Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
22q11.2 deletion syndrome is a genomic disorder with a broader clinical and genetic spectrum. To exclude the presence of 22q11.2 microdeletion, we optimize a PCR-RFLP analysis of three SNP located in the typically proximal 22q11.21 deleted region of 1.5 Mb. PCR reactions, optimized with a Touch-Down program, were performed using three pairs of primers. The amplicons were cleaved by three restrictive enzymes: HaeIII, CviAII, and BsrI applied respectively, for rs4819523, rs4680, and rs5748411. The efficiency of this PCR RFLP assay was confirmed in the light of its application in a small cohort of 10 Tunisian patients, having a congenital heart defect and a known status of 22q11 deletion by FISH and MLPA. The principle of the proximal 22q11.2 microdeletion, applied with exclusion technique seems to be interesting but further population studies for the determination of the heterozygosity rate of the polymorphic 22q11 region markers are needed, particularly in North Africa.
Genomics of Signalopathies at the Service of Precision Medicine UR17ES36/LR23ES07, Faculty of Medicine, University of Sfax, Tunisia
Samir Aloulou
Genomics of Signalopathies at the Service of Precision Medicine UR17ES36/LR23ES07, Faculty of Medicine, University of Sfax, Tunisia
Sonda Kammoun
Genomics of Signalopathies at the Service of Precision Medicine UR17ES36/LR23ES07, Faculty of Medicine, University of Sfax, Tunisia
Aymen Damak
Department of Thoracic and Cardiovascular Surgery, Faculty of Medicine, Habib Bourguiba Hospital, Sfax, Tunisia
Mona Rekik
Genomics of Signalopathies at the Service of Precision Medicine UR17ES36/LR23ES07, Faculty of Medicine, University of Sfax, Tunisia
Saloua Ben Amor
Genomics of Signalopathies at the Service of Precision Medicine UR17ES36/LR23ES07, Faculty of Medicine, University of Sfax, Tunisia
Oldez Kaabi
Genomics of Signalopathies at the Service of Precision Medicine UR17ES36/LR23ES07, Faculty of Medicine, University of Sfax, Tunisia
Balkiss Abdelmoula
Genomics of Signalopathies at the Service of Precision Medicine UR17ES36/LR23ES07, Faculty of Medicine, University of Sfax, Tunisia
*Address all correspondence to: nouha_abdelmoulabouayed@yahoo.fr
1. Introduction
22q11.2 deletion syndrome is a genomic disorder with a broader clinical and genetic spectrum [1, 2, 3, 4, 5, 6, 7, 8]. Microalterations at the 22q11.2 locus include the most frequent 3 Mb deletion and the smallest nested 1.5 Mb deletion [2, 4, 5]. Non-allelic recombination events between LCRs A and H are responsible for 22q11.2 genomic rearrangements [6]. Molecular diagnosis of 22q11.2 microdeletion is usually made by fluorescence in situ hybridization (FISH), multiplex ligation-dependent probe amplification (MLPA), and comparative genomic hybridization (CGH) microarray analysis, or by real-time polymerase chain reaction (qPCR) [3].
Since 2003, it has been shown that a three-step PCR–RFLP analysis can be useful to exclude the presence of 22q11.2 microdeletion in the typically deleted 22q11 region of 1.5 Mb with less cost, high specificity, and with the benefit of reducing the number of the patients who are supposedly screened for 22q11.2 microdeletion (from 100 to 1%) [9]. This method was developed on the basis of the presence of polymorphism in consecutive selected SNP markers, located in the 1,5 Mb 22q11.2 microdeleted region, with a high heterozygosity rate in Brazilian population. Heterozygosity excluded 22q11.2 deletion. However, homozygosity is unable to exclude deletion and leads to a subsequent analysis of 22q11 region by FISH or MLPA [9, 10, 11].
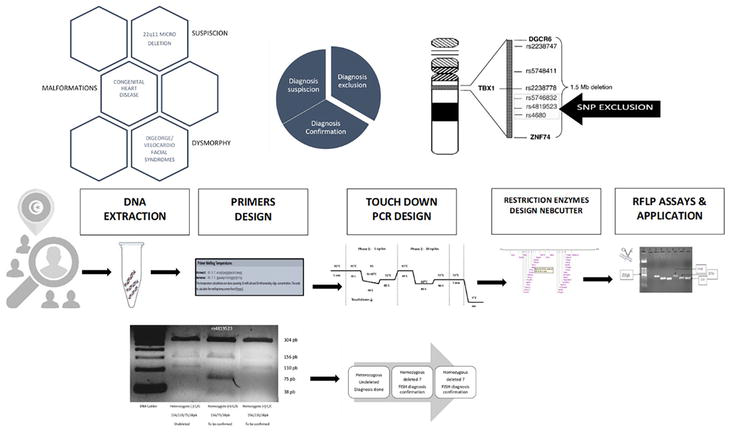
Here, an efficient PCR-RFLP for 22q11.2 microdeletion exclusion was set up. It was evaluated using the technique of a small cohort of known deleted and undeleted Tunisian patients (Figure 1).
Figure 1.
Graphical illustration of our study design of 22q11 microdeletion exclusion.
The application of the optimized PCR-RFLP technique was performed on 10 Tunisian patients having congenital heart diseases (CHD), and 22q11 known status. All patients were explored by karyotyping. The presence or the absence of 22q11.2 microdeletion was confirmed by FISH, using two LSI probes, which are respectively, LSI D22S75 (N25 region) SpectrumOrange/LSI ARSA SpectrumGreen Probe and DiGeorge Region Probe - LSI TUPLE 1 SpectrumOrange/LSI ARSA SpectrumGreen Probe. For two patients, the confirmation of 22q11.2 microdeletion was conducted by MLPA, which was performed using the MRC-Holland SALSA MLPA P300 control kit, and designed synthetic probes targeting chromosomal regions, including the classical and distal 22q11.22 microdeletion region of DiGeorge/Velo-cardio-facial syndrome (Not shown).
2.2 Methods
2.2.1 Cytogenetic analysis
Cytogenetic analysis was carried out using lymphocyte cultures for 72h with phytohemagglutinin induction. Metaphase chromosomes were prepared in accordance with the standard cytogenetic protocols. Chromosome identification and karyotyping analysis were conducted by using RHG and GTG banding. Chromosomal abnormalities were interpreted and written in conformity with the international standard nomenclature (ISCN) 2005–2009.
2.2.2 Simplex PCR-RFLP: Technical optimization and analysis
A simplex PCR-RFLP was optimized for a rapid negative diagnosis of proximal microdeletions of chromosome 22, based on the method of molecular exclusion described by Pereira (2003) and Gioli-Pereira (2006) [9, 10].
To exclude the presence of 22q11.2 microdeletion, the method was conducted by the analysis of the polymorphism of the three loci located in the typically 22q11.21 deleted region of 1.5 Mb. These loci were rs4819523, rs5748411, and rs4680. They are characterized by a high rate of heterozygosity.
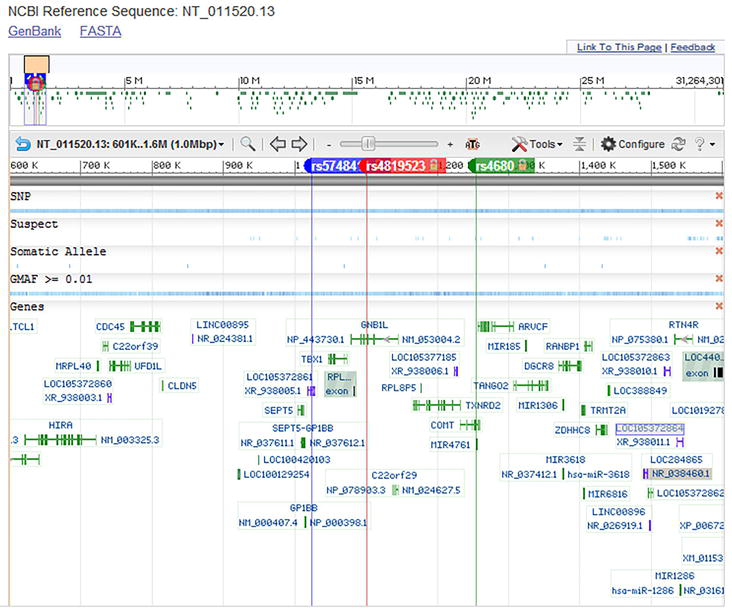
The interrogation of the SNP database (Figure 2) showed that these polymorphic markers included the ancestral alleles reported in some populations as the European, the Asian, and the Sub-Saharan African people.
Figure 2.
Alignment of rs4819523, rs5748411, and rs4680 at the Homo sapiens chromosome 22 NCBI reference sequence: NT_011520.13 (https://www.ncbi.nlm.nih.gov/snp/).
The rs4819523 is a SNP (SNV) at the position: 19810549 with two alleles C/G (C is the ancestral one) (Table 1). It is an intronic variant of GNB1L gene (G-protein beta subunit-like protein ID 54584 location: 22q11.21 exon count: 8).
Marker
Alleles and sequences of the ancestral alleles with the colored base in red
Sequences of ancestral alleles of the analyzed 22q11 polymorphic markers.
The rs4680 is a SNP (SNV) at the position: 1254184 with two alleles G/A (the G allele is the ancestral one) (Table 1) localized in COMT gene (Catechol-O-methyltransferase ID 1312 location 22q11.21 exon count: 8).
The rs5748411 is a SNP (SNV) at the position: 19733950 with two alleles C/A (C is the ancestral allele) (Table 1).
In addition, the interrogation of the SNP database showed that there is no information about the characteristics and the frequencies of the allelic polymorphisms of the three selected SNP (rs4819523, rs4680, and rs5748411) in the North African population.
PCR reactions were performed using three pairs of primers as described by Huber (2014) (Table 2) [11].
Marker
Forward PRIMERS
Reverse PRIMERS
Amplicon size
Alleles
rs4819523
ACAGTGAGGGGACATCAAGG
GGAAAGCTCATGGGTGTCTG
304 pb
C/G
rs4680
GTGATTCAGGAGCACCAGCC
GCCCTTTTTCCAGGTCTGAC
217 pb
G/A
rs5748411
ATCAGCCCACAGAGCAGACC
GCATAGAGAAAAGCAACTCC
202 pb
C/A
Table 2.
Sequences of primers used for PCR-RFLP validated by PCR in Silico.
They were optimized by a Touch-Down program. Reactions were done, in a final volume of 25 μl, by using 10 pmol of each primer, 1x PCR buffer, 2 mmol/l of MgCl2, 0,2 mmol/l of dNTP, 200 ng DNA, 1 unit Taq DNA polymerase. Amplification conditions were optimized using 35 cycles Touch-Down program with initial melting step at 95°C for 5 min and final extension step at 72°C for 7 min in all reactions: five first cycles with 95°C for 40 s/ 65 to 60°C for 40 s and 72°C for 40 s and then 30 cycles with 95°C for 40 s/60°C for 40 s and 72°C for 40 s (Table 3).
Thermocycling Conditions
Initial Denaturation
95°C
5 min
1 cycle
5 cycles
Denaturation
95°C
40 s
Annealing
65 °C → 60°C
40 s
Extension
72°C
40s
30 cycles
Denaturation
95°C
40 s
Annealing
60°C
40 s
Extension
72°C
40 s
Final extension
72°C
7 min
1 cycle
Hold
4%
∞
Table 3.
The thermocycling conditions of the touch-down PCR reaction.
For the RFLP analysis, the amplicons were cleaved by three restriction enzymes as described by Huber (2014) [11], in conformity with the manufacturer’s recommendations (NEBiolabs) (Table 4).
SNP
Restriction Enzyme
Restriction Sequence
Cleavage Site
rs4819523
HaeIII
GGCC
5′…GG↓CC…3′ 3’…CC↑GG…5′
rs4680
BclI
TGATCA
5′…T↓G A T C A…3′ 3′…A C T A G↑T…5’
rs5748411
BsrI
ACTGG
5′… ACTGGN↓…3′ 3′… TGAC↑CN…5’
Table 4.
Restriction enzymes of RFLP and electrophorese profiles after cleavage.
The digestion of amplicons by the three restriction enzymes was optimized using three digestion programs (X1, X2, X3) and restriction fragments (Table 5) were visualized on 4% agarose gel. For X1 program: Reaction was performed, in a final volume of 20 μl, with 10 μl of PCR product, 2 μl of enzymatic tampon, 1 μl enzyme, and 7 μl H2O. Digestion was carried out at 37°C during 15 hours. For X2 program: Reaction was performed in a final volume of 30 μl in which there were 15 μl of PCR product, 3 μl of enzymatic tampon, 1 μl of enzyme, and 11 μl H2O. Digestion was carried out at 37°C during 1 hour. For X3 program, in a final volume of 20 μl, there were 14 μl of PCR product, 5 μl of enzymatic tampon, and 1 μl of enzyme (without H2O). Digestion was carried out at 37°C during 1 hour.
Marker
Restriction Enzyme
Cleavage fragments in the presence of the cleavage site
Cleavage fragments in the absence of the cleavage site
rs4819523
HaeIII
Allele C: 4 bands 156pb 75pb 38pb 35pb
Allele G: 3 bands 156pb 110pb 38pb
rs4680
BclI
Allele A: 2 bands 102pb 115pb
Allele G: 1 band 217pb
rs5748411
BsrI
Allele C: 2 bands 64pb 138pb
Allele A: 1 band 202pb
Table 5.
Electrophoretic profiles after marker digestion according to Huber et al. 2014.
PCR amplification reactions and conditions were optimized using in Silico PCR tools and gradient melting assays.
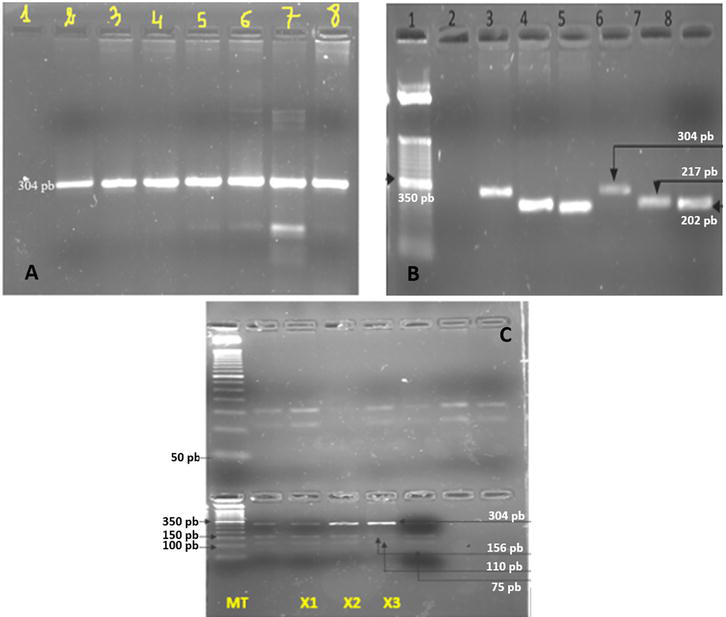
One touch-down PCR reaction was finally designed for a simultaneous amplification of the three markers. The optimization of the PCR Touch-Down program and the optimization of the digestion protocol are shown in Figure 3.
Figure 3.
A: Electrophoresis profile of amplification product of rs4819523 using a melt gradient B: Electrophoresis profile of the PCR products of the 3 selected STS C: Electrophoresis profile of the PCR-RFLP products of the rs4819523 using for the digestion, the HaeIII restriction enzyme. A-legend: Line 1: H2O, line 2: Tm 64,6°C, line 3: Tm 63,7°C, line 4: Tm 62,1°C, line 5: Tm 60,2°C, line 6: Tm 57,5°C, line 7: Tm 55°C, line 8: Amplification using touch-down PCR program B-legend: Line 1: 50 bp ladder DNA marker, line 2: H2O, lines 3,4, and 5: DNA control for the three SNP rs4819523 (304 bp), rs4680 (217 bp), and rs5748411 (202 bp) C-legend: Digestion of amplification product of a DNA control sample for rs4819523 using HaeIII permits to obtain three fragments: 156pb, 110pb, 75pb. This profile is compatible with the heterozygous status C/G. MT: 50 bp ladder DNA marker, X1, X2, X3 are three digestion programs.
It was possible via Nebcutter to validate virtual rs4819523 and rs5748411 digestion profiles, using respectively HaeIII and BsrI restriction enzymes. However, the digestion of rs4680 by BclI, as described by Huber (2014) [11], was not possible and another enzyme was then considered. Indeed, the verification of the digestion profile of rs4680 by BclI, showed both manually and by Nebcutter, confirmed the absence of the restriction site at the level of the sequence of the amplicons generated in silico PCR for both the ancestral G allele and the A allele. The design of other enzymes via the same Biolabs Nebcutter tool was then performed. Three other enzymes can be used, FatI, CviAII, and NLaIII, which are isoschizomers (Figure 4).
Figure 4.
Restriction enzymes that cut the rs4680.
They highlight the selected SNP. The digestion of the rs4680 by CviAII gives the homozygous alleles GG (2 restriction sites and 3 bands) and AA (3 restriction sites and 4 bands). The heterozygous profile is the combination of the two previous ones (Table 6).
SNP
Restriction Enzyme
Restriction Sequence
Cleavage Site
rs4680
CviAII
CATG
5′…C↓ATG…3′ 3′…G TA↑C…5’
Cleavage fragments in the presence of the cleavage site
Cleavage fragments in the absence of the cleavage site
Allele A: 4 bands 96pb 69pb 34pb 18pb
Allele G: 3 bands 114pb 69pb 34pb
Table 6.
Electrophoretic profiles after rs4680 digestion using CviAII restriction enzyme.
Fragments generated after restriction/digestion were visualized on 4% agarose gel. Figure 3 shows the electrophoresis profile of amplification product of rs4819523 using a melt gradient (Figure 3A), the electrophoresis profile of the PCR products of the three selected STS (rs4819523, rs4680, and rs5748411) (Figure 3B), and finally the electrophoresis profile of the PCR-RFLP products of the rs4819523 using for the digestion, the HaeIII restriction enzyme (Figure 3C).
3.1.1 Application
The results of the 22q11.2 exclusion using the optimized PCR-RFLP technique in our sample of 10 patients known as having or not a 22q11 microdeletion are shown in Table 7.
No
Sex
Age range (years)
Congenital Heart Disease
Karyotype
FISH/MLPA results of 22q11 microdeletion
PCR-RFLP exclusion of microdeletion 22q11
1
F
[2–5]
Double outlet right ventricle
46,XX
Negative
Positive
2
M
[2–5]
Coarctation of Aorta
46,XY
Negative
Positive
3
F
[2–5]
Pulmonic Valve Stenosis
46,XX
Negative
Positive
4
F
[5–10]
Tetralogy of Fallot
46,XX
Positive
Negative
5
M
[0–2]
Coarctation of Aorta/Persistent Ductus Arteriosus
46,XY
Negative
Positive
6
F
[5–10]
Tetralogy Of Fallot
46,XX
Negative
Positive
7
M
[0–2]
Coarctation of Aorta
Failure of culture
Negative (MLPA)
Positive
8
M
[0–2]
Tetralogy Of Fallot
46,XY
Negative
Positive
9
F
[0–2]
Atrial septal defect/Persistent Ductus Arteriosus
Failure of culture
Negative (MLPA)
Positive
10
M
[10–15]
Muscular Ventricular Septal Defect
45,XY,-22 der(11)t(11;22)(q23;q11)
Positive
Negative
Table 7.
PCR-RFLP results for exclusion of 22q11 microdeletion in our 10 Tunisian CHD patients.
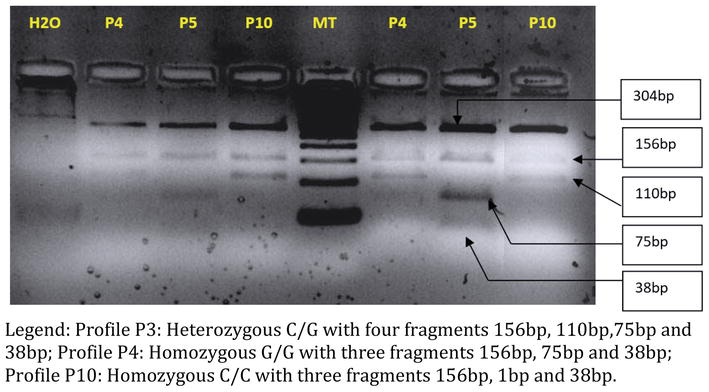
Figure 5 shows the PCR-RFLP electrophoresis profile of rs4819523 digested amplicons in our 10 patients as well as the rs4819523 alleles of all patients (Figure 5 and Table 8). Table 8 Shows the genotypes of the rs4819523 SNP of all patients (Table 8).
Figure 5.
RFLP electrophoresis profile of rs4819523 amplicons in the 10 patients. Profile P3: Heterozygous C/G with four fragments 156 bp, 110 bp, 75 bp, and 38 bp; profile P4: Homozygous G/G with three fragments 156 bp, 75 bp, and 38 bp; profile P10: Homozygous C/C with three fragments 156 bp, 1 bp, and 38 bp.
Patient number
rs4819523 alleles
Genotype
P1
C/G
Heterozygote
P2
C/G
Heterozygote
P3
C/G
Heterozygote
P4
G/G
Homozygote
P5
C/G
Heterozygote
P6
C/G
Heterozygote
P7
C/G
Heterozygote
P8
C/G
Heterozygote
P9
C/G
Heterozygote
P10
C/C
Homozygote (ancestral allele)
Table 8.
rs4819523 genotypes of our 10 Tunisian CHD patients.
Application of our technique for our 10 patients involving two deleted and eight undeleted patients proved that the PCR-RFLP 22q11.2 exclusion results were concordant with FISH/MLPA results (Figure 6). The status of the deleted subjects was homozygous while the profile of the undeleted ones was heterozygous. PCR-RFLP analysis of rs4819523 showed heterozygote status C/G in the eight undeleted patients with a consequent exclusion of the microdeletion 22q11. Comparatively, in microdeleted patients which was already confirmed by FISH (Figure 6) or MLPA, PCR-RFLP analysis of rs4819523 showed a homozygous G/G status in patient P4 and a homozygous C/C status in the one of P10 (Table 8 and Figure 5).
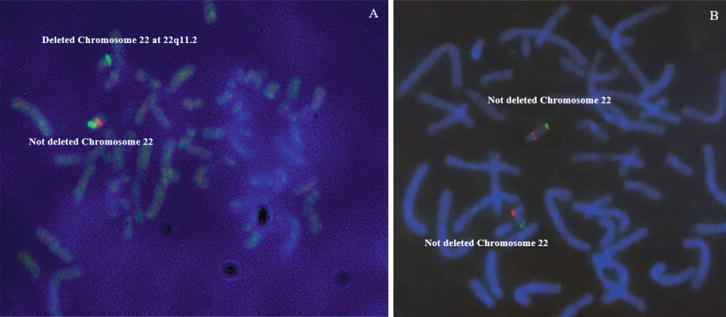
Figure 6.
FISH results showing deleted (A) and not deleted (B) patients (P4 and P6 respectively) (12).
Microdeletions and microduplications, known as segmental aneusomies are defined by the presence of a sub-cytogenetic chromosomal anomaly, less than 5 Mb in size, classically detectable by the molecular cytogenetic technique of fluorescent in situ hybridization (FISH) [12, 13].
Molecular cytogenetics has made it possible to link microdeletions to syndromes that have long been described clinically but whose genetic origin was suspected. The two oldest entities studied are the 22q11 microdeletion syndrome associated with Digeorge (DG: MIM 188400) and velocardiofacial (VCF: MIM 192430) syndromes and the 7q11 microdeletion syndrome associated with Williams syndrome (WBS: MIM 194050) [14, 15]. It should be noted that there are several synonyms for DG syndrome, namely CATCH 22, Shprintzen syndrome, Takao syndrome, and conotruncal anomaly face (CAF) syndrome [16, 17, 18].
Mechanistically, microdeletions and microduplications occur during meiosis as a result of illegal crossovers with mismatches and non-allelic homologous recombination (NAHR) between DNA sequences with high homology. These are in particular segmental duplications or LCRs for Low Copy Repeats [19]. These are sequences of 10 to 300 kb, with more than 95% homology, often associated with Alu sequences. They are located particularly in the pericentromeric and subtelomeric regions and represent 5 to 10% of the genome, which explains the presence of recombination hotspots. NAHR during meiosis with LCRs in direct orientation leads to deletions/duplications.
Several genes located and cloned in these regions are specifically responsible for certain phenotypes, in particular by haploinsufficiency mechanisms [20]. Recent advances in molecular biology have led to the discovery of a progressively increasing number of microdeletional and microduplicational syndromes through the search for imbalances in the regions delimited by the LCRs using CGH array [21, 22]. The same advances have made it possible to broaden the spectrum of techniques enabling their detection and screening on a large scale, namely CGH array, real-time PCR, and the MLPA technique [20, 23].
The existence of two recurrent types of 22q11 microdeletion has been demonstrated. The first deletion extends over 3 Mb with a commonly deleted region or TDR found in 90% of patients. The second, always proximal, extends over 1.5 Mb and is found in 7% of patients [24, 25]. Both deletions occur by NAHR between LCR22 (A, B, C, and D) with LCRA/D and LCRA/B respectively [26]. Atypical deletions, often distal, sparing the minimal critical region, which is the smallest region responsible for the DG phenotype in its absence, were also reported.
22q11.2 microdeletions, now correspond, thanks to the advent of high-resolution molecular techniques, to a very heterogeneous genomic entity, that of CNVs, linked to recurrent rearrangements in the 22q11. Two region, secondary to too frequent non-allelic homologous recombination due to the richness of the region in LCRs [24]. In fact, mapping of the 22q11 region has revealed the presence of 8 LCR22s identified at the 22q11.2 region, named LCR22-A to LCR22-H or LCR22-2 to LCR22-8 [21, 22]. The four centromeric LCR22s from LCR-A to LCR-D, known for a long time, are involved in the proximal classical recurrent microdeletions specific to DG/VCF syndromes and in reciprocal 22q11.21 microduplication (MIM 608363). The 4 other LCR22 with telomeric disposition, named LCR22-E to LCR22-H located in the region immediately distal to 22q11.21 TDR (i.e. 22q11.22-q11.23), are involved in recurrent distal microdeletions associated with particular phenotypes (MIM 611867) as well as in reciprocal microduplications.
Study of the LCR22-D, −E, and −F region showed the presence of the BLCR module in each of these 3 LCR22s, suggesting the involvement of the NAHR mechanism in distal 22q11.2 microdeletions. In this particular region, Mikhail et al. (2014) demonstrated the presence of microdeletions variable in size, challenging the notion that microdeletion in this region is associated with a single clinical entity [21, 24]. The same author proposed a new classification of distal 22q11.2 microdeletions into three major types I, II, and III emanating from three respective types of NAHR: LCR22-D/−E or -F; LCR22-E/-F and LCR22-F/−G. Molecular studies based on high-resolution techniques have also identified 22q11.2 microdeletions with atypical breakpoints that do not involve any of the known LCR22s [21, 24]. These authors suggested the involvement of SINE/Alu elements and highlighted the extreme complexity of the 22q11.2 genomic region, its rearrangements, and relative phenotypes [27].
Microdeletion and microduplication syndromes are currently referred to as CNVs [21]. A CNV for recurrent copy-number variations, is defined as a segment of DNA whose size varies from 1 kb to several Mb and whose copy number is different from that of a control for which it is fixed at 2. These polymorphisms are distributed throughout the genome and listed on a website: Database of genomic variants. The number of polymorphisms has grown steadily, reaching thousands, spread across all the chromosomes, and representing around 5% of the human genome. They are included in genes, but also in highly conserved regions of the genome and regions that regulate gene expression. Their effect can take the form of a gene dosage effect, corresponding to deletions and duplications in cytogenetic nomenclature, or be linked to the interruption of a gene.
The more than 40 genes in the 22q11.2 region have been extensively studied to delineate the phenotypes associated with 22q11.2 microdeletions and microduplications and their pathophysiological mechanisms, including neural crest migration abnormalities [28]. The TBX1 gene (MIM 602054) belonging to the proximal 22q11.2 region remains the most incriminated gene in the cardiac manifestations of 22q11.2 microdeletions, with apparently clinical heterogeneity secondary to modifying factors such as gene environment [21, 24].
Despite these molecular advances and the overly broad spectrum of phenotypic manifestations associated with 22q11.2 microdeletions classified into several types, the group of proximal 22q11.2 microdeletions with its two subtypes, common 3 Mb microdeletion and central 1.5 Mb microdeletion, remain those most frequently reported to date. Compared to distal microdeletions, proximal 22q11.2 microdeletions are the entities that most include, in their phenotypic spectrum, cardiac malformations, in particular, conotruncal heart disease (MIM 217095) [11, 12, 23, 29, 30]. According to three landmark studies in which the phenotype of 810 patients with microdeletions at 22q11 was determined, conotruncal heart disease was reported in 75% of cases [31, 32, 33]. Tetralogy of Fallot is the most common heart disease, with a prevalence of 22–35%. The frequency of CHD also differs according to the involved clinical syndrome. It should be noted that cardiac involvement seems to be rare in distal microdeletions except for deletions classified as type II according to Mikhail et al. (2014) [24] i.e. those resulting from NAHR LCR22-D/E, where the frequency of CHD exceeds 40% and where the CHD seems to correspond to atrial septal defects (ASD) and ventricular septal defects (VSD), patent ductus arteriosus (PCA), and bicuspid aortic valve. With the exception of PCA, these CHD are rarely involved in proximal microdeletions for which the most frequent (but not exclusive) are conotruncal heart defects.
Fluorescence in situ hybridization (FISH), which remains the gold standard test to determine 22q11.2 microdeletion and detects more than 95% cases, is very expensive, time-consuming, and not widely available. It usually requires chromosomal preparations. Settings, such technique for widespread screening of microdeletions, are limited in the medical background with a shortage of resources [4, 34, 35, 36].
Given the financial cost of 22q11.21 microdeletion detection techniques and the impossibility of applying them on a large scale, we have tried in this work to optimize a simple and inexpensive molecular technique using PCR-RFLP in order to allow the exclusion of 22q11.21 microdeletion syndrome. This negative screening is helpful to the reduction of the number of patients who are candidates for the confirmation of the 22q11 syndrome by FISH, MLPA, qPCR, or array CGH. The principle of this exclusion is based on the use of three markers located at 22q11.21, each of which has two alleles. The heterozygosity profiles for these CNV confirm the absence of a microdeletion. Only the homozygous profiles would be likely to be deleted. However, it requires confirmation. Experimentally, the PCR-RFLP technique was firstly developed for the rs4819523 marker and subsequently for the two other ones. After optimization, the technique was applied to 10 patients whose microdeletional status was previously known using the two techniques of reference, notably, FISH and MLPA. All patients had CHD, primarily consisting of conotruncal CHD.
The current study allowed us to validate the technique by showing the heterozygous status for the eight undeleted patients and the homozygous status for the two patients with a confirmed 22q11.2 microdeletion. Accordingly, we have demonstrated that this technique is promising for the 22q11.21 region as well as for other chromosomal regions prone to microdeletions such as the distal 22q11.2 and the 7q11 regions.
For an appropriate application in our population, and may be others, it will be necessary to conduct preliminary population studies in order to identify the polymorphic markers whose characteristics and allelic frequencies are accurately estimated. It requires further improvement, notably, the selection of other biallelic markers to cover more widely the 22q11.21 region of 3 Mb as well as the identification of more polymorphic markers of the selected regions in our Tunisian population, to define the allelic forms and ultimately, to estimate their alleles frequencies.
In conclusion, the principle of the proximal 22q11.2 microdeletion, applied with the exclusion technique, is apparently interesting and possibly used for several applications. Furthermore, this principle could be used to target patients with distal microdeletions, which are becoming more and more frequent for an increasingly rapid confirmation. In this vein, it could be used for excluding these distal 22q11.22-q11.23 microdeletions. However, it is necessary to choose the appropriate polymorphic markers for each type of distal microdeletion and to develop the technique. Further studies of the population for the determination of the heterozygosity rate of 22q11 region markers are needed before setting up such interesting methods of deletion screening in our population and may be others.
1.Rooney K, Levy MA, Haghshenas S, Kerkhof J, Rogaia D, Tedesco MG, et al. Identification of a DNA methylation episignature in the 22q11.2 deletion syndrome. International Journal of Molecular Sciences. 2021;22(16):8611. DOI: 10.3390/ijms22168611
2.Molck MC, Vieira TP, Simioni M, Sgardioli IC, dos Santos AP, Xavier AC, et al. Distal 22q11.2 microduplication combined with typical 22q11.2 proximal deletion: A case report. American Journal of Medical Genetics. Part A. 2015;167A(1):215-220. DOI: 10.1002/ajmg.a.36809 Epub 2014 Oct 30
3.McDonald-McGinn DM, Hain HS, Emanuel BS, Zackai EH. 22q11.2 deletion syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, LJH B, Gripp KW, Mirzaa GM, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1999 Sep 23 [updated 2020 Feb 27], 1993–2022
4.Morrow BE, McDonald-McGinn DM, Emanuel BS, Vermeesch JR, Scambler PJ. Molecular genetics of 22q11.2 deletion syndrome. American Journal of Medical Genetics. Part A. 2018;176(10):2070-2081. DOI: 10.1002/ajmg.a.40504
5.Ben-Shachar S, Ou Z, Shaw CA, Belmont JW, Patel MS, Hummel M, et al. 22q11.2 distal deletion: A recurrent genomic disorder distinct from DiGeorge syndrome and velocardiofacial syndrome. American Journal of Human Genetics. 2008;82(1):214-221. DOI: 10.1016/j.ajhg.2007.09.014
6.Vervoort L, Dierckxsens N, Pereboom Z, Capozzi O, Rocchi M, Shaikh TH, et al. 22q11.2 low copy repeats expanded in the human lineage. Frontiers in Genetics. 2021;12:706641. DOI: 10.3389/fgene.2021.706641
7.Carli D, Moroni A, Eleonora DG, Zonta A, Montin D, Licciardi F, et al. Atypical microdeletion 22q11.2 in a patient with tetralogy of Fallot. Journal of Genetics. 2021;100:5
8.Motahari Z, Moody SA, Maynard TM, LaMantia AS. In the line-up: Deleted genes associated with DiGeorge/22q11.2 deletion syndrome: Are they all suspects? Journal of Neurodevelopmental Disorders. 2019;11(1):7:1-28. DOI: 10.1186/s11689-019-9267-z
9.Pereira AC, Corrêa RF, Mota GF, Kim CA, Mesquita SF, Krieger JE. High specificity PCR screening for 22q11.2 microdeletion in three different ethnic groups. Brazilian Journal of Medical and Biological Research. 2003;36:1359-1365
10.Gioli-Pereira L, Pereira AC, Mesquita SM, Lopes AA, Krieger JE. PCR screening for 22q11.2 microdeletion: Development of a new cost-effective diagnostic tool. Clinica Chimica Acta. 2006;369:78-81
11.Huber J, Catarino Peres V, Luz de Castro A, Jeronimo dos Santos T, Fontoura D, Beltraõ L, et al. Molecular screening for 22Q11.2 deletion syndrome in patients with congenital heart disease. Pediatric Cardiology. 2014;35:56-62
12.Gargouri B, Abdelmoula N, Trabelsi Sahnoun I, Gargouri B, Rebaï T, Amouri A. Screening for cytogenetic and molecular chro-mosome rearrangements in Tuni-sian children with Conotruncal heart defects. Laboratory Medi-cine. 2009;40:357-361
13.Abdelmoula NB, Portnoi MF, Vialard F, Amouri A, VandenAkker J, Taillemite JL. Molecular cytogenetic techniques: principles and advances. Médecine/Sciences. 2000;16:1405-1441
14.Lowery MC, Morris CA, Ewart A, Brothman LJ, Zhu XL, Leonard CO, et al. Strong correlation of elastin deletions, detected by FISH, with Williams syndrome: Evaluation of 235 patients. American Journal of Human Genetics. 1995;57:49-53
15.Schluth-Bolard C, Till M, Edery P, Sanlaville D. Syndromes chromosomiques émergents. Pathologie et Biologie. 2008;56:380-387
16.Driscoll DA, Budarf ML, Emanuel BS. A genetic etiology for DiGeorge syndrome: Consistent deletions and microdeletions of 22q11. American Journal of Human Genetics. 1992;50:924-933
17.Scambler PJ, Kelly D, Lindsay E, Williamson R, Goldberg R, Shprintzen R, et al. Velocardio-facial syndrome associated with chromosome 22 deletions encompassing the DiGeorge locus. Lancet. 1992;339:1138-1139
18.Burn J, Takao A, Wilson D, Cross I, Momma K, Wadey R, et al. Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22q11. Journal of Medical Genetics. 1993;30:822-824
19.Baumer A, Dutly F, Balmer D, Riegel M, Tükel T, Krajewska-Walasek M, et al. High level of unequal meiotic crossovers at the origin of the 22q11. 2 and 7q11.23 deletions. Human Molecular Genetics. 1998;7:887-894
20.Monteiro FP, Vieira TP, Sgardioli IC, Molck MC, Damiano AP, Souza J, et al. Defining new guidelines for screening the 22q11.2 deletion based on a clinical and dysmorphologic evaluation of 194 individuals and review of the literature. European Journal of Pediatrics. 2013;172:927-945
21.Mikhail FM. Copy number variations and human genetic disease. Current Opinion in Pediatrics. 2014;26:646-652
22.Béri-Dexheimer M, Bonnet C, Chambon P, Brochet K, Grégoire MJ, Jonveaux P. Microarray-based comparative genomic hybridization in the study of constitutional chromosomal abnormalities. Pathologie-Biologie (Paris). 2007;55:13-18
23.Frigerio M, Passeri E, de Filippis T, Rusconi D, Valaperta R, Carminati M, et al. SNPs and real-time quantitative PCR method for constitutional allelic copy number determination, the VPREB1 marker case. BMC Medical Genetics. 2011;12:61
24.Mikhail FM, Burnside RD, Rush B, Ibrahim J, Godshalk R, Rutledge SL, et al. The recurrent distal 22q11.2 microdeletions are often de novo and do not represent a single clinical entity: A proposed categorization system. Genetics in Medicine. 2014;16:92-100
25.Scambler PJ. The 22q11 deletion syndromes. Human Molecular Genetics. 2000;9:2421-2426
26.Vergés L, Molina O, Geán E, Vidal F, Blanco J. Deletions and duplications of the 22q11.2 region in spermatozoa from DiGeorge/velocardiofacial fathers. Molecular Cytogenetics. 2014;7:86
27.Beaujard MP, Chantot S, Dubois M, Keren B, Carpentier W, Mabboux P, et al. Atypical deletion of 22q11.2: Detection using the FISH TBX1 probe and molecular characterization with high-density SNP arrays. European Journal of Medical Genetics. 2009;52:321-327
28.Karayiorgou M, Simon TJ, Gogos JA. 22q11.2 microdeletions: Linking DNA structural variation to brain dysfunction and schizophrenia. Nature Reviews. Neuroscience. 2010;11:402-416
29.Abdelmoula NB. Contribution of molecular cytogenetics in the diagnosis of chromosomal abnormalities. Annales de biologie clinique. 2004;62:1-9
30.Wozniak A, Wolnik-Brzozowska D, Wisniewska M, Glazar R, Materna-Kiryluk A, Moszura T, et al. Frequency of 22q11.2 microdeletion in children with congenital heart defects in western Poland. BMC Pediatrics. 2010;10:88
31.McDonald-McGinn DM, Kirschner R, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, et al. The Philadelphia story: The 22q11.2 deletion: Report on 250 patients. Genetic Counseling. 1999;10:11-24
32.McDonald-McGinn DM, LaRossa D, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, et al. The 22q11.2 deletion: Screening, diagnostic workup, and outcome of results; report on 181 patients. Genetic Testing. 1997;1:99-108
33.Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: A European collaborative study. Journal of Medical Genetics. 1997;34(10):798-804
34.Tomita-Mitchell A, Mahnke DK, Larson JM, Ghanta S, Feng Y, Simpson PM, et al. Multiplexed quantitative real-time PCR to detect 22q11.2 deletion in patients with congenital heart disease. Physiological Genomics. 2010;42A(1):52-60
35.Maran S, Faten SA, Lim SE, Lai KS, Ibrahim WPW, Ankathil R, et al. Screening of 22q11.2DS using multiplex ligation-dependent probe amplification as an alternative diagnostic method. BioMed Research International. 2020;2020:6945730
36.Campos-Garcia FJ, Castillo-Espinola AM, Medina-Escobedo CE, Zenteno JC, Lara-Riegos JC, Rubio-Zapata H, et al. Screening method for 22q11 deletion syndrome involving the use of TaqMan qPCR for TBX1 in patients with Conotruncal congenital heart disease. Cardiogenetics. 2022;12(3):253-260
Written By
Nouha Bouayed Abdelmoula, Samir Aloulou, Sonda Kammoun, Aymen Damak, Mona Rekik, Saloua Ben Amor, Oldez Kaabi and Balkiss Abdelmoula
Submitted: 28 April 2023Reviewed: 10 July 2023Published: 28 July 2023
 Open access peer-reviewed chapter
Open access peer-reviewed chapter