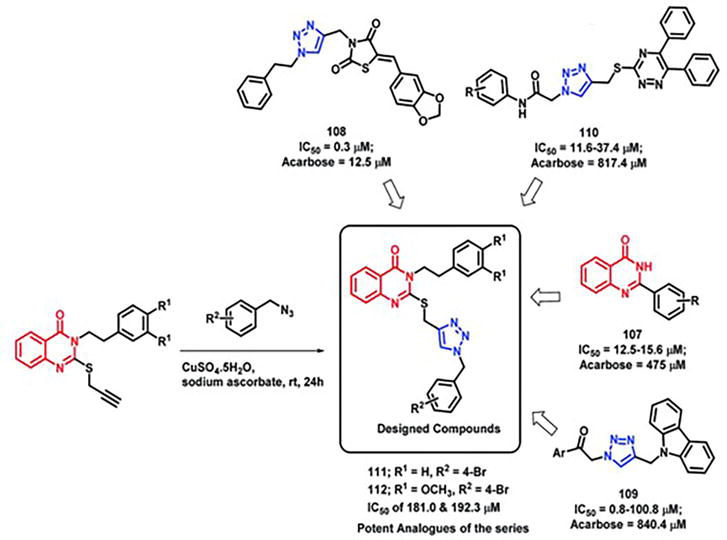
2.11 Hybrids that fight diabetes
An essential enzyme called α-glucosidase breaks down carbohydrates so that glucose and other monosaccharides can be absorbed [12]. By inhibiting this enzyme, the risk of postprandial hyperglycemia is lowered by lowering the postprandial blood glucose level. Prior studies have illustrated the significance of quinazolinone and triazole in inhibiting α-glucosidase. Considering these aspects, Saeedi et al. [12] created hybrids based on quinazolinone and triazole and assessed how well they inhibited α-glucosidase. Using acarbose as the reference drug (IC50 of 750 μM), it was found that all the synthesized hybrid analogs inhibited the α-glucosidase enzyme through competitive inhibition, with IC50 values falling within the range of 181–474.5 μM. Compounds 111 and 112, which exhibited the highest potency, had IC50 values of 181.0 and 192.3 μM, in that order (Figure 31).
Figure 31.
Triazole-linked quinazolinone hybrids: synthesis and design strategy focused on α-glucosidase.
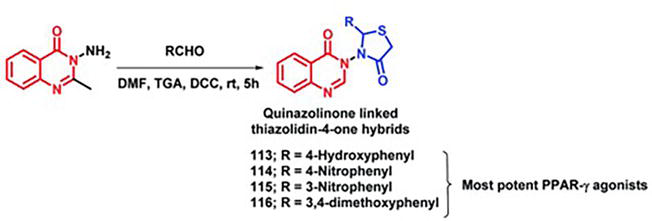
Jangam et al. [12] 122 hybridized thiazolidinediones (rosiglitazone, pioglitazone, and troglitazone) with the medicinally significant quinazolinone scaffold, taking into account the significance of these compounds as PPAR-γ agonists. All of the designed compounds demonstrated good binding at the PPAR-γ receptor’s active site (PDB: 4PRG) according to the docking study. When the synthesized compounds were tested on diabetic rats induced by streptozotocin, compounds 113, 114, 115, and 116 demonstrated a significant anti-diabetic potential (Figure 32). The compounds under investigation underwent testing for a range of biochemical parameters, including glycated hemoglobin (HbA1C), insulin, alanine transaminase (ALT), aspartate transaminase (AST), as well as the levels of total protein, triglycerides, cholesterol, high-density lipoprotein (LDL), and low-density lipoprotein (LDL). It was discovered that each of these parameters was significant.
Figure 32.
Creation of hybrids between quinazolinone and thiazolidinone.
2.12 Hybrid anti-convulsant
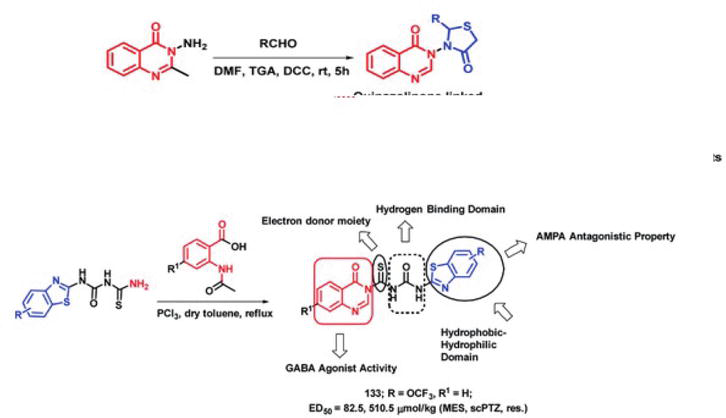
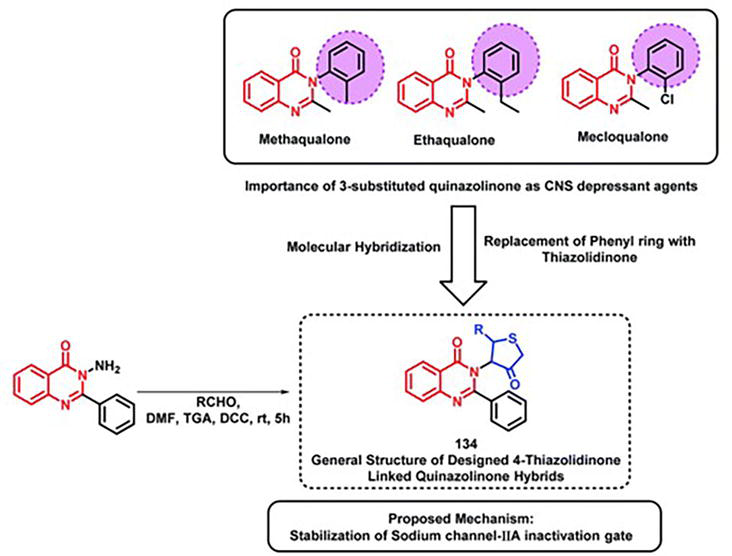
A category of disorders known as epilepsy is characterized by neuronal hyperexcitability [12], often accompanied by episodes of motor, sensory, or autonomic phenomena, with or without a loss of consciousness. Derivatives of quinazolinone, including methaqualone, afloqualone, and mecloqualone, are clinically approved medications with the potential for treating epilepsy. Because of their antagonistic action against AMPA, benzothiazole derivatives (e.g., Riluzole) are also attracting interest for their anti-convulsant properties. In order to create some hybrid analogs, Malik et al. [12] combined the AMPA antagonistic and GABA agonistic properties of benzothiazoles and quinazolinones, respectively, into a single hybrid scaffold. The following pharmacophoric characteristics were added to the molecule to increase the anti-convulsant potential of the created analogs. The distal hydrophobic domain (d), electron donor moiety (c), hydrogen bonding domain (b), and hydrophobic domain (a) are examples of these characteristics. In this manner, hybrids based on benzothiazoles and quinazolinones were created (Figure 33). In mice, seizure models in relation to subcutaneous pentylenetetrazole (scPTZ) and maximal electroshock (MES) were created in order to screen the synthesized hybrids for anti-convulsants.
Figure 33.
Quinazolinone-benzothiazole hybrid synthetic approach highlighting the significance of structure.
Compound 133 exhibited greater potency compared to the positive controls, with an ED50 of 92 μmol kg−1 for MES (Maximal Electroshock Seizure) and >3540 μmol kg−1 for scPTZ (subcutaneous pentylenetetrazole), surpassing the effects of phenytoin and ethosuximide.
Anti-convulsant analogs containing quinazolinone (methaqualone, ethaqualone, and mecloqualone) suggest that quinazolinone’s third aromatic ring substitution is necessary for its CNS depressant and anti-convulsant properties. To create powerful CNS-acting medications, numerous researchers have attempted to replace a number of aromatic ring substituents. Taking these facts into account, Jangam et al. [12] connected 4-oxothiazolidine at the quinazolinone backbone’s third position to create hybrids based on quinazolinone (Figure 34). Using MES-induced mice as a screening model, many of the synthesized compounds demonstrated moderately to very effectively anticonvulsant. The SAR study demonstrated the significance of groups that withdraw electrons for anti-convulsant activity at the para, meta, and ortho positions of the phenyl ring attached at the second place of thiazolidine. The sodium channel IIA inactivation gate’s active site was docked with these designed analogs (PDB ID: 1BYY), and their binding energy ranged from −5.15 to −6.13 kcal mol−1, indicating appropriate binding at the active site of the receptor.
Figure 34.
The synthesis and design methodology for quinazolinone and 4-oxothiazolidine hybrids.
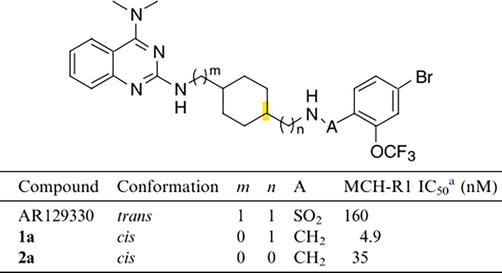
It has been demonstrated that quinazoline derivatives that bind to MCHR1 have unique anti-obesity properties. Sasmal et al. explored the potential anti-obesity properties of quinazoline derivatives, identifying them as antagonists for the melanin-concentrating hormone receptor 1 (MCHR1) [13]. By switching out the substituent groups, a number of compounds were produced, such as 4-morpholinyl-quinazoline, 4-hydroxypiperidine-quinazoline, 4-propyl-quinazolinone, 4-pyrrolidin-quinazolinone, and so forth. First, these compounds’ solubility and metabolic stability in blood were investigated. Subsequently, these derivatives were evaluated for their potential in preventing obesity. A prototype molecule, 4-Morpholinyl-quinazoline (Figure 35), was selected due to its favorable oral pharmacokinetic (PK) profile for further investigation of its effects in diet-induced obese (DIO) C57BL/6 J mice. And after receiving that compound orally for 14 days (30 mg/kg, b.i.d.), the tested mice clearly lost 12% of their body weight. According to the findings, 4-morpholinyl-quinazoline, a representative compound, had a pronounced anti-obesity effect. However, it was also noted that additional stability enhancement of the compound in plasma concerning the oxymethylene linker was required [14, 18, 19].
Figure 35.
A prototype molecule, 4-morpholinyl-quinazoline.
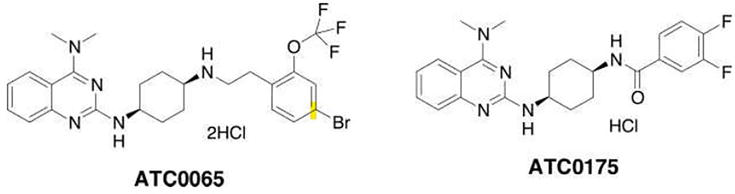
In addition, a variety of other quinazoline derivatives, such as 4-amino-2-cyclohexyl aminoquinazoline and 4-dimethylamino quinazoline, exhibit strong inhibitory activity for MCHR1. ATC0065 (N2-[cis-4-({2-[4-bromo-2 (trifluoromethoxy)phenyl]ethyl}amino)cyclohexyl]- N4,N4-dimethylquinazoline-2,4-diamine dihydrochloride) and ATC0175 (N-(cis-4-{[4-(dimethylamino)quinazolin-2-yl]amino}cyclohexyl)-3,4-difluoro-benzamide hydrochloride) are two of these compounds that function as MCHR1 antagonists. We now present profiles of ATC0065 and ATC0175 in vitro and in vivo in a range of rodent tests indicative of anxiolytic and antidepressant action (Figure 36) [19].
Figure 36.
As 4-amino-2-cyclohexyl aminoquinazoline and 4-dimethylamino quinazoline, exhibit strong inhibitory activity for MCHR1.
Numerous proliferative illnesses, including liver cirrhosis, pulmonary fibrosis, cancer, glomerulonephritis, glomerulosclerosis, atherosclerosis, and restenosis following PTCA, are brought on by the aberrant platelet-derived growth factor receptor (PDGFR)-induced cell proliferation. One possible treatment option for these proliferative diseases is to use PDGFR phosphorylation inhibitors [20].
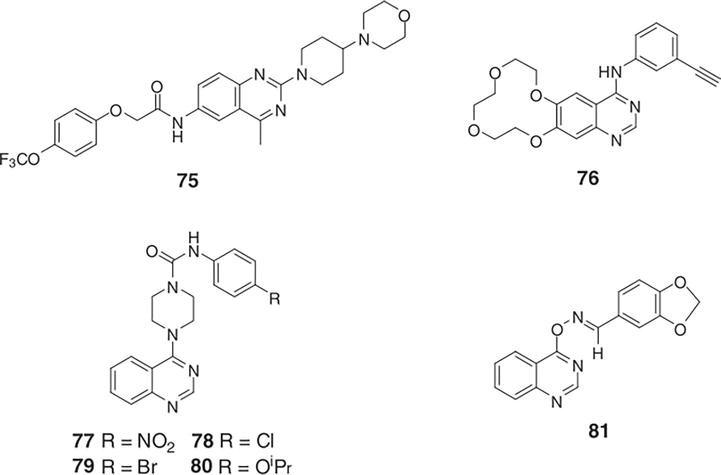
Matsuno et al. screened PDGFR phosphorylation inhibitors and discovered a number of compounds with a 4-piperazinyl substituted quinazoline core [20]. In the analysis of SAR, KN1022 was selected as the prototype inhibitor. The 4-nitrobenzene urea moiety was scrutinized, revealing that compounds with substitutions at position 4 of the benzene ring exhibited optimal performance. However, compounds with more than two substituents on the benzene ring showed diminished activity. In their study, various derivatives of KN1022 (77) with promising activity were synthesized, including 4-(4-methylphenoxy)phenyl, 4-tert-butylphenyl, and 4-phenoxyphenyl. These three compounds were orally administered to Sprague-Dawley rats at a dose of 30 mg/kg, twice daily, for in vivo assessments. Next, it was shown that 4-isopropoxyphenyl (80), 4-chlorophenyl (78), and 4-bromophenyl (Figure 37) (79) comparable had clear inhibitory action against the development of neointima in the carotid artery of the balloon catheter de-endothelialized vessel in the rats [21].
Figure 37.
Typical 4-substituted quinazoline structures with bioactivity.
2.15 Germicide
In an effort to find new acaricides, Li et al. [12] combined and assessed a number of 4-quinazoline oxime ether compounds biologically [24]. The compounds found in this study were shown to suppress the phytovirus TMV to varying degrees. Of these compounds, compound 81 (Figure 37) exhibited strong activity both in vivo and in vitro against TMV, with 65% and 61% of the total, correspondingly. Additionally, following virus vaccination, compound 81 demonstrated positive inhibitory activities against PVX, PVY, and CMV, according to bioassays [21].
2.16 Anti-TB activity
In the quest for new anti-TB drugs, quinazoline has already been used to create a large number of unique compounds, some of which have shown encouraging anti-TB activity. This review offers a comprehensive overview of recent progress in the development of quinazoline-based medications for the treatment of tuberculosis (TB) 4-Alkylthioquinazoline derivatives and their synthesis. Shashikant et al. [22] synthesized a series of N3[4-(4-chlorophenyl thiazole2-yl)-2-aminomethyl] quinazoline-4(3H)-one analogs in 2006, and different spectroscopic analyses, including FT-IR and 1H NMR spectra, confirmed the structure of the novel compounds. The in vitro anti-TB activity of the compounds was assessed on Lowenstein-Jensen egg medium (LJ Medium) using the H37RV strain. All of the synthesized compounds showed moderate to excellent anti-TB activity, with MIC ranges of 100–10 μM.
Among the compounds, 3f, 3 h, and 3j exhibited the most significant anti-TB activity at a concentration of 10 μg mL−1. Compounds 3c, 3d, and 3 g demonstrated moderate activity, whereas compounds 3a, 3b, and 3e did not exhibit noticeable activity. Compound 3i, on the other hand, showed no antitubercular activity at 100 μg mL−1. The potent anti-TB activity of 3f, 3 h, and 3j was attributed to the combination of quinazolinone with isoniazid, pyrazinamide, and para-aminosalicylic acid, respectively (Figure 38) [23].
Figure 38.
AntiTB.
2.17 Allosteric modulators of glutamate receptors
Lindsley and colleagues introduced a new class of substituted pyrazolo[1,5-a]quinazolin-5(4H)-ones, acting as negative allosteric modulators of metabotropic glutamate receptors 2 and 3 (mGlu2 and mGlu3, respectively). The SAR profile of these substances was relatively steep, with slight framework alterations resulting in notable efficacy losses. Introducing a 3-sulfonylphenyl or 3-pyridyl group at R2 resulted in inhibitors with low-micromolar to high nanomolar IC50 values for both mGlu2 and mGlu3, as long as R1 remained a phenyl ring and R3 remained a methyl group. Installing a phenyl or 4-methoxyphenyl at this location, on the other hand, produced compounds that had very little effect. Significantly reduced activity at both receptors was also the outcome of truncating a methyl group in this position. A number of substances were discovered to be strong inhibitors overall, such as quinazolin-5(4H)-one, 4-methyl-2-phenyl-8-(pyrimidin-5-yl)pyrazolo[1,5-a],which was found to have excellent selectivity against the other mGluRs and to have potent in vitro activity as dual mGlu2 /mGlu3 NAMs (Figure 39) [24].
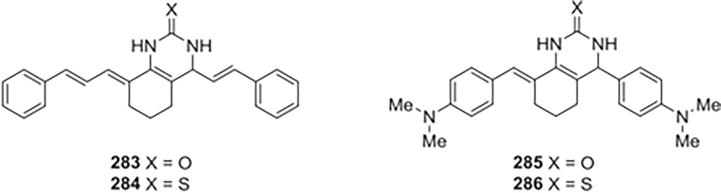
New analogs based on quinazoline-2(1H)-ones and quinazoline-2(1H)-thiones, bischalcones, were conceptualized and synthesized by Raghav and Singh. The compounds that were produced were evaluated for their potential as novel inhibitors of cathepsin B and H. 3-Hexahydro-3phenylallylidene)-4-styrylquinazoline-2(1H)-one 283 the compound was found to exhibit the highest level of inhibition among the others, exhibiting 100% inhibition at 1.0104 M concentration and 50% inhibition at ∼0.1104 M concentration. The assessment of cathepsin B activity was also conducted with regard to substituted benzylidene-3,4,5,6,7,8-hexahydro-quinazoline-2(1H) thione derivatives. That 3,4,5,6,7,8hexahydro-3-phenylallylidene)-4-styrylquinazoline-2(1H)-one 284 had the greatest inhibitory effect, showing 100% inhibition at 0.50104 M concentration and half maximum inhibition at approximately 0.01104 M concentration. Similar to this, benzylidene-3,4,5,6,7,8-hexahydro-quinazoline2(1H)-one and its derivatives at various concentrations were synthesized and used to calculate the activities of cathepsin H. Within quinazoline derivatives, 2,6-bis(4′(dimethyl amino) benzylidene) cyclohexanone 285 exhibited the most potent inhibition, reaching half-maximum inhibition at approximately 0.15 × 10−4 M concentration and achieving 100% inhibition at 1.0 × 10−4 M concentration. Benzylidene-3,4,5,6,7,8hexahydro-quinazoline-2(1H)-one derivatives were also found to exhibit 100% maximum inhibition of 2,6-bis(4′(dimethyl amino) benzylidene) cyclo hexanone 286 at ∼0.25104 M concentration (Figure 40) [25].
Figure 40.
New analogs based on quinazoline-2(1H)-ones and quinazoline-2(1H)-thiones, bischalcones.
2.19 Phosphodiesterase inhibitors
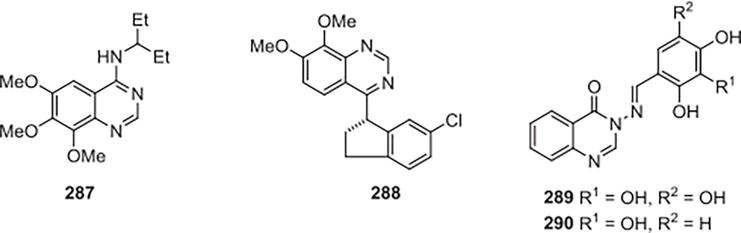
A novel class of quinazoline PDE1 inhibitors that are selective and penetrate the central nervous system was revealed by Humphrey et al. [26] based on the two file screen hits’ SAR development. After structurally refining the lead compounds using a combination of conventional and parallel organic synthesis, molecular modeling and X-ray crystallography, the resultant compounds were identified as PF-04471141) for aminoquinazoline 287 and PF-04822163) for indanylquinazoline 288. These compounds are among the best-defined and most potent PDE1 inhibitors discovered to date. In rodents, studies on pharmacokinetics show that all compounds reach systemic concentrations higher than their IC50 figures. Therefore, compounds 288 and 287 present a great deal of promise as chemical probes for additional research into the biological processes of the central nervous system that are affected by PDE1 function, as well as for evaluating the pan-PDE1 inhibition’s potential as a therapeutic in order to address neuropsychiatric disorders. Abdel-Aziz and associates [27] created and produced a novel series of hybrids between the Schiff base and quinazolin-4(3H)-one. The in vitro activity of the synthesized compounds in inhibiting phosphodiesterase 4 (PDE4) was evaluated, with Rolipram employed as a positive reference for PDE4 inhibition. When compared to rolipram, a few of them displayed good-to-moderate activity. Compound 289, with an IC50 of 1.60 mM, was the most potent in this series of PDE4 inhibition. The outcome of the activity showed that the inhibitory activity requires the Schiff base part’s hydroxyl-substituted phenyl to exist. The base of the Schiff 290 demonstrated a modest degree of inhibition with a 29.3 mM IC50. Trihydroxyphenyl Schiff base 289, however, demonstrated in this series’ maximum PDE4B inhibitory activity. The Schiff bases with methoxyphenyl groups were the least active in this series, highlighting the importance of the free OH groups on the aryl ring of the Schiff base. Furthermore, the inhibitory action of the compounds that were created was not enhanced by the 5-methyl substitution. The compounds displaying PDE4 inhibition were subsequently evaluated for their antiproliferative potential against various human tumor cell lines. Compound 290 was found to have noteworthy antiproliferative activity in breast, lung, and colon tumor cells, with IC50 values of 140, 79, and 320 nM, respectively. Additionally, compound 289 was docked in the PDE4B active site to determine a potential binding mode, which offers information for future improvements to this unique scaffold that inhibits PDE4 (Figure 41).
Figure 41.
A novel class of quinazoline PDE1.
2.20 Antihistamine agents
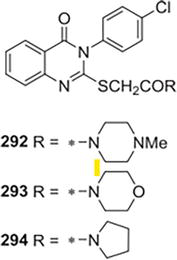
Alagarsamy and colleagues [28] created and combined a number of unique four(3H)-quinazolinone compounds by reacting 2-(3-(4chlorophenyl)-2-(2-(4-substituted)-2oxoethylthio)acetyl chloride of 4-oxo-3,4-dihydroquinazolin-2-ylthio) with different amines. For assessing the in vivo H1-antihistaminic activity on conscious guinea pigs at a dose level of 10 mg/kg, chlorpheniramine maleate served as the reference standard. It was discovered that every tested compound had strong antihistaminic properties. Every test substance in the series demonstrated substantial defense in the 65–771% range, according to percentage protection data. According to biological research, the biological activity of substituents placed over the quinazoline ring’s third position varies. Significant activity was demonstrated by the existence of the group of piperazinyl (292, Log P1/4 3.43). Potency is retained as soon as the heteroatom piperazinyl’s nitrogen is substituted with an oxygen compound (Log P1/4 3.27, morpholinyl substitution, 293), and after piperazinyl’s nitrogen is eliminated (pyrrolidinyl substitution, 294, Log P1/4 3.99). Activity decreased when ethyl and diethyl substituents were added. The most effective of the series was, compound 3-(4-chlorophenyl) in general-2-(2-(1-methylpiperazin)292.) quinazolin-4(3H)-one-2-oxoethylthio. It is evident from Figure 42 that lipophilicity (Log P) is a significant factor in their antihistaminic action.
Figure 42.
Unique Four(3H)-quinazolinone with antihistamine activity.
2.21 Cholinesterase inhibitors
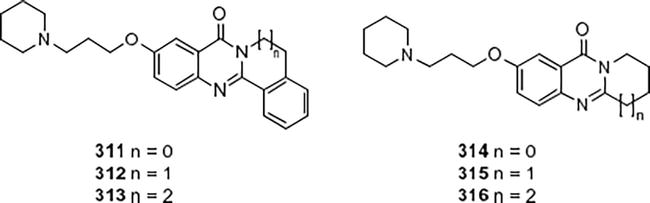
Utilizing 1-H-benzo-6-(benzyloxy)[d] [1, 3], a simple 6-substituted quinazolinone synthesis was developed. Oxazine-2,4-dione was the initial substance by Decker and associates [29]. The ability of each target compound to inhibit butyryl and acetylcholinesterase was examined. Compared to AChE, tacrine exhibited a potency of 3 times lower (IC50 1/4 71.5 nM) for the enzyme in humans, while AChE showed an IC50 of 24.1 nM. The inhibitors of piperidinyl 311–316 exhibited inhibitory activities against the AChE derived from electric eels that were 7e 39 times higher. Additionally, docking studies were performed to look into a potential binding mode for AChE (Figure 43).
Figure 43.
Cholinesterase inhibitors.
2.22 Inhibitors of chitin synthase
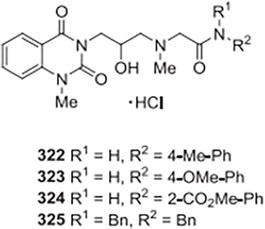
Ji et al. [30] created, produced, and described a number of unique derivatives of quinazoline-2,4-dione that are 1-methyl-3-substituted. One-step ELISA analysis of chitin generated from UDP utilized wheat germ agglutinin (WGA) labeled with horseradish peroxidase (HRP) as the probe. -Glc-NAc was used to measure the prepared compounds’ inhibitory activity against CHS. The assay’s positive control in this case was polyoxin B. Each compound’s half-inhibition concentration (IC50) was found. Out of all the substances that were assessed, a few analogs showed strong inhibitory effects in comparison to polyoxin B, and their IC50 values were lower against CHS, which was 0.18 millimoles. Compound 322 is the most potent inhibitor among these, with an IC50 value of 0.08 mmol/L. 323–325’s inhibitory actions were similar to those of polyoxin B. Other substances exhibited modest to negligible inhibition. Using aryl R2 as a substituent in these active compounds resulted in greater inhibitory activities against CHS compared to using alkyl R2. To increase inhibitory activities in aryl R2, a substituent that donates electrons on the aromatic ring is preferable over an electron-withdrawing substituent (Figure 44).
Figure 44.
Chitin synthase inhibitors.
2.23 Anti-asthmatic properties
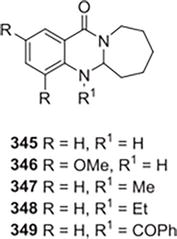
Using a murine model of asthma, Rayees et al. synthesized several derivatives of azepino [2,1-b] quinazoline [31] and assessed their ability to prevent asthma. In animals with asthma, the compounds 345–349 significantly reduced the secretion of Th2 cytokines and eosinophilia. However, in the case of the animals treated with 349, the decline was quite noteworthy. Compound 349 was the subject of molecular modeling studies involving the transcription factors GATA3 and STAT6, which are primarily responsible for Th2 cell differentiation. Additionally, after oral and intravenous administrations, the pharmacokinetics of 349 were studied in mice (Figure 45).
Figure 45.
Anti-asthmatic activity.
2.24 Bronchodilator activity
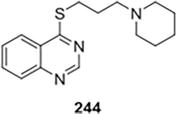
Taking vasicinone and theophylline as prescribed, many quinazoline and quinazolinone derivatives were reported by Špulak et al., who also tested them for their ability to act as in vitro bronchodilators on isolated rat trachea [32]. Remarkably, nearly every derivative had a greater biological impact than theophylline. Compared to their alkoxy and alkylamino analogs, the 4alkylsulfanyl derivatives were more potent and showed the most noticeable effect. With an ED50 in the micromolar range, among the synthesized derivatives, compound 244, which contains the 1-piperidylpropyl fragment, exhibited the highest level of activity. When considered collectively, these results make the compounds intriguing targets for additional methodical research and development (Figure 46).
Figure 46.
Bronchodilator activity.
2.25 Glucuronidase inhibitors
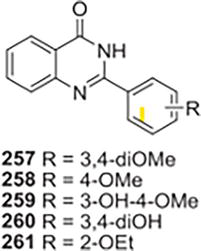
Using CuCl2.2H2O as a catalyst, Khan and colleagues created a fresh run of analogs of 2-arylquinazolin-4(3H)-one derived from anthranilamide and various benzaldehydes. The b-glucuronidase inhibitory potential of the synthesized 2-arylquinazolin-4(3H)-ones were assessed utilizing 1,4-lactone of D-saccharic acid (IC50 1/4 45.75 ± 2.16 mM). Considering the activity results that were obtained, the link between structure and activity for these compounds was looked into, suggesting that the phenyl ring substitutions, located at C-2 of the quinazolin4(3H)-one skeleton, are the primary source of the compounds in this class’s ability to inhibit b-glucuronidase [33] Compound 257 (IC50 1/4 0.6 ± 0.45 mM) demonstrated the highest b-glucuronidase inhibition among all the structures evaluated; 76 times higher than the standard D-saccharic acid 1,4-lactone. It was shown that the two methoxy groups at positions C-30 and C-40 were in the best configuration to interact with the enzyme. The activity was reduced to nearly half when a hydrogen atom was substituted for one methoxy group at C-30, compound 258 (IC50 1/4 1.1 ± 0.05 mM) as an example. The level of activity dropped five times when the hydroxyl group at C-30 was replaced with a methoxy group, compound 259 (IC50 1/4 2.8 ± 0.05 mM) as an example. Compound 261 exhibited 65-fold greater potent activity than the reference (IC50 1/4 0.7 ± 0.01 mM). Substituting hydroxyl groups for both methoxy groups in compound 257 caused the activity to decrease by two times; exchanging ethoxy groups for hydroxyl groups reduced the activity by a factor of 14. In total, an IC50 trend of inhibition for the enzyme within the range of 0.6–198.2 mM was noted and contrasted with the reference (Figure 47).
Figure 47.
Glucuronidase inhibitors.
2.26 Kinase inhibitors
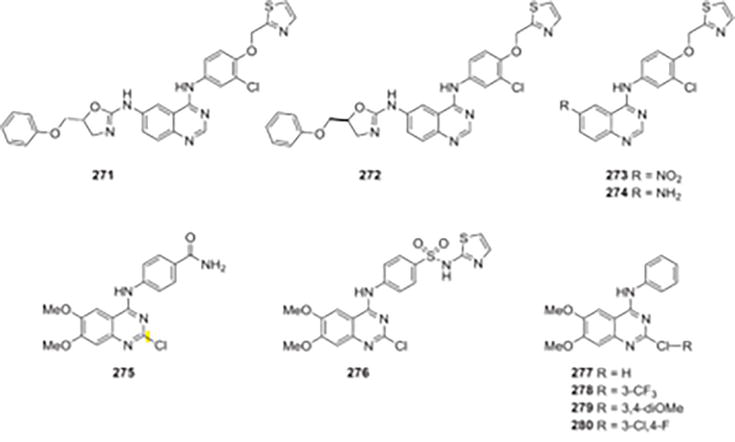
Additives 271 and 272, which have an alternative oxazole scaffold showed stronger inhibitory effects on EGFR at the 7-positions (IC50D 1.21 and 0.95 mmol/L) in comparison to compounds 273 and 274, which only include amino or nitro groups where they are found. Hou et al. created and produced a novel category of quinazoline compounds, through the fusion of oxazole and quinazoline fundamental structures into a single heteroaromatic unit. The EGFR inhibitory potency and anti-proliferative properties of the synthesized structures were examined [34]. Their actions followed the same trends and had a positive correlation with antiproliferative actions. In order to find new EGFR inhibitors, it is feasible and required to continue investigating the bioactivity and alteration of quinazoline analogs substituted through an oxazole scaffold, even though the outcomes did not possess a structure reminiscent of the positive control Erlotinib (EGFR IC50 1/4 0.03 mmol/L). A new class of quinazolines (2-chloro-4-anilino) was designed and synthesized by de Castro Barbosa and colleagues [35] as dual inhibitors of EGFR and VEGFR-2, and their inhibitory effects were assessed. The acquired biological data indicated that derivatives of 2-chloro-4-anilino-quinazoline have the potential to act as dual inhibitors of VEGFR-2 and EGFR. The IC50 values for the derivatives featuring a hydrogen bond donor at the para position of the aniline moiety were lower. Compound 275 was identified as the most active, with an IC50 of 0.90 mM for EGFR and 1.17 mM for VEGFR-2. Compared to the prototype 276, this compound was about 7 times more potent on VEGFR-2 and about 11 times more powerful on EGFR. Pharmacophoric groups for both kinases were identified through SAR and docking studies, revealing the critical importance of a hydrogen bond donor for interaction with conserved Glu and Asp amino acids in EGFR and VEGFR-2 binding sites, particularly at the para position of the aniline moiety. A new series of unique 4-anilinoquinazoline derivatives was designed and synthesized by Waiker and colleagues [36] who then assessed them as possible inhibitors of protein kinases linked to Alzheimer’s disease. Five distinct kinases were tested for potential inhibitory effects of the 6,7-dimethoxy-N-phenylquinazolin-4amines produced artificially: CK1d/ε (casein kinase 1), GSK-3a/b (Glycogen Synthase Kinase 3a/b), DYRK1A (dual-specificity, tyrosine phosphorylation regulated kinase), CDK5/p25 (CDK5/p25), and CLK1 (cdc2-like kinase 1). At the highest concentration tested (10 mM), none of the synthesized anilino quinazolines showed inhibitory activity against CDK5/p25, DYRK1A, or CK1d/ε, based on the results of kinase inhibitory assays. Given that four compounds (277–280) exhibited inhibitory activity on CLK1, it appeared that the 4anilinoquinazolines were the most effective against the enzyme. At concentrations below 10 mM, two of the ten anilinoquinazolines that were produced—279 and 280—exhibited notable strength of the inhibitor against CLK1. Remarkably, compound 279, which features a 3,4-dimethoxy substitution on the aryl ring of the aniline moiety, inhibits the enzymes CLK1 (IC50 = 1.5 mM) and GSK-3a/b (IC50 = 3 mM) at concentrations less than 5 mM. Interestingly, compound 280 exhibited no inhibition on GSK-3a/b and a 5-fold reduction in inhibition on CLK1 (IC50 = 7.6 mM) due to the presence of 3-fluoro and 4-chloro substitution in the aryl ring. Docking studies were conducted to elucidate how the compounds bind to the active sites of GSK-3β and CLK1. According to the study’s findings, compound 279 might be a useful model for the creation of dual inhibitors of the GSK-3a/b and CLK1 enzymes that could be used as a treatment for Alzheimer’s disease (Figure 48).
In conclusion, the quinazolines are heterocyclic stable nucleus that is susceptible to different chemical reactions to give biologically active compounds which treat a different life-threatening diseases such as cancer, bacterial, fungal, viral, infections, hypertension, and other pandemic disease such as malaria. In addition, these compounds represent a goal for hybridization approach for drug design as they are biologically active pharmacophores. They give a variety of hydride compounds with other bioactive nuclei that could be heterocyclic, alicyclic, benzylic, and allylic potentiate its activity and altered pharmacokinetics and pharmacodynamics of the combined drugs and increase the biological targets react with, results in a variety of chemical bioactive compounds for different diseases such as antidiabetic, anticholinesrase, dihydrofolate reductase inhibition, and cellular phosphorylation inhibition.
References
1.Zayed MF. Medicinal chemistry of quinazolines as anticancer agents targeting tyrosine kinases. Scientia Pharmaceutica. 2023;91(2):18
2.Srivastava S, Srivastava S. Biological activity of quinazoline: A review. International Journal of Pharmaceutical Sciences and Research. 2015;6(9):1206-1213
3.Jafari E, Khajouei MR, Hassanzadeh F, Hakimelahi GH, Khodarahmi GA. Quinazolinone and quinazoline derivatives: Recent structures with potent antimicrobial and cytotoxic activities. Research in Pharmaceutical Sciences. 2016;11(1):1
4.Khan I, Ibrar A, Ahmed W, Saeed A. Synthetic approaches, functionalization and therapeutic potential of quinazoline and quinazolinone skeletons: The advances continue. European Journal of Medicinal Chemistry. 2015;90:124-169
5.Zayed MF. Medicinal chemistry of quinazolines as analgesic and anti-inflammatory agents. ChemEngineering. 2022;6(6):94
6.Lei Z, Yao J, Liu H, Bai X, Gao X, Pan Q, et al. Design, synthesis, and bioactivity of novel quinazolinone scaffolds containing pyrazole carbamide derivatives as antifungal agents. Current Issues in Molecular Biology. 2022;44(11):5605-5621
7.Magesh M, Keerthika B, Bharathi D, Premavathi K, Vijayalakshmi M, Lavanya M. Overview of the biological activities of quinazolines. Journal of Coastal Life Medicine. 2023;11:1879-1887
8.Das RK, Singh A, Sharma D, Rai M, Paul S, Gaur R, et al. Microwave synthesized hydrophilic 4-Oxo-2-phenylquinazoline-3 (4H)–carboximidamide and–carboxamide as privileged small molecule scaffolds with anti-HIV-1 activities. ChemistrySelect. 2023;8(37):e202302115
9.Modh RP, De Clercq E, Pannecouque C, Chikhalia KH. Design, synthesis, antimicrobial activity and anti-HIV activity evaluation of novel hybrid quinazoline–triazine derivatives. Journal of Enzyme Inhibition and Medicinal Chemistry. 2014;29(1):100-108
10.De A, Sarkar S, Majee A. Recent advances on heterocyclic compounds with antiviral properties. Chemistry of Heterocyclic Compounds. 2021;57(4):410-416
11.Abdelmonsef AH, Omar M, Rashdan HR, Taha MM, Abobakr AM. Design, synthetic approach, in silico molecular docking and antibacterial activity of quinazolin-2, 4-dione hybrids bearing bioactive scaffolds. RSC Advances. 2023;13(1):292-308
12.Auti PS, George G, Paul AT. Recent advances in the pharmacological diversification of quinazoline/quinazolinone hybrids. RSC Advances. 2020;10(68):41353-41392
13.Sasmal S, Balaji G, Reddy HRK, Balasubrahmanyam D, Srinivas G, Kyasa S, et al. Design and optimization of quinazoline derivatives as melanin concentrating hormone receptor 1 (MCHR1) antagonists. Bioorganic & Medicinal Chemistry Letters. 2012;22(9):3157-3162
14.Kanuma K, Omodera K, Nishiguchi M, Funakoshi T, Chaki S, Nagase Y, et al. Identification of 4-amino-2-cyclohexylaminoquinazolines as metabolically stable melanin-concentrating hormone receptor 1 antagonists. Bioorganic & Medicinal Chemistry. 2006;14(10):3307-3319
15.Metri S, Melavanki S, Dashyal B, Bhandarkavthe M, Iliyas M, Kotnal R. Recent advances in synthesis of quinazolines: A review. International Journal of Pharmaceutical Science Invention. 2022;11:32-44
16.Raghu M, Swarup H, Shamala T, Prathibha B, Kumar KY, Alharethy F, et al. Design, synthesis, anticancer activity and docking studies of novel quinazoline-based thiazole derivatives as EGFR kinase inhibitors. Heliyon. 2023;9(9):4-6
17.Chatturong U, Chootip K, Martin H, Tournier-Nappey M, Ingkaninan K, Temkitthawon P, et al. The new quinazoline derivative (N2-methyl-N4-[(thiophen-2-yl) methyl] quinazoline-2, 4-diamine) vasodilates isolated mesenteric arteries through endothelium-independent mechanisms and has acute hypotensive effects in Wistar rats. European Journal of Pharmacology. 15 August 2023;953:175829
18.Kanuma K, Omodera K, Nishiguchi M, Funakoshi T, Chaki S, Semple G, et al. Lead optimization of 4-(dimethylamino) quinazolines, potent and selective antagonists for the melanin-concentrating hormone receptor 1. Bioorganic & Medicinal Chemistry Letters. 2005;15(17):3853-3856
19.Chaki S, Funakoshi T, Hirota-Okuno S, Nishiguchi M, Shimazaki T, Iijima M, et al. Anxiolytic-and antidepressant-like profile of ATC0065 and ATC0175: Nonpeptidic and orally active melanin-concentrating hormone receptor 1 antagonists. Journal of Pharmacology and Experimental Therapeutics. 2005;313(2):831-839
20.Matsuno K, Ichimura M, Nakajima T, Tahara K, Fujiwara S, Kase H, et al. Potent and selective inhibitors of platelet-derived growth factor receptor phosphorylation. 1. Synthesis, structure− activity relationship, and biological effects of a new class of quinazoline derivatives. Journal of Medicinal Chemistry. 2002;45(14):3057-3066
21.Wang D, Gao F. Quinazoline derivatives: Synthesis and bioactivities. Chemistry Central Journal. 2013;7:1-15
22.Pattan SR, Krishna Reddy VV, Manvi FV, Desai BG, Bhat AR. Synthesis of N-3 (4-(4-Chlorophenyl Thiazole-2-Yl)-(2-(Amino) Methyl)-Quinazoline-4 (3H)-One and their Derivatives for Antitubercular Activity. 2006
23.Sahu V, Kushwaha V, Sahu V, Shukla S, Khan I. Review on synthesis schemes of first line drugs of antitubercular drugs. Indian Journal of Science and Research. 2022;2(2):53-59
24.Wenthur CJ, Morrison RD, Daniels JS, Conn PJ, Lindsley CW. Synthesis and SAR of substituted pyrazolo [1, 5-a] quinazolines as dual mGlu2/mGlu3 NAMs. Bioorganic & Medicinal Chemistry Letters. 2014;24(12):2693-2698
25.Raghav N, Singh M. Design, synthesis and docking studies of bischalcones based quinazoline-2 (1H)-ones and quinazoline-2 (1H)-thiones derivatives as novel inhibitors of cathepsin B and cathepsin H. European Journal of Pharmaceutical Sciences. 2014;54:28-39
26.Humphrey JM, Yang E, am Ende CW, Arnold EP, Head JL, Jenkinson S, et al. Small-molecule phosphodiesterase probes: Discovery of potent and selective CNS-penetrable quinazoline inhibitors of PDE1. MedChemComm. 2014;5(9):1290-1296
27.Abdel-Rahman HM, Abdel-Aziz M, Canzoneri JC, Gary BD, Piazza GA. Novel Quinazolin-4 (3H)-one/Schiff Base hybrids as antiproliferative and phosphodiesterase 4 inhibitors: Design, synthesis, and docking studies. Archiv der Pharmazie. 2014;347(9):650-657
28.Alagarsamy V, Narendhar B, Sulthana M, Solomon VR. Design and synthesis of 3-(4-chlorophenyl)-2-(2-(4-substituted)-2-oxoethylthio) quinazolin-4 (3 H)-one as antihistamine agents. Medicinal Chemistry Research. 2014;23(11):4692-4699
29.Darras FH, Wehle S, Huang G, Sotriffer CA, Decker M. Amine substitution of quinazolinones leads to selective nanomolar AChE inhibitors with ‘inverted’binding mode. Bioorganic & Medicinal Chemistry. 2014;22(17):4867-4881
30.Ji Q, Yang D, Wang X, Chen C, Deng Q, Ge Z, et al. Design, synthesis and evaluation of novel quinazoline-2, 4-dione derivatives as chitin synthase inhibitors and antifungal agents. Bioorganic & Medicinal Chemistry. 2014;22(13):3405-3413
31.Rayees S, Satti NK, Mehra R, Nargotra A, Rasool S, Sharma A, et al. Anti-asthmatic activity of azepino [2, 1-b] quinazolones, synthetic analogues of vasicine, an alkaloid from Adhatoda vasica. Medicinal Chemistry Research. 2014;23:4269-4279
32.Špulák M, Pourová J, Vopršálová M, Mikušek J, Kuneš J, Vacek J, et al. Novel bronchodilatory quinazolines and quinoxalines: Synthesis and biological evaluation. European Journal of Medicinal Chemistry. 2014;74:65-72
33.Khan KM, Saad SM, Shaikh NN, Hussain S, Fakhri MI, Perveen S, et al. Synthesis and β-glucuronidase inhibitory activity of 2-arylquinazolin-4 (3H)-ones. Bioorganic & Medicinal Chemistry. 2014;22(13):3449-3454
34.Hou X, Zhang J, Zhao X, Chang L, Hu P, Liu H. Design, synthesis and bioactivities evaluation of novel quinazoline analogs containing oxazole units. Chinese Journal of Chemistry. 2014;32(6):538-544
35.de Castro Barbosa ML, Lima LM, Tesch R, Sant'Anna CMR, Totzke F, Kubbutat MH, et al. Novel 2-chloro-4-anilino-quinazoline derivatives as EGFR and VEGFR-2 dual inhibitors. European Journal of Medicinal Chemistry. 2014;71:1-14
36.Waiker DK, Karthikeyan C, Poongavanam V, Kongsted J, Lozach O, Meijer L, et al. Synthesis, biological evaluation and molecular modelling studies of 4-anilinoquinazoline derivatives as protein kinase inhibitors. Bioorganic & Medicinal Chemistry. 2014;22(6):1909
Written By
Ali Gamal Al-Kaf and Rana Abdullah Al-Robaidi
Submitted: 14 December 2023Reviewed: 14 December 2023Published: 21 February 2024
 Open access peer-reviewed chapter
Open access peer-reviewed chapter