 Open access peer-reviewed chapter
Open access peer-reviewed chapter
Abstract
This chapter provides an overview of the complex biological processes involved in bone development and regeneration. The skeletal system serves crucial functions such as structural support, mineral storage, and organ protection. Bone development encompasses diverse cell types, matrices, and signals from embryonic stages to adulthood, with age-related decline in regeneration requiring additional support for large defects. Intramembranous and endochondral ossification processes are explored, involving differentiation of mesenchymal cells into osteoblasts and cartilage formation replaced by bone, respectively. Collagen and proteoglycans, particularly collagen I and II and heparan sulfates, play vital roles in the microenvironment for bone formation and mineralization. Signaling molecules such as BMPs, FGFs, IGFs, and PDGFs important for proliferation and differentiation of bone precursors, embryonic development, growth and maintenance of mature bone include regeneration and angiogenesis. Cell-based approaches, microenvironment-based technologies, and signal-based technologies utilizing growth factors are explored as bone regeneration strategies. Understanding these processes, factors, and technologies is pivotal in improving the treatment of conditions such as osteoporosis, fractures, and bone reconstruction, ultimately developing new technologies.
Keywords
- osteogenesis
- osteoinductive signals
- microenvironment
- bone development
- bone regeneration
1. Introduction
A remarkable cascade of biological events takes place from mesenchymal condensation during early embryonic development to postnatal maturation, providing the body with the skeletal system for structural support and stability, mineral storage, and protection of vital organs. The process involves various cell types, extracellular matrices (“microenvironment”), and biological signals that orchestrate the formation of the bone in the microenvironment throughout early developmental and maturation stages, even into adulthood. This process may provide a unique insight into developing novel technologies for regenerating and repairing bone defects or bridging bony gaps to achieve a solid fusion as required in fusion surgeries. Bone is able to regenerate and repair itself in healthy individuals as long as the defect is less than a critical size although the regenerative potential declines with aging. While there is no clear definition of a critical-sized bone defect [1], it is generally referred to as the distance of the boney gap that the body is unable to bridge with new bone spontaneously. A critical-sized bone defect would need an additional osteogenic (stem and progenitor cells), osteoinductive (biological signals), and/or osteoconductive (microenvironment) support for full bone regeneration and repair. Obesity, diabetes, osteoporosis, metabolic disorders, extended use of steroids, poor nutrition, advancing age, and drinking and smoking may lead to pseudoarthrosis, which is defined as a chronic condition where a broken bone has failed to heal after a fracture or surgical procedure. Patients with these risk factors and/or those who have been diagnosed with pseudoarthrosis may require a surgical intervention that includes a regenerative approach. Thus, it may be critical to understand how the body utilizes different biological components for developing the skeletal system and apply such understanding in designing an optimal regenerative approach for patients in need.
2. Cells in embryonic bone development
2.1 Intramembranous ossification
Embryonic epithelial and mesenchymal cells undergo a series of sequential and reciprocal communications to initiate mesenchymal condensation through a signaling pathway, which induces the expression of neural cell adhesion molecules (NCAM) and N-cadherin and mediates cell-cell adhesion during intramembranous ossification [2, 3]. The epithelial-mesenchymal crosstalk at the cellular and molecular levels appears to be the first event that leads to the condensation and migration of presumptive skeletogenic cells that are derived from the neural crest or mesoderm. The role of epithelial cells in mesenchymal condensation is clearly identified for intramembranous ossification [4, 5, 6]. Interestingly, mandibular bones failed to form in the absence of mandibular epithelium in the chicken (HH 24) and mouse (day 10) embryos, suggesting the crucial role of the epithelium in early bone development. Mesenchymal cells derived from the neural crest proliferate and differentiate directly into the osteogenic lineage without going through the intermediary chondrogenic transition by condensing into compact nodules during intramembranous ossification (Figure 1). These condensing mesenchymal cells differentiate into osteoblasts with activation of transcription factors such as CBFA1, which appears to trigger the expression of genes associated with the production of osteopontin, osteocalcin, and extracellular matrices that support bone development [7]. Additional factors such as extracellular signal-regulated kinase (ERK), p38 (MAPK), Akt, p65 (NF-kB), and bone morphogenetic proteins (BMPs) in addition to the activation of transcription factors such as CBFA1 [8].
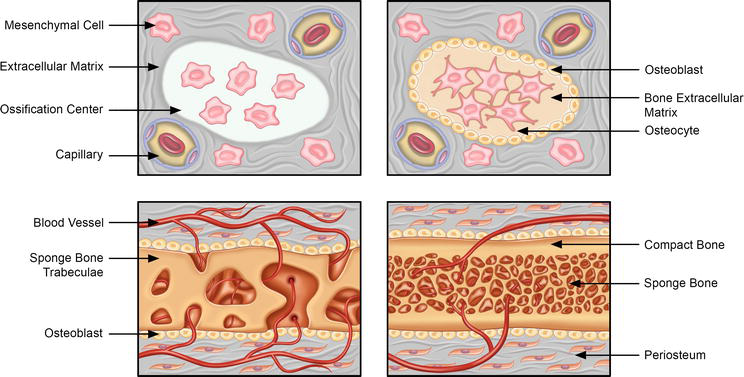
Figure 1.
Intramembranous ossification. The stages of intramembranous ossification include: (i) undifferentiated mesenchymal cells cluster and differentiation into osteoblasts. The ossification centers also emerge wherein the osteoblasts secrete osteoid, containing collagen precursors and other proteins, and entrapping the osteoblasts, which then transform into osteocytes. The clusters of osteoid are invaded by the blood vessels to form the trabecular matrix, whereas osteoblasts on the surface of the newly formed spongy bone form the periosteum. The periosteum then leads to the development of the compact bone superficial to the spongy trabecular bone.
Mesenchymal cells originating from the neural crest undergo a series of biochemical and cellular events, including proliferation and condensation, leading to the formation of compact nodules (Figure 2). Within these nodules, the cells undergo differentiation toward either capillaries or osteoblasts—specialized bone precursor cells responsible for generating collagen and proteoglycan matrix, crucial for calcium binding [9]. Through this process, the osteoid matrix, an initial form of bone, is generated. Over time, this matrix becomes calcified, resulting in the formation of primitive bone. While actively producing the osteoid matrix, osteoblasts maintain proximity to the calcification site. As they become embedded within the calcified matrix, osteoblasts undergo further differentiation and transform into osteocytes. Meanwhile, mesenchymal cells aggregate around the developing calcified bone matrix, forming the periosteum—an outer layer surrounding the bone [10]. The periosteum contributes to bone growth by generating additional osteoblasts and producing osteoid matrix, positioned adjacent to the bone’s spicules.
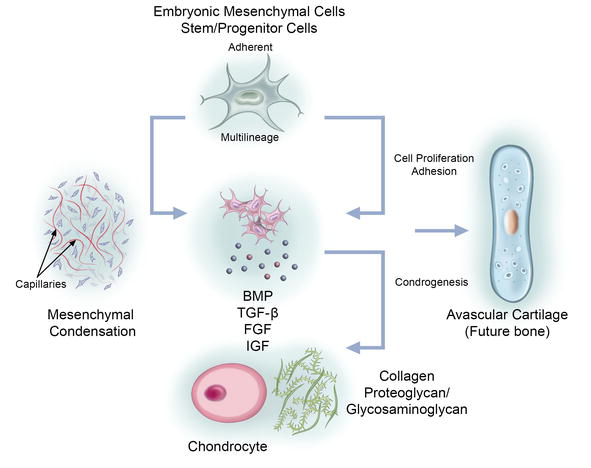
Figure 2.
Embryonic bone development. Multilineage embryonic stem/progenitor cells aggregate into chondrogenic clusters driven by cellular processes including mesenchymal condensation, proliferation, and adhesion. These events are driven by growth factors such as bone morphogenetic proteins (BMP), transforming growth factor-β (TGF-β), fibroblast growth factors (FGF), and insulin-like growth factor (IGF). The developing chondrocytes secrete or modify matrix proteins including collagen, proteoglycans or glycosaminoglycans to initiate avascular cartilage formation, forming a template for future bone development.
2.2 Endochondral ossification
Endochondral ossification is a highly intricate process involving the differentiation of mesenchymal cells into chondrocytes, leading to the formation of cartilage that is subsequently replaced by bone (Figure 3). The initiation of this process is mediated by paracrine signaling from neighboring mesodermal cells, which induces the expression of key transcription factors such as Scleraxis (SCX) and Pax 1 in mesenchymal cells [11, 12]. These transcription factors play a crucial role in driving the differentiation of mesenchymal cells into chondrocytes, initiating the formation of cartilage. Notably, SCX is particularly important during the early stages of cartilaginous condensation and any mutations in this gene can result in skeletal deformities [11]. The condensation and maintenance of chondrocytes is facilitated by the involvement of N-cadherin and N-CAM [13], which play significant roles in initiating and sustaining the cellular condensation process. During the proliferative and condensation stage, chondrocytes undergo a remarkable transformation, transitioning from actively dividing cells to hypertrophic chondrocytes. As they undergo hypertrophy, these chondrocytes undergo profound changes in the composition of extracellular matrix (ECM). They modify ECM by incorporating additional fibronectin and collagen X [14], which are crucial for enabling the subsequent mineralization of the cartilage matrix through the deposition of calcium carbonate. The lifecycle of hypertrophic chondrocytes culminates in apoptosis, creating space for the invasion of blood vessels and the establishment of bone marrow [15].
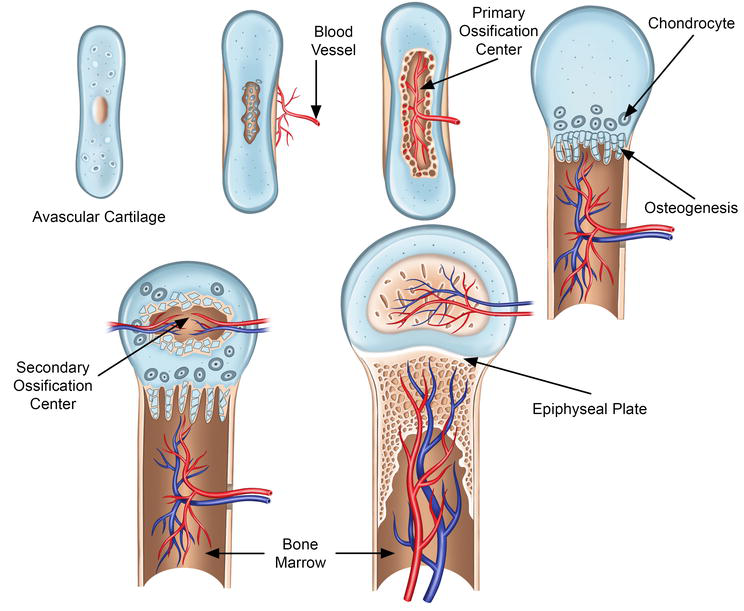
Figure 3.
Endochondral ossification. Endochondral ossification is marked by the formation of cartilage, which in later stages is replaced by bone. Bone formation process is initiated in the primary ossification center by the differentiation of mesenchymal cells into chondrocytes. The primary ossification center leads to the development of the diaphysis of the long bones, and the secondary ossification center leads to the development of the epiphysis. During late differentiation, the chondrocytes undergo apoptosis, forming a mineralized scaffold, which is invaded by the osteoblasts (brought in by the supplying blood vessels). This results in osteogenesis and lengthening of the bone along with the formation of the bone marrow cavity. Endochondral ossification occurs at the growth plate, which is responsible for longitudinal growth; it is also vital for the elongation of the long bones.
Simultaneously, surrounding cells undergo differentiation into osteoblasts, which are responsible for synthesizing and depositing the bone matrix onto the remnants of degraded cartilage. This gradual replacement of cartilage by bone matrix ultimately leads to the complete substitution of the original cartilaginous template. In the context of long bones, endochondral ossification occurs in a directional manner, spreading outward from the center of the bone. At the ends of long bones, specialized regions known as epiphyseal growth plates play a pivotal role in enabling continued bone growth [16]. These growth plates consist of distinct regions, including a zone of chondrocyte proliferation, a zone of mature chondrocytes, and a zone of hypertrophic chondrocytes. Chondrocytes within the growth plates undergo a carefully orchestrated series of events involving proliferation, maturation, and hypertrophy, contributing to the expansion of the bone structure. As long as the epiphyseal growth plates retain their capacity to produce chondrocytes, the process of bone growth persists, allowing for the longitudinal growth of bones.
3. Microenvironment
3.1 Collagen
The precise arrangement and composition of collagen types play a fundamental role in the intricate process of bone development (Figure 2). During embryonic bone formation, two main mechanisms are involved: endochondral ossification and intramembranous ossification, originating from distinct cell sources such as the paraxial and lateral plate mesoderm or neural crest. Collagen I is the predominant type found in mature bone, while Collagen II is predominantly present in developing cartilage [17]. In endochondral ossification, bone tissue replaces a pre-existing cartilaginous scaffold, whereas intramembranous ossification occurs directly from mesenchymal cells [18]. Notably, during the initiation phase and subsequent differentiation into osteoblasts, Collagen II and Collagen IX mRNA expression is observed in osteoblasts of mice and chicks [19, 20]. Furthermore, Collagen X is expressed during the terminal differentiation of chondrocytes, serving as a hallmark of hypertrophy in endochondral ossification.
In the case of intramembranous ossification, Collagen I is a primary collagen type involved [18]. Collagen X production primarily occurs in hypertrophic chondrocytes within the region of the endochondral growth plate dedicated to matrix mineralization. Additionally, Collagen X expression is observed at the edges of epiphyseal cartilage during fracture repair, osteoarthritis, and within the intervertebral disc [21]. As the cartilaginous extracellular matrix (ECM) rich in Collagen X is replaced by the collagenous ECM abundant in Collagen I, a crucial transition from cartilage to bone occurs. Collagen XIII, another collagen type, influences bone formation and potentially plays a role in connecting the regulation of bone mass to mechanical use [22]. Collagen XXIV, in turn, acts as a marker for osteoblast differentiation and bone formation. Collagen XXVII is predominantly found in cartilage even in adulthood [23]. It is associated with cartilage calcification and is believed to contribute to the conversion of cartilage to bone during skeletogenesis. This involvement in skeletogenesis has been established in zebrafish, where Collagen XXVII is essential for vertebral mineralization and postembryonic axial growth [24].
Matrix metalloproteinases (MMPs) are a group of zinc-dependent endopeptidases classified within the metzincin superfamily. These enzymes play a vital role in the degradation of the extracellular matrix (ECM) and collectively possess the ability to degrade various ECM proteins [17]. MMPs are involved in both physiological processes, such as development and tissue repair, and pathological conditions, including tumorigenesis and metastasis. Particularly, they are the primary enzymes responsible for collagen degradation [17]. Understanding the roles and interactions of these collagen types and MMPs provides valuable insights into the intricate processes involved in bone development and maintenance.
3.2 Proteoglycan
Proteoglycans, situated within the extracellular matrix (ECM) among collagen fibrils, play a significant role by providing a high negative charge, which generates osmotic pressure and attractive forces for cations like calcium [25]. Furthermore, glycosaminoglycans, particularly heparan sulfates, facilitate the binding of growth factors (Figure 2). The ECM consists of approximately three dozen proteoglycans that contribute to the filling and lubrication of the ECM space. In adult bone, biglycan and decorin promote the formation of bone and collagen fibrils, while keratocan enhances the rate of mineral deposition [9]. During embryonic ossification, proteoglycans also play crucial roles in osteoblast proliferation and differentiation. Poole et al. [26] discovered that proteoglycans become encapsulated within calcified cartilage during mineralization, providing a scaffold for osteoid and bone formation. Fisher et al. [27] identified a small proteoglycan localized to developing bone trabeculae and dentin, as well as osteoblasts and osteoprogenitor cells adjacent to areas undergoing rapid osteogenesis. Proteoglycan desulfation, as identified by Settembre et al. [28], serves as a critical regulator of chondrogenesis preceding endochondral bone formation. Simonet et al. [29] identified osteoprotegerin, a novel secreted glycoprotein acting as an anti-osteoclastogenic decoy receptor, aiding in bone formation by reducing bone resorption.
Perlecan, a ubiquitous and multifunctional molecule, is upregulated in hypertrophic chondrocytes that establish the primary and secondary ossification centers. Functioning as a heparan sulfate proteoglycan (HS) or HS/chondroitin sulfate hybrid, perlecan acts as a pressure sensor in bone development and remodeling [30]. Depletion of chondroitin sulfate results in irregular deposition and aggregation of collagen fibers, impairing connective tissue organization and impeding intramembranous ossification [31]. Matrix metalloproteinases (MMPs) and A disintegrin and metalloproteinase with thrombospondin motifs (ADAMTSs) are involved in the degradation of proteoglycans such as aggrecan, versican, and brevican. Heparan sulfate proteoglycan binds to various growth factors and facilitates ligand-receptor interactions, while betaglycan (TGF-β type III receptor), an integral membrane proteoglycan, binds to TGF-β and presents it to the core type II receptor [32]. Decorin, a small leucine-rich proteoglycan, provides a physical linkage that enhances the adhesion and assembly of aggrecan [33]. In the process of endochondral ossification, chondrocytes synthesize proteoglycans such as aggrecan and decorin, which are then deposited into the ECM along with collagen type II, forming a cartilaginous template for bone formation. These proteoglycans regulate chondrocyte proliferation, differentiation, and ECM mineralization. In summary, proteoglycans and related molecules play essential roles in embryonic ossification, including osteoblast proliferation and differentiation, and are crucial for bone development and remodeling throughout life.
4. Osteoinductive signals
4.1 Bone morphogenetic proteins
Bone morphogenetic proteins (BMPs) are the signaling molecules or growth factors belonging to the transforming growth factor-β (TGF-β) superfamily of proteins. They act as morphogens inducing embryogenesis and development (such as cardiogenesis, somite formation, somatic chondrogenesis, eye formation, digit apoptosis, neurogenesis, and musculoskeletal development). Thus, BMPs play a significant role in bone and cartilage formation (Figure 4). They are also multi-functional and are continuously expressed in adulthood to regulate the maintenance of tissue homeostasis (such as joint integrity, vascular remodeling, intramembranous and endochondral ossification) [34, 35]. BMPs occur in different subtypes and play critical roles in bone development. BMP-2 is crucial for chondrocyte proliferation and maturation during endochondral bone development; it is essential for appropriate osteogenesis, chondrogenesis, and adipogenesis. BMP-2 also promotes angiogenesis and is essential for initiating fracture healing [34, 35, 36]. It can also stimulate the differentiation of multipotent fibroblastic C3H10T1/2 cells into osteoblasts, chondrocytes, and adipocytes [37]. BMP-2 also enhances the expression of osteogenic markers including alkaline phosphatase, osteocalcin, and osteopontin [36]. BMP-4 coordinates the formation of the apical ectodermal ridge and digit patterning. BMP-5 mutant mice have shorter and weaker bones whereas BMP-6 mutant mice show delayed sternal ossification and shorter bones (specifically the long bones). BMP-7 and BMP-11 have a significant role in skeletal patterning during development. BMP-12, BMP-13, and BMP-14 are also crucial in the formation of bones, joints, tendons, and ligaments. BMP-12 is important in maintaining the structural integrity of the bone, and BMP-14 regulates bone and joint formation during digit development. In contrast to other BMPs, BMP-3 acts as a negative regulator of bone density. Along with bone formation, BMPs also regulate the development of cartilage [34].
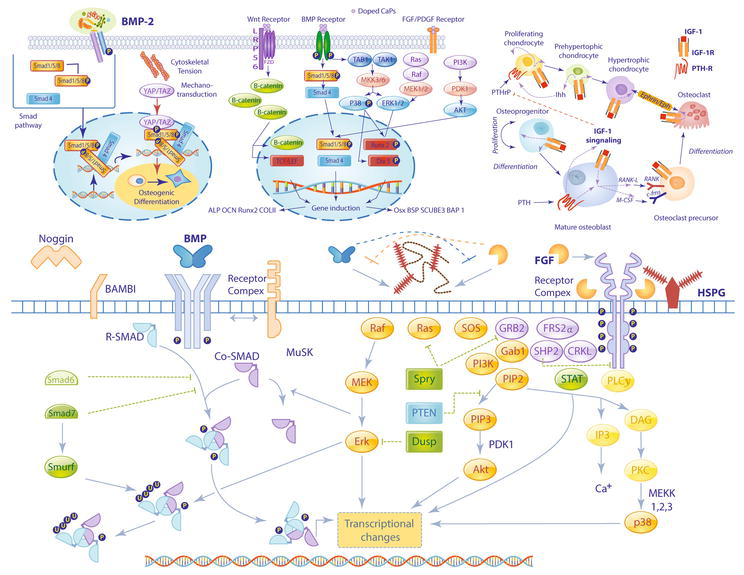
Figure 4.
Growth factors for bone formation. Growth factors and other signaling factors act on osteogenic precursors to drive bone morphogenesis and repair. Bone morphogenetic proteins (BMPs) play critical roles in chondrocyte proliferation and maturation and are essential to the formation of bones, joints, ligaments, and tendons. BMPs bind to cell-surface receptors driving intracellular signaling processes in tandem with SMAD and Wnt signaling pathways to induce transcription of osteogenic genes. Fibroblast growth factors (FGFs) and their receptors (FGFRs) are expressed in specific spatiotemporal patterns during development. The FGF-FGFR complex, in coordination with heparin sulfate proteoglycans at the cell surface, act via intracellular tyrosine kinase signaling to influence osteoblast proliferation, differentiation, and survival. Other anabolic factors, such as growth hormone (GH) and insulin-like growth factor (IGF), act as potent mitogens and differentiation signals which promote bone ossification during early development and bone maintenance and remodeling throughout growth, maturation, and aging processes.
During the formation of a new bone, mesenchymal stem cells (MSCs) differentiate into osteoblasts (under the influence of BMP-2) and secrete the organic matrix of the bone. Some osteoblasts get embedded in the bone (as osteocytes), leading to the development of the bone structure. The BMP signal transduction occurs via type I and II BMP receptors and Smad1, 5, and 8 proteins [situated downstream to the BMP receptors (BMPR)]. BMP-2 is secreted by pre-existing osteocytes, osteoblasts, and endothelial cells into the bone matrix or bloodstream. Upon secretion, BMP-2 binds with type I or type II serine/threonine kinase receptors present on the MSCs, activating either canonical (Smad) or non-canonical (non-Smad) signaling pathways that lead to bone, cartilage, and fat development. Upon phosphorylation, Smad1, 5, and 8 molecules get complexed with Smad4. This complex is then translocated into the nucleus, activating the osteogenic genes such as Runt-Related Transcription Factor 2 (RUNX2) and Osterix (Osx) in osteoblasts [35, 38, 39]. Alternative non-canonical (non-Smad) signaling via BMP/BMPR interaction may be mediated through the activation of p38, or c-Jun N-terminal kinases via mitogen-activated protein kinase (MAPK), or extracellular signal-related kinase (ERK), phosphatidylinositol 3-kinase (PI3K), and the transforming growth factor-β-activated kinase 1/binding protein 1 (TAB1/TAK1) pathways. All of these pathways result in an increased differentiation of MSCs into osteoblasts [35, 39]. BMPs also show tremendous potential for their use as therapeutic agents to prevent osteoporosis, heal bone fractures, treat periodontal bone defects, and heal patients requiring bone reconstruction [40]. Studies show that subcutaneous injection of BMP-2 can stimulate the formation of ectopic bone nodules, and its expression in fibroblasts may help to repair acute bone defects in the calvaria [41]. Recombinant human BMP-2 has been approved by the Food and Drug Administration (FDA) for clinical use and is available as therapeutic in lumbar spinal fusions (within tapered or cylindrical interbody cages), but the use is limited to due cost and concerns for adverse reactions. Research suggests that developing novel therapeutics targeting BMP-signaling pathways may have clinical implications in the treatment of various disorders (osteoporosis, cardiovascular disease, cancer, etc.) without any adverse side effects [35].
4.2 Fibroblast growth factors
Fibroblast growth factors (FGFs) are a family of 18 signaling molecules known for their role in many developmental processes including skeletal development (Figure 4) [42, 43]. As a family, the FGFs have a homologous core region flanked by variations in the amino (N-) and carboxyl (C-) terminal tails which provide the structural basis for varying biological effects which are directed by activation of FGF receptors (FGFRs) in paracrine or endocrine mechanisms [44]. Most of the FGFs are expressed in specific spatiotemporal patterns in the developing embryo where diffusion and FGFR activation is regulated by the presence of heparan sulfate proteoglycans [45]. FGFR1 and FGFR2 are expressed in undifferentiated distal limb bud mesenchyme where they are activated by FGFs produced by the apical ectodermal ridge to induce promixo-distal outgrowth of the limb bud [46].
In endochondral mesenchymal condensation, FGFR3 expression is first activated as the condensed mesenchyme differentiates into chondrocytes, then decreased as central chondrocytes begin hypertrophy [47]. FGFR2 expression is decreased with chondrocyte differentiation while FGFR1 expression increases as chondrocytes mature and hypertrophy [47]. In trabecular bone, mesenchymal progenitors express FGFR1, differentiating osteoblasts express FGFR2, while chondrocyte progenitors express FGFR3 [48]. Early stages of intramembranous bone formation are characterized by expression of FGF2, FGF4, FGF9, FGF18, FGFR1, FGFR2, and FGFR3 [49]. The interaction of FGF18 with FGFR2 regulates early intramembranous ossification [50], while at later stages FGF18 and FGF9 are expressed in differentiating osteoblasts [49, 51]. Osteoblast proliferation, differentiation, and survival are dependent on FGF9 and FGF18 interactions with heparin sulfate proteoglycans, FGFR1, and FGFR2 through downstream tyrosine kinase signaling. Mutations in FGFRs1-3 have been identified in craniosynostosis, while mutations of FGFR3 are implicated in achondroplasia or hypochondroplasia (skeletal dwarfism), and skeletal overgrowth and scoliosis syndromes are associated with inactivation of FGFR3 [52, 53]. Mutations in FGFR2 affect the appendicular skeleton and are implicated in bent bone dysplasia, as well as premature cranial ossification due to accelerated maturation of osteoblasts [54].
4.3 Insulin-like growth factor
Growth Hormone (GH) and Insulin-like growth factors (IGF-1 and IGF-2) are the anabolic hormones that play an important role as potent mitogens and differentiation factors; consequently, promoting bone ossification (during early development), and bone remodeling or maintenance (throughout growth and aging) (Figure 4) [55, 56]. The mode of action of GH and IGF-1 occurs either in an endocrine, autocrine, or paracrine manner. GH controls the IGF-1 serum concentrations via its action in the liver, which contributes 75% of serum IGF-1. Elevated serum IGF-1 concentration further regulates GH concentration via the negative feedback control mechanism. IGF-1 and IGF-2 are the most abundant growth factors secreted by osteoblasts, and stored in bone. Serum GH and IGF-1 concentrations rise and peak during postnatal growth and puberty (phases witnessing peak bone mass accrual) [56, 57]. GH and IGF-1 predominantly promote the proliferation of osteoprogenitors and mesenchymal stem cells and stimulates their differentiation into osteoblasts and chondrocytes by inhibiting lipogenic genes, activating the PI3K/PDK-1/Akt pathway, and increasing Wnt (Wingless and INT-1) signaling [55, 57]. In addition to promoting osteoblastogenesis, IGF-1 also decreases osteoblast apoptosis by stabilizing β-catenin; thus, increasing Wnt-dependent activity. The Wnt proteins have a significant impact on embryonic development as well as postembryonic tissue homeostasis [55].
GH and IGF-1 also stimulate the expression of bone morphogenetic proteins that function in skeletal patterning. A signaling cascade is stimulated via IGF-1 binding and activation of the IGF-1 receptor present on mesenchymal stem cells that eventually elevates
4.4 Platelet-derived growth factor
Platelet-derived growth factors (PDGFs) are potent angiogenesis factors as well as the mitogenic, chemo-attractive, and vascular docking agents that regulate processes such as embryonic development and tissue regeneration (Figure 4) [61, 62, 63]. They are expressed by platelets, osteoblasts, macrophages, and fibroblasts. PDGFs also function as osteoanabolics and enhance bone regeneration. They are secreted in various isoforms (AA, BB, AB, CC, and DD) that mediate their action via two distinct dimerized receptors [PDGFRs (α and β): members of receptor tyrosine kinases]. PDGF-AA predominantly activates PDGFR-α whereas PDGF-BB predominantly activates PDGFR-β [64]. Among all isoforms, PDGF-BB is considered the most common PDGF due to its varied physiological roles attributed to its ability to bind with all receptor isotypes [62, 63]. PDGF-BB is actively involved in angiogenesis, osteogenesis, and mesengenesis. It functions as a significant connector of these pathways and has the potential to be utilized as a potent therapeutic agent for bone regeneration and repair [63].
The formation of bone involves the formation of microvasculature. The rapidly dividing endothelial cells (present in the microvasculature) stimulate angiogenesis and secrete PDGF-BB that mediates its signal via PDGFR-β interaction, recruiting pericyte precursors into the region of new vessel formation. Pericytes are the perivascular or mural cells of mesenchymal origin. A majority of pericytes are mesenchymal stem cells (MSCs) having osteogenic potential. The PDGF-BB/PDGFR-β signaling pathway also regulates pericyte attachment on the vasculature, their maturation, destabilization, and detachment [62, 63].
PDGF-BB also stimulates vascular endothelial growth factor (an angiogenic factor) expression by pericytes, further promoting angiogenesis. PDGF-BB thus mediates the release of pericytes and MSCs from their abluminal sites in the active angiogenic locations; consequently, giving rise to free MSCs. These local or free MSCs divide rapidly under the influence of PDGF-BB; thus, elevating the pool of osteochondral precursors. In the presence of osteogenic factors such as Wnt signaling and BMPs, these osteochondral progenitors further differentiate into osteoblasts; this signaling process is modulated by PDGF-BB. Furthermore, in the presence of PDGF-BB, the remaining non-differentiated MSCs are repositioned in the perivascular space, stabilizing the newly formed blood vessels and leading to efficient bone formation (during embryonic bone formation and bone injury/fracture repair). In summary, PDGF-BB mediates the production of osteoprogenitor cells at a specific site, induces their multiplication, modifies their responsiveness to osteogenic factors, and assures the structural stability of the newly formed blood vessels [62, 63].
The isoform PDGF-AA is primarily produced and secreted by epithelial cells. It mediates its action on mesoderm-derived cells (via PDGFR-α), and stimulates mesenchyme expansion and angiogenesis. In contrast to PDGF-BB, PDGF-AA can stimulate MSC osteogenic differentiation by the BMP-Smad1/5/8-Runx2/Osx signaling pathway. The activation of BMP-Smad1/5/8 signaling by PDGF-AA occurs by down-regulating PDGFR-α (feedback control), which allows the free BMPR-I to get complexed with BMPR-II. The BMPR-I-BMPR-II complex further activates smad1/5/8 using BMP molecules, thereby promoting MSC differentiation and migration [64].
The isoform PDGF-BB has also been recognized as a significant paracrine factor involved in early bone healing. A study conducted on osteoblast-like cell-cultures also proposed the potential use of a combination of PDGF-BB and synthetic peptides (AC-100, p-15, TP508) for accelerating bone healing [65]. Another study conducted on animal models and periosteal cell cultures showed that PDGF-BB/PDGFR-β signaling inhibits BMP-2-induced osteogenesis (by attenuating Smad1/5/8 phosphorylation) [62]. Both PDGF-BB and BMP-2 are anabolic agents; however, when used in combination, they inhibit osteogenesis, suggesting a sequential use of both growth factors for better bone healing [62]. Contrary to PDGF-BB, there exists a link between PDGF-AA and BMP pathways that stimulates MSC osteogenic differentiation and migration [64]. These findings suggest the potential clinical implications of PDGF-AA along with BMP.
5. Bone growth and maturation
During fetal development, mineralization of the skeleton is a critical process, facilitated by the active involvement of the placenta in transporting essential minerals like calcium, magnesium, and phosphorus from the maternal circulation [66]. By the 8th week of gestation, the human fetus establishes a complete cartilaginous framework, and primary ossification centers begin to form between the 8th and 12th weeks, primarily in vertebrae and long bones. However, it is during the third trimester when the majority of mineralization occurs, with approximately 80% of the ash weight and mineral content being accreted [67]. Additional secondary ossification centers develop in the femur around the 34th week. The differentiation of osteoblasts into osteocytes occurs as they become embedded within the extracellular matrix, while other osteoblasts align along the bone surface, increasing in size. The interconnected trabeculae gradually form woven bone as growth progresses [66]. The primary center of ossification serves as the focal point for bone growth, and osteons, the fundamental units of compact bone, play a significant role in bone structure. Osteoblasts establish interconnections through cytoplasmic processes, which transform into the canaliculi of osteons. The transformation to compact bone involves the reduction of the perivascular space due to bone spicule formation around blood vessels. Consequently, the blood vessel assumes the role of the central canal within the osteon [68]. Adequate mineral delivery is crucial for normal skeletal development and mineralization in the fetus and neonate. The rate of mineral accretion intensifies significantly, with a notable surge from around 60 mg per day at week 25 to over 300 mg per day from the 35th through the 38th weeks. This acceleration slightly diminishes in the final 2 weeks before birth [67]. The turnover of the skeleton helps maintain a robust calcium level in the fetal circulation, as evident in cases of severe maternal hypocalcemia resulting from hypoparathyroidism, where skeletal resorption increases in response [17, 69].
Mineral and bone metabolism operate differently during the fetal development compared to adulthood [67]. The placenta plays a crucial role in providing essential minerals from the maternal circulation to the fetus, resulting in higher concentrations of these minerals in the fetal bloodstream for proper skeletal development. Hormones like parathyroid hormone (PTH) and PTH-related protein are crucial in regulating fetal bone development and serum mineral levels, while factors like vitamin D/calcitriol, fibroblast growth factor-23, calcitonin, and sex steroids have minimal impact [69, 70]. In the neonatal phase, there is a transition in the regulation of mineral homeostasis. Initially, serum calcium levels decrease while phosphorus levels rise, gradually reaching adult values within 24–48 hours [67, 70]. During this phase, the neonate’s intestines become the primary source of minerals, kidneys reabsorb minerals, and bone turnover contributes to mineral supply. This transition is triggered by the loss of the placenta and a decline in serum calcium levels after birth. Subsequently, there is an increase in PTH secretion, followed by an elevation in calcitriol levels [67, 70]. Intestinal calcium absorption shifts from a passive process facilitated by lactose to an active and calcitriol-dependent process. However, increasing dietary calcium intake or administering calcium through parenteral routes can bypass the role of calcitriol in regulating mineral and bone metabolism [71].
Endochondral ossification is a fundamental process responsible for the development of the majority of bones in the skeletal system, known as endochondral bones. It involves several key stages that span over several weeks. Initially, the bones take the form of hyaline cartilage models. The process begins with the formation of a bone collar around the middle of the cartilage model, followed by the degeneration of the underlying cartilage. Concurrently, capillaries and osteoprogenitor cells invade the resulting ossification center from the periosteum. Osteoblasts subsequently deposit osteoid, which undergoes calcification, leading to the formation of woven bone. This woven bone is later remodeled into compact bone. Osteoblasts secrete bone matrix, while osteoclasts in the ossification center remove a portion of the newly formed bone, contributing to the creation of the bone marrow cavity [72, 73].
The primary ossification center initially forms in the diaphysis, the middle region of each developing bone. Subsequently, secondary ossification centers develop in the epiphyses through a similar process. Following ossification, the primary and secondary ossification centers are separated by a segment of cartilage called the epiphyseal growth plate, which remains between the epiphysis and diaphysis, enabling continued bone elongation. Ossification of the growth plate persists until around the age of 20 years. The growth plate facilitates bone lengthening by sustaining the growth of cartilage. The two ossification centers eventually merge when the epiphyseal plate disappears, which typically signifies the attainment of full stature [74]. The epiphyseal plates exhibit lower biomechanical properties compared to adjacent mature bone, ligaments, and tendons. A fracture in the growth plate can occur in children from a similar mechanical insult that would result in a join sprain in adults. Growth plate fractures impact the layer of developing tissue near the ends of a child’s bones, with higher prevalence observed in the fingers, forearm, and lower leg. The majority of growth plate fractures heal successfully without negatively impacting future bone growth. However, in some cases, the fracture may cause alterations in the growth plate, leading to potential complications later on. These complications can include mild misalignment of the bone, resulting in slight deviation or marginal differences in length compared to the expected outcome. Growth plate fractures are significantly common among children, accounting for approximately 15–30% of all fractures in this age group. Moreover, they occur twice as frequently in boys compared to girls.
6. Aging bone changes
Skeletal mass increases with growth and maturational changes in bone density and dimensions. The most rapid period of bone growth occurs during adolescence with skeletal mass doubling during the pubertal growth spurt. Males showing greater bone strength with increased volume, trabecular number and thickness than females from mid-puberty onward due to the action of testosterone on periosteal apposition [75, 76]. Peak total bone mineral content and total bone mineral density is 22 years for females and 23–26 years for males [77]. The amount of bone present in the skeleton at the end of the maturation process, known as peak bone mass, is a key indicator of bone mass in the elderly [78, 79]. Normal bone homeostasis, a delicate balance of bone formation and resorption is disrupted with aging. Declines in bone mineral density are observed before midlife in trabecular and cortical bone in women but several decades later in men [80, 81]. Noninvasive imaging studies demonstrate greater loss of cortical bone and increases in cortical bone porosity compared with trabecular bone with advancing age [82, 83]. Bone loss in both sexes is driven by declines in estrogen and other steroid hormones. Women experience a period of marked cancellous bone loss with the onset of menopause, followed by a slower phase of loss that continues through the lifespan [84]. Estrogen loss is thought to increase osteoclast recruitment and activation and decrease osteoclast apoptosis, creating a shift toward increased bone resorption [85]. Cellular senescence, a factor in reduced osteoblast proliferation and differentiation, may contribute to a shift in production of osteogenic progenitors in the bone marrow, further exacerbating the shift toward bone resorption in the aged population [86, 87].
7. Factors affecting bone health
Bone health is determined by many factors including genetics, gender, nutrition, hormonal status, bodyweight, age, exercise, metabolic diseases such as diabetes, and lifestyle choices including smoking and alcohol consumption. From birth to adulthood, and perhaps most importantly in the aged, bone health is essential to well-being and quality of life. Bone mineral density is a key factor linked with healthy, normal bones in adulthood. Low bone mineral density is associated with reduced strength of bones and conditions such as fractures and osteoporosis. In contrast, abnormal and excessive growth of new bone tissue on the other end of the spectrum may be just as debilitating. Heterotopic ossification, the formation of extra-skeletal bone in muscle and soft tissues, or Paget’s disease characterized by large bones with irregular structure prone to fractures, bowing and deformities are examples of bone overgrowth conditions.
7.1 Smoking
While genetics account for 50–90% of bone mineral density variation in humans [88, 89], many lifestyle factors are controllable and exert a strong influence on bone health. Cigarette smoking is one of the most prevalent and preventable risk factors for osteoporosis and bone fractures and has significant adverse effects on bone healing [90, 91]. Smoking is associated with lower bone mineral density in both men and women over age 50, increased risk of fractures [92, 93], and slower rates of bone repair [94]. The mechanisms by which smoking affects bone health are poorly understood but may be related to changes in hormone status (estrogen, adrenal cortical hormones), impaired calcium absorption, reduced vitamin D availability, impaired vascularization, and/or increased free radical production [95].
7.2 Alcohol consumption
Another widespread and avoidable risk factor with negative consequences for bone health is alcohol consumption [96, 97, 98], although many epidemiological studies find minimal effects on fracture risk or bone mineral density with moderate alcohol use [99, 100, 101]. Chronic alcohol abuse or heavy binge-drinking is associated with an increased incidence of fractures from falls and delays in fracture healing [97, 102]. In rodent models, chronic alcohol administration reduced serum concentrations of the active form of vitamin D3 [103]. This finding was mitigated with vitamin D supplementation [104]. Loss of bone mineral density, trabecular volume and compressive strength were observed in a study of binge-alcohol treatment in rats [105] as well as chronic alcohol consumption [106]. Further, excessive drinking is associated with an increase in bone resorption that disrupts normal bone homeostatic turnover and leads to increases in cortical bone porosity [107, 108]. Studies to date appear to implicate alterations in bone remodeling, via increase in osteoclast activity and inhibition of osteoblast differentiation as possible mechanisms through which alcohol influences bone health [109, 110].
7.3 Diabetes
The long-term exposure to metabolic changes related to diabetes has profound effects on bone metabolism and molecular alterations in bone structure. Type 1 diabetes mellitus (T1DM) patients experience reduced cortical bone size and inadequate peak bone mass most likely attributable to suppression of osteoblast differentiation and activity. Younger patients at the time of onset were observed with greater deficits in bone accrual, bone mineral density, and compressive bone strength in the radius and tibia [111, 112]. Hip fracture risk increases 5–8% in T1DM patients compared with non-diabetic controls, despite similar measures of bone mineral density in the lumbar spine [113]. The proposed mechanisms underlying these changes include insulin deficiency, accumulation of advanced glycation end products, reduced bone deposition and turnover, inflammation, and osteocyte dysfunction [114]. Patients with type 2 diabetes mellitus (T2DM) are frequently observed with normal or even increased bone mineral density but are predisposed to fragility fractures due to increased cortical porosity, smaller cortical area, and decreased bone material strength. Serum markers of bone turnover were reduced compared with non-diabetic controls, suggesting impairment of normal bone remodeling mechanisms [115]. Another hypothesis proposes that prolonged high circulating glucose concentrations lead to advanced glycation end-products in circulation and resulting in collagen crosslinks in bone. Like bisphosphonate therapy, prolonged low bone turnover and changes in the biomechanical properties in T2DM create a more brittle bone that contributes to increased fracture risk [115, 116].
7.4 Genetics
Senescent cells show genetic alterations like telomere shortening, impaired DNA repair and damage response [117]. The up-regulation of GATA4 by DNA damage response (DDR) stimulates NF-κB and senescence-associated secretory phenotype (SASP) production [118]. The bone microenvironment in aged cells like osteoblast precursors and osteocytes develops a SASP, tissue degeneration, and enhanced expression of senescence-associated markers like p16Ink4a, p21 and p53 [119]. The expression of cytokines like IL1 also activates SASP in osteocytes of older bones. Accumulation of Poly [ADP-ribose] polymerase 1 (PARP1) in senescent cells causes bone mineralization, vasodilation triggered by DDR, and excessive extracellular matrix calcification [120]. Ataxia-telangiectasia-mutated (ATM) gene encoding for a Ser/Thr kinase is involved in DNA repair of double-strand break and its inactivation causes short telomeres, decreased bone formation, and defective osteoblasts with enhanced bone resorption [121]. Xeroderma pigmentosum-type D (XPD) gene is found to be crucial for DNA repair and its alteration causes osteoporosis and kyphosis [122]. Mutation in autophagy-related 7 (ATG7) gene, a key component of autophagy can lead to bone loss. Mutations in the mtDNA polymerase gamma (Polg), the DNA polymerase in mitochondria causes osteoporosis with reduced osteogenesis and increased osteoclastogenesis [123].
7.5 Obesity
Obesity is defined at a body mass index ≥30 kg/m2 with excessive body fat and has increasingly been affecting global health. Specifically, obesity has been shown to result in higher blood loss during lumbar fusion procedures, longer hospital stay, greater complication rates, and worse functional outcomes compared to nonobese patients [124]. While various factors such as environment, metabolism, genetics, and behavior interplay in the onset and development of obesity, the imbalance between high food intake and low energy expenditure ultimately lead to the accumulation of excessive body fat. Proinflammatory factors such as TNF-a, IL1b, IL6, resistin, and leptin has been shown to upregulate in the adipose tissue, resulting in their release to the bloodstream and a systemic low-grade inflammatory state in other organs including the skeletal system [125]. In addition, hyperlipidemia decreases the osteogenic capability of bone precursors, potentially due to the disturbance in the balance between adipogenesis and osteogenesis toward adipogenesis [126]. Obese patients show a significantly lower lumbar fusion rate as well as a series of post-operative complications compared to nonobese patients [127]. As with patients with other risk factors as described above, a more advanced approach may be needed for successful bone repair and regeneration, which includes appropriate cells, microenvironment, and biological signals.
8. Bone regeneration technologies
Bone regeneration technologies and approaches aims at repairing or replacing damaged or lost bone tissue. These technologies can help restore the structure, function, and integrity of bones affected by trauma, disease, or congenital abnormalities. A comprehensive approach to an efficient bone repair and regeneration may include stem and progenitor cells (exogenous, endogenous), proper microenvironment (collagen, proteoglycan), and signals (growth factors) (Figure 5).
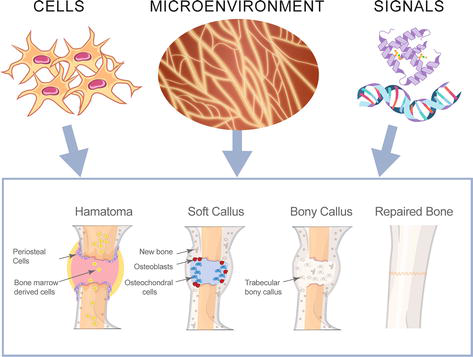
Figure 5.
Stages of bone repair and regeneration. 1. Hematoma formation: The broken blood vessels and hemorrhage at the break site result in the formation of a hematoma or clotted blood. 2. The soft callus formation: Phagocytic cells invade the hematoma, and clear the dead cells. Fibroblasts and osteoblasts also invade the region and initiate the bone reformation process. Fibroblasts synthesize collagen fibers whereas osteoblasts form the spongy bone. The resultant repaired tissue (between the broken bone ends) is known as the soft or fibrocartilaginous callus. 3. Bony callus formation: The soft callus is converted into a bony callus of spongy bone. This process is similar to the endochondral ossification of bone. 4. Repaired bone: The osteoblasts and osteoclasts then carry out bone remodeling to convert the bony callus to create bone tissue similar to the original bone. The entire bone repair process is influenced by the infiltration of the bone cells (osteoblast and osteoclast), the microenvironment of the bone, and the availability of external signals (mediated by growth factors and cytokines).
8.1 Cell-based approaches
Cell-based bone regeneration technologies involve the use of living cells to repair and regenerate damaged or lost bone tissue. These approaches aim to overcome the limitations of traditional bone grafting techniques by harnessing the regenerative potential of cells. Cell-based bone regeneration technologies utilize a variety of cells, including mesenchymal stem cells (MSCs) [128, 129], osteoprogenitors [130], induced pluripotent stem cells (iPSCs) [131, 132], endothelial progenitor cells (EPCs), and periosteal cells [133], to facilitate the regeneration and repair of bone tissue. MSCs, derived from sources like bone marrow or adipose tissue, can differentiate into bone-forming cells and secrete regenerative molecules. Stem cells are classically defined as cells that can self-renew and differentiate into multiple cellular lineages. Osteoprogenitors, found in the bone marrow, contribute to bone formation and repair. iPSCs, generated by reprogramming adult cells, can differentiate into osteoblasts. EPCs promote vascularization, while periosteal cells, obtained from the outer bone layer, possess regenerative potential and can differentiate into various bone-related cell types. Collectively, these cells offer promising approaches for advancing cell-based bone regeneration if challenges in delivery and retention of cells can be overcome.
However, it is important to note that a typical cell population cultured in a laboratory may contain different cell types, which can significantly confound the identification of “true” stem cells. For example, only about 7% of cells in a typical MSC culture possess the stem cell-like characteristics at a clonal level, suggesting 93% of cells in a typical MSC culture are not stem/progenitor cells [134]. Interestingly, there has been no stem cell-based treatment modality approved by the U.S. FDA for the repair and regeneration of damaged bone. While MSCs may need further identification and characterization to obtain a cell population that can provide a better clinical outcome, it is feasible to postulate that stem and progenitor cells may need a niche resembling the environment of early bone development. MSCs, for example, may not differentiate into functional bone without going through the developmental stages (intramembranous or endochondral ossification) through mesenchymal condensation in a properly formulated microenvironment as discussed above. This may be particularly true as MSCs have been known to be very responsive to their microenvironment [135].
8.2 Microenvironment-based technologies
Microenvironment-based bone regeneration technologies utilize a range of materials, including hydroxyapatite/calcium phosphate, polymers, ceramics, bioglasses, metals, and composites, to create an ideal environment for bone regeneration. Hydroxyapatite and calcium phosphate mimic the structure of natural bone mineral and support osteogenesis. Polymers like poly(lactic-co-glycolic acid) (PLGA) and polycaprolactone (PCL) provide biocompatible scaffolds that mimic the extracellular matrix and promote cell growth. Ceramics such as calcium phosphate ceramics offer excellent biocompatibility and act as bone substitutes. Bioglasses stimulate bone regeneration by releasing bioactive ions. Metals like titanium and its alloys provide mechanical support and can be modified to enhance bioactivity. Composites combine different materials to optimize properties. These microenvironment-based approaches, in the form of scaffolds, coatings, or fillers, aim to enhance bone regeneration and restore function in patients with bone defects or injuries.
In a therapeutic context, the condition of the implant site can have a significant impact on the fate of transplanted stem and progenitor cells [136]. Factors such as cytokines, growth factors, endogenous cells, enzymes, and mechanical stimuli play a role in maintaining the balance between anabolic and catabolic processes in a healthy intervertebral disc. However, a degenerating disc characterized by decreased proteoglycans and collagen II, increased proteinases and cytokines, and decreased pH can create an unfavorable microenvironment for stem cells used in therapy [136]. The success of interbody fusion surgeries, such as anterior cervical discectomy and fusion (ACDF) or lumbar spine fusion procedures, depends on a favorable microenvironment at the disc [135]. If fusion fails, patients may experience persistent or new pain, and the implanted devices may fail, necessitating additional surgeries. The microenvironment in posterolateral fusion in the lumbar spine can pose even more challenges to successful fusion [135]. Autologous bone grafting, where bone is harvested from a non-load bearing site like the iliac crest, is considered the gold standard for bone repair due to the presence of both stem cells and their native microenvironment [135]. However, this approach has limitations and risks, including donor site morbidity, fracture, infection, increased blood loss, prolonged operative time, and nerve damage. As a result, bone graft substitutes have gained popularity as alternatives to autografts. These substitutes, which include allografts, ceramics, polymers, and biologics, have shown potential in bone regeneration [135].
Corticocancellous allograft and demineralized bone matrix (DBM) are examples of allograft-based bone graft substitutes that have osteoconductive and osteoinductive properties [137]. DBM, in particular, provides a scaffold for bone stem and progenitor cells and has been shown to upregulate osteogenic genes and mineralization [137]. Mechanical stress, vascularity, and surface characteristics of graft materials can influence the microenvironment and stimuli experienced by mesenchymal stem cells (MSCs) and affect their osteogenic differentiation [138]. Ceramic-based substitutes like hydroxyapatite (HA) and β-tricalcium phosphate (β-TCP) have demonstrated varying degrees of ectopic bone formation and cell attachment [139]. Surface modifications and coating can improve the compatibility and performance of calcium phosphate ceramics [140]. Synthetic substitutes such as poly-l-lactic acid (PLLA), polyglycolic acid (PGA), and PLGA have shown compatibility with MSCs but may degrade too quickly to bridge critical-sized bone defects [141, 142]. Polysaccharide-based materials, including cellulose, alginate, chitosan, and glycosaminoglycans, have been investigated for bone regeneration and have shown promise as scaffolds for tissue engineering [143, 144, 145, 146]. Recent studies have highlighted the potential of carbohydrate-based polymers including the hyper-crosslinked carbohydrate polymer in enhancing bone formation (Figure 6) [147, 148]. While bone graft substitutes aim to create an conducive environment for bone regeneration, they may not be sufficient for high-risk patient populations, such as those with diabetes, who have shown lower rates of bone regeneration [149, 150]. Additional therapeutic approaches and considerations such as inclusion of growth factors and cell-based approaches are necessary to address the microenvironmental challenges in these cases.

Figure 6.
Carbohydrate-based microenvironment. The hyper-crosslinked carbohydrate polymer (HCCP) scaffold is characterized by high-density, interconnected pores with extraordinary surface area for cell adhesion and space for nutrient transfer (left panel, SEM image, scale bar = 200 μm). Mesenchymal stem cells (MSC) and CD34+ hematopoietic cells show exceptional adherence to HCCP. Biocompatibility is demonstrated via normal cellular growth, migration, and expansion patterns (left panel, SEM image, scale bar = 50 μm).
8.3 Signal-based technologies
Signals, such as growth factors, can also be used to stimulate bone regeneration. Growth factors are proteins that promote the growth, division, and differentiation of cells. Growth factors can be delivered to the site of injury using a variety of methods, including injections, gels, and membranes. Infuse™ Bone Graft (Medtronic, Minneapolis, MN) is a bone regeneration technology that utilizes the bone morphogenetic protein-2 (BMP-2) signal to stimulate bone healing. This technique involves implanting a collagen sponge soaked with recombinant human BMP-2 into the site of bone defect or fracture. The advantage of this approach includes enhanced bone healing, minimally invasive delivery, elimination of autograft harvesting, reduced pain and morbidity. However, there are also cons to consider, such as potential side effects like excessive bone growth and inflammation, concerns with off-label use, higher cost compared to traditional grafting, and the need for more long-term safety data. Careful evaluation by medical professionals is necessary to weigh the benefits and risks to ensure its appropriate use in clinical practice. Augment Bone Graft (Wright Medical Technology, Inc) employs recombinant human PDGF-BB with a β-tricalcium phosphate carrier to improve healing in ankle and foot fusion surgeries [151, 152]. Approved by the FDA since 2015, this product is also in use in dental and neuropathic ulceration applications. In a sheep model, Augment Bone Graft was comparable with autograft bone for promoting spinal fusion suggesting this combination may be a viable alternative in the clinical setting [153].
9. Conclusion
Bone development, growth, and maturation are complex processes dependent on progenitor cells, hormones, and growth factors. Under normal physiological conditions, peak bone mass is achieved in early adulthood. Thereafter, bone homeostasis is maintained through a balance of resorption and deposition that is influenced by many environmental factors including nutrition, age, disease status, drinking, and smoking, to name a few. Loss of bone mineral density begins about menopause in women, several decades earlier than men, but continues throughout the aging process in both sexes. Bone fragility in the elderly is most likely due to a physiological shift in favor of bone resorption activity creating less bone density, as well as reduced activity of the progenitor cell populations. Bone regeneration technologies are still in their early stages of development but have the potential to revolutionize the way we treat bone diseases and injuries. New technologies patterned using cues from embryonic bone development may provide patients with new and improved options for bone repair and regeneration.
Conflict of interest
Charles Lee is an inventor of the hyper-crosslinked carbohydrate polymer discussed in this chapter and founder of Molecular Matrix, Inc.
References
- 1.
Nauth A, Schemitsch E, Norris B, Nollin Z, Watson JT. Critical-size bone defects: Is there a consensus for diagnosis and treatment? Journal of Ortopaedic Trauma. 2018; 32 (Suppl. 1):S7-S11 - 2.
Long F, Ornitz DM. Development of the endochondral skeleton. Cold Springer Harbour Perspectives Biology. 2013; 5 (1):a008334 - 3.
Giffin JL, Gaitor D, Franz-Odendaal TA. The forgotten Skeletogenic condensations: A comparison of early skeletal development amongst vertebrates. Journal of Developmental Biology. 2019; 7 (1):4 - 4.
Hall BK, Tremaine R. Ability of neural crest cells from the embryonic chick to differentiate into cartilage before their migration away from the neural tube. The Anatomical Record. 1979; 194 (3):469-475 - 5.
Dunlop LL, Hall BK. Relationships between cellular condensation, preosteoblast formation and epithelial-mesenchymal interactions in initiation of osteogenesis. The International Journal of Developmental Biology. 1995; 39 (2):357-371 - 6.
Hall BK. Tissue interactions and the initiation of osteogenesis and chondrogenesis in the neural crest-derived mandibular skeleton of the embryonic mouse as seen in isolated murine tissues and in recombinations of murine and avian tissues. Journal of Embryology and Experimental Morphology. 1980; 58 :251-264 - 7.
Huang W, Yang S, Shao J, Li YP. Signaling and transcriptional regulation in osteoblast commitment and differentiation. Frontiers in Bioscience. 2007; 12 :3068-3092 - 8.
Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell. 1997; 89 (5):747-754 - 9.
Lin X, Patil S, Gao YG, Qian A. The bone extracellular matrix in bone formation and regeneration. Frontiers in Pharmacology. 2020; 11 :757 - 10.
Breeland G, Sinkler MA, Menezes RG. Embryology, Bone Ossification. Treasure Island, FL: StatPearls Publishing; 2023. Available from: http://www.ncbi.nlm.nih.gov/books/NBK539718/ - 11.
Alberton P, Popov C, Prägert M, Kohler J, Shukunami C, Schieker M, et al. Conversion of human bone marrow-derived mesenchymal stem cells into tendon progenitor cells by ectopic expression of scleraxis. Stem Cells and Development. 2012; 21 (6):846-858 - 12.
Wallin J, Wilting J, Koseki H, Fritsch R, Christ B, Balling R. The role of Pax-1 in axial skeleton development. Development. 1994; 120 (5):1109-1121 - 13.
Tavella S, Raffo P, Tacchetti C, Cancedda R, Castagnola P. N-CAM and N-cadherin expression during in vitro chondrogenesis. Experimental Cell Research. 1994; 215 (2):354-362 - 14.
Wilson R, Norris EL, Brachvogel B, Angelucci C, Zivkovic S, Gordon L, et al. Changes in the chondrocyte and extracellular matrix proteome during post-natal mouse cartilage development. Molecular & Cellular Proteomics. 2012; 11 (1):M111 - 15.
Yang L, Tsang KY, Tang HC, Chan D, Cheah KSE. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111 (33):12097-12102 - 16.
Karimian E, Chagin AS, Sävendahl L. Genetic regulation of the growth plate. Frontiers in Endocrinology (Lausanne). 2011; 2 :113 - 17.
Ricard-Blum S. The collagen family. Cold Spring Harbor Perspectives in Biology. 2011; 3 (1):a004978 - 18.
Galea GL, Zein MR, Allen S, Francis-West P. Making and shaping endochondral and intramembranous bones. Developmental Dynamics. 2021; 250 (3):414-449 - 19.
Nah HD, Pacifici M, Gerstenfeld LC, Adams SL, Kirsch T. Transient chondrogenic phase in the intramembranous pathway during normal skeletal development. Journal of Bone and Mineral Research. 2000; 15 (3):522-533 - 20.
Kulyk WM, Coelho CN, Kosher RA. Type IX collagen gene expression during limb cartilage differentiation. Matrix. 1991; 11 (4):282-288 - 21.
Kielty CM, Grant ME. The collagen family: Structure, assembly, and Organization in the Extracellular Matrix. In: Royce PM, Steinmann B, editors. Connective Tissue and its Heritable Disorders [Internet]. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2002. pp. 159-221. DOI: 10.1002/0471221929.ch2 - 22.
Ylönen R, Kyrönlahti T, Sund M, Ilves M, Lehenkari P, Tuukkanen J, et al. Type XIII collagen strongly affects bone formation in transgenic mice. Journal of Bone and Mineral Research. 2005; 20 (8):1381-1393 - 23.
Plumb DA, Dhir V, Mironov A, Ferrara L, Poulsom R, Kadler KE, et al. Collagen XXVII is developmentally regulated and forms thin fibrillar structures distinct from those of classical vertebrate fibrillar collagens. The Journal of Biological Chemistry. 2007; 282 (17):12791-12795 - 24.
Gaudreault P, Beland M, Girodias JB, Thivierge RL. Single daily doses of trimethoprim/sulphadiazine for three or 10 days in urinary tract infections. Acta Paediatrica. 1992; 81 (9):695-697 - 25.
Hynes RO, Naba A. Overview of the matrisome: An inventory of extracellular matrix constituents and functions. Cold Spring Harbor Perspectives in Biology. 2012; 1 (1):a004903 - 26.
Poole AR, Pidoux I, Rosenberg L. Role of proteoglycans in endochondral ossification: Immunofluorescent localization of link protein and proteoglycan monomer in bovine fetal epiphyseal growth plate. The Journal of Cell Biology. 1982; 92 (2):249-260 - 27.
Fisher LW, Termine JD, Dejter SW, Whitson SW, Yanagishita M, Kimura JH, et al. Proteoglycans of developing bone. The Journal of Biological Chemistry. 1983; 258 (10):6588-6594 - 28.
Settembre C, Arteaga-Solis E, McKee MD, de Pablo R, Al Awqati Q , Ballabio A, et al. Proteoglycan desulfation determines the efficiency of chondrocyte autophagy and the extent of FGF signaling during endochondral ossification. Genes & Development. 2008; 22 (19):2645-2650 - 29.
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lüthy R, et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell. 1997; 89 (2):309-319 - 30.
Hayes AJ, Farrugia BL, Biose IJ, Bix GJ, Melrose J. Perlecan, a multi-functional, cell-instructive, matrix-stabilizing proteoglycan with roles in tissue development has relevance to connective tissue repair and regeneration. Frontiers in Cell and Development Biology. 2022; 10 :856261 - 31.
Ida-Yonemochi H, Morita W, Sugiura N, Kawakami R, Morioka Y, Takeuchi Y, et al. Craniofacial abnormality with skeletal dysplasia in mice lacking chondroitin sulfate N-acetylgalactosaminyltransferase-1. Scientific Reports. 2018; 8 (1):17134 - 32.
Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Springer Harbor Perspective Biology. 2011; 3 (7):a004952 - 33.
Han B, Li Q , Wang C, Patel P, Adams SM, Doyran B, et al. Decorin regulates the Aggrecan network integrity and biomechanical functions of cartilage extracellular matrix. ACS Nano. 2019; 13 (10):11320-11333 - 34.
Wang RN, Green J, Wang Z, Deng Y, Qiao M, Peabody M, et al. Bone morphogenetic protein (BMP) signaling in development and human diseases. Genes Diseases. 2014; 1 (1):87-105 - 35.
Halloran D, Durbano HW, Nohe A. Bone morphogenetic Protein-2 in development and bone homeostasis. Journal of Developmental Biology. 2020; 8 (3):19 - 36.
Chen D, Harris MA, Rossini G, Dunstan CR, Dallas SL, Feng JQ , et al. Bone morphogenetic protein 2 (BMP-2) enhances BMP-3, BMP-4, and bone cell differentiation marker gene expression during the induction of mineralized bone matrix formation in cultures of fetal rat calvarial osteoblasts. Calcified Tissue International. 1997; 60 (3):283-290 - 37.
Date T, Doiguchi Y, Nobuta M, Shindo H. Bone morphogenetic protein-2 induces differentiation of multipotent C3H10T1/2 cells into osteoblasts, chondrocytes, and adipocytes in vivo and in vitro. Journal of Orthopaedic Science. 2004; 9 (5):503-508 - 38.
Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors. 2004; 22 (4):233-241 - 39.
Ingwersen LC, Frank M, Naujokat H, Loger K, Bader R, Jonitz-Heincke A. BMP-2 long-term stimulation of human pre-osteoblasts induces osteogenic differentiation and promotes Transdifferentiation and bone Remodeling processes. International Journal of Molecular Sciences. 2022; 23 (6):3077 - 40.
Sykaras N, Opperman LA. Bone morphogenetic proteins (BMPs): How do they function and what can they offer the clinician? Journal of Oral Science. 2003; 45 (2):57-73 - 41.
Chen G, Xu H, Yao Y, Xu T, Yuan M, Zhang X, et al. BMP Signaling in the development and regeneration of cranium bones and maintenance of Calvarial stem cells. Frontiers in Cell and Development Biology. 2020; 8 :135 - 42.
Ornitz DM, Marie PJ. Fibroblast growth factor signaling in skeletal development and disease. Genes & Development. 2015; 29 (14):1463-1486 - 43.
Beenken A, Mohammadi M. The FGF family: Biology, pathophysiology and therapy. Nature Reviews. Drug Discovery. 2009; 8 (3):235-253 - 44.
Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine & Growth Factor Reviews. 2005; 16 (2):107-137 - 45.
Wu ZL, Zhang L, Yabe T, Kuberan B, Beeler DL, Love A, et al. The involvement of heparan sulfate (HS) in FGF1/HS/FGFR1 signaling complex. The Journal of Biological Chemistry. 2003; 278 (19):17121-17129 - 46.
Kimura I, Pancho LR, Isoi Y, Kimura M. Diabetes-induced enhancement of prostanoid-stimulated contraction in mesenteric veins of mice. Japanese Journal of Pharmacology. 1989; 51 (3):403-410 - 47.
Peters KG, Werner S, Chen G, Williams LT. Two FGF receptor genes are differentially expressed in epithelial and mesenchymal tissues during limb formation and organogenesis in the mouse. Development. 1992; 114 (1):233-243 - 48.
Jacob AL, Smith C, Partanen J, Ornitz DM. Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Developmental Biology. 2006; 296 (2):315-328 - 49.
Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes & Development. 2002; 16 (12):1446-1465 - 50.
Eswarakumar VP, Monsonego-Ornan E, Pines M, Antonopoulou I, Morriss-Kay GM, Lonai P. The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development. 2002; 129 (16):3783-3793 - 51.
Reinhold MI, Naski MC. Direct interactions of Runx2 and canonical Wnt signaling induce FGF18. The Journal of Biological Chemistry. 2007; 282 (6):3653-3663 - 52.
Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdisciplinary Reviews: Developmental Biology. 2015; 4 (3):215-266 - 53.
Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: The achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocrine Reviews. 2000; 21 (1):23-39 - 54.
Holmes G, Rothschild G, Roy UB, Deng CX, Mansukhani A, Basilico C. Early onset of craniosynostosis in an Apert mouse model reveals critical features of this pathology. Developmental Biology. 2009; 328 (2):273-284 - 55.
Kawai M, Rosen CJ. The insulin-like growth factor system in bone: basic and clinical implications. Endocrinology Metabolic Clinical North America. 2012; 41 (2):323-333 - 56.
Dixit M, Poudel SB, Yakar S. Effects of GH/IGF axis on bone and cartilage. Molecular and Cellular Endocrinology. 2021; 519 :111052 - 57.
Locatelli V, Bianchi VE. Effect of GH/IGF-1 on bone metabolism and Osteoporsosis. International Journal of Endocrinology. 2014; 2014 :235060 - 58.
Kawane T, Qin X, Jiang Q , Miyazaki T, Komori H, Yoshida CA, et al. Runx2 is required for the proliferation of osteoblast progenitors and induces proliferation by regulating Fgfr2 and Fgfr3. Scientific Reports. 2018; 8 (1):13551 - 59.
Kim WJ, Shin HL, Kim BS, Kim HJ, Ryoo HM. RUNX2-modifying enzymes: Therapeutic targets for bone diseases. Experimental & Molecular Medicine. 2020; 52 (8):1178-1184 - 60.
Liu Q , Li M, Wang S, Xiao Z, Xiong Y, Wang G. Recent advances of Osterix transcription factor in osteoblast differentiation and bone formation. Frontiers in Cell and Development Biology. 2020; 8 :601224 - 61.
Balamurugan K, Koehler L, Dürig JN, Hempel U, Rademann J, Hintze V, et al. Structural insights into the modulation of PDGF/PDGFR-β complexation by hyaluronan derivatives. Biological Chemistry. 2021; 402 (11):1441-1452 - 62.
Novak S, Madunic J, Shum L, Vucetic M, Wang X, Tanigawa H, et al. PDGF inhibits BMP2-induced bone healing. NPJ. Regenerative Medicine. 2023; 8 (1):3 - 63.
Caplan AI, Correa D. PDGF in bone formation and regeneration: New insights into a novel mechanism involving MSCs. Journal of Orthopaedic Research. 2011; 29 (12):1795-1803 - 64.
Li A, Xia X, Yeh J, Kua H, Liu H, Mishina Y, et al. PDGF-AA promotes osteogenic differentiation and migration of mesenchymal stem cell by down-regulating PDGFRα and derepressing BMP-Smad1/5/8 signaling. PLoS One. 2014; 9 (12):e113785 - 65.
Vordemvenne T, Paletta JRJ, Hartensuer R, Pap T, Raschke MJ, Ochman S. Cooperative effects in differentiation and proliferation between PDGF-BB and matrix derived synthetic peptides in human osteoblasts. BMC Musculoskeletal Disorders. 2011; 12 :263 - 66.
Kovacs CS. Bone development in the fetus and neonate: Role of the calciotropic hormones. Current Osteoporosis Reports. 2011; 9 (4):274-283 - 67.
Kovacs CS. Bone development and mineral homeostasis in the fetus and neonate: Roles of the calciotropic and phosphotropic hormones. Physiological Reviews. 2014; 94 (4):1143-1218 - 68.
Florencio-Silva R, da Sasso GR, Sasso-Cerri E, Simões MJ, Cerri PS. Biology of bone tissue: Structure, function, and factors that influence bone cells. BioMed Research International. 2015; 2015 :421746 - 69.
Viljakainen HT, Korhonen T, Hytinantti T, Laitinen EKA, Andersson S, Mäkitie O, et al. Maternal vitamin D status affects bone growth in early childhood—A prospective cohort study. Osteoporosis International. 2011; 22 (3):883-891 - 70.
Zhang R, Lu Y, Ye L, Yuan B, Yu S, Qin C, et al. Unique roles of phosphorus in endochondral bone formation and osteocyte maturation. Journal of Bone and Mineral Research. 2011; 26 (5):1047-1056 - 71.
Katagiri T, Takahashi N. Regulatory mechanisms of osteoblast and osteoclast differentiation. Oral Diseases. 2002; 8 (3):147-159 - 72.
Seeman E. Bone modeling and remodeling. Critical Reviews in Eukaryotic Gene Expression. 2009; 19 (3):219-233 - 73.
Szulc P, Seeman E, Duboeuf F, Sornay-Rendu E, Delmas PD. Bone fragility: Failure of periosteal apposition to compensate for increased endocortical resorption in postmenopausal women. Journal of Bone and Mineral Research. 2006; 21 (12):1856-1863 - 74.
Burr DB, Allen MR, editors. Basic and Applied Bone Biology. Amsterdam: Elsevier/Academic Press; 2013. p. 373 - 75.
Kirmani S, Christen D, van Lenthe GH, Fischer PR, Bouxsein ML, McCready LK, et al. Bone structure at the distal radius during adolescent growth. Journal of Bone and Mineral Research. 2009; 24 (6):1033-1042 - 76.
Farr JN, Khosla S. Skeletal changes through the lifespan—From growth to senescence. Nature Reviews. Endocrinology. 2015; 11 (9):513-521 - 77.
Lu J, Shin Y, Yen MS, Sun SS. Peak bone mass and patterns of change in Total bone mineral density and bone mineral contents from childhood into young adulthood. Journal of Clinical Densitometry. 2016; 19 (2):180-191 - 78.
Hernandez CJ, Beaupré GS, Carter DR. A theoretical analysis of the relative influences of peak BMD, age-related bone loss and menopause on the development of osteoporosis. Osteoporosis International. 2003; 14 (10):843-847 - 79.
Rizzoli R, Bianchi ML, Garabédian M, McKay HA, Moreno LA. Maximizing bone mineral mass gain during growth for the prevention of fractures in the adolescents and the elderly. Bone. 2010; 46 (2):294-305 - 80.
Riggs BL, Melton LJ, Robb RA, Camp JJ, Atkinson EJ, McDaniel L, et al. A population-based assessment of rates of bone loss at multiple skeletal sites: Evidence for substantial trabecular bone loss in young adult women and men. Journal of Bone and Mineral Research. 2008; 23 (2):205-214 - 81.
Riggs BL, Melton Iii LJ, Robb RA, Camp JJ, Atkinson EJ, Peterson JM, et al. Population-based study of age and sex differences in bone volumetric density, size, geometry, and structure at different skeletal sites. Journal of Bone and Mineral Research. 2004; 19 (12):1945-1954 - 82.
Zebaze RMD, Ghasem-Zadeh A, Bohte A, Iuliano-Burns S, Mirams M, Price RI, et al. Intracortical remodelling and porosity in the distal radius and post-mortem femurs of women: A cross-sectional study. Lancet. 2010; 375 (9727):1729-1736 - 83.
Nicks KM, Amin S, Atkinson EJ, Riggs BL, Melton LJ, Khosla S. Relationship of age to bone microstructure independent of areal bone mineral density. Journal of Bone and Mineral Research. 2012; 27 (3):637-644 - 84.
Riggs BL, Khosla S, Melton LJ. Sex steroids and the construction and conservation of the adult skeleton. Endocrine Reviews. 2002; 23 (3):279-302 - 85.
Clarke BL, Khosla S. Physiology of bone loss. Radiologic Clinics of North America. 2010; 48 (3):483-495 - 86.
Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nature Medicine. 2017; 23 (9):1072-1079 - 87.
Khosla S, Farr JN, Kirkland JL. Inhibiting cellular senescence: A new therapeutic paradigm for age-related osteoporosis. The Journal of Clinical Endocrinology and Metabolism. 2018; 103 (4):1282-1290 - 88.
Eisman JA. Genetics of osteoporosis. Endocrine Reviews. 1999; 20 (6):788-804 - 89.
Ralston SH, de Crombrugghe B. Genetic regulation of bone mass and susceptibility to osteoporosis. Genes & Development. 2006; 20 (18):2492-2506 - 90.
Hernigou J, Schuind F. Tobacco and bone fractures: A review of the facts and issues that every orthopaedic surgeon should know. Bone and Joint Research. 2019; 8 (6):255-265 - 91.
Niu S, Lim F. CE: The effects of smoking on bone health and healing. The American Journal of Nursing. 2020; 120 (7):40-45 - 92.
Kanis JA, Johnell O, Oden A, Johansson H, De Laet C, Eisman JA, et al. Smoking and fracture risk: A meta-analysis. Osteoporosis International. 2005; 16 (2):155-162 - 93.
Scolaro JA, Schenker ML, Yannascoli S, Baldwin K, Mehta S, Ahn J. Cigarette smoking increases complications following fracture: A systematic review. The Journal of Bone and Joint Surgery. American Volume. 2014; 96 (8):674-681 - 94.
Wong PKK, Christie JJ, Wark JD. The effects of smoking on bone health. Clinical Science (London, England). 2007; 113 (5):233-241 - 95.
Al-Bashaireh AM, Haddad LG, Weaver M, Chengguo X, Kelly DL, Yoon S. The effect of tobacco smoking on bone mass: An overview of pathophysiologic mechanisms. Journal of Osteoporosis. 2018; 2018 :1206235 - 96.
Hannan MT, Felson DT, Dawson-Hughes B, Tucker KL, Cupples LA, Wilson PW, et al. Risk factors for longitudinal bone loss in elderly men and women: The Framingham Osteoporosis Study. Journal of Bone and Mineral Research. 2000; 15 (4):710-720 - 97.
Chakkalakal DA. Alcohol-induced bone loss and deficient bone repair. Alcoholism, Clinical and Experimental Research. 2005; 29 (12):2077-2090 - 98.
Kelsey JL. Risk factors for osteoporosis and associated fractures. Public Health Reports. 1989; 104 :14-20 - 99.
Williams FMK, Cherkas LF, Spector TD, MacGregor AJ. The effect of moderate alcohol consumption on bone mineral density: A study of female twins. Annals of the Rheumatic Diseases. 2005; 64 (2):309-310 - 100.
Hansen MA, Overgaard K, Riis BJ, Christiansen C. Potential risk factors for development of postmenopausal osteoporosis—Examined over a 12-year period. Osteoporosis International. 1991; 1 (2):95-102 - 101.
Yuan S, Michaëlsson K, Wan Z, Larsson SC. Associations of smoking and alcohol and coffee intake with fracture and bone mineral density: A Mendelian Randomization Study. Calcified Tissue International. 2019; 105 (6):582-588 - 102.
Santori C, Ceccanti M, Diacinti D, Attilia ML, Toppo L, D’Erasmo E, et al. Skeletal turnover, bone mineral density, and fractures in male chronic abusers of alcohol. Journal of Endocrinological Investigation. 2008; 31 (4):321-326 - 103.
Turner RT, Aloia RC, Segel LD, Hannon KS, Bell NH. Chronic alcohol treatment results in disturbed vitamin D metabolism and skeletal abnormalities in rats. Alcoholism, Clinical and Experimental Research. 1988; 12 (1):159-162 - 104.
Mercer KE, Wynne RA, Lazarenko OP, Lumpkin CK, Hogue WR, Suva LJ, et al. Vitamin D supplementation protects against bone loss associated with chronic alcohol administration in female mice. The Journal of Pharmacology and Experimental Therapeutics. 2012; 343 (2):401-412 - 105.
Callaci JJ, Juknelis D, Patwardhan A, Wezeman FH. Binge alcohol treatment increases vertebral bone loss following ovariectomy: Compensation by intermittent parathyroid hormone. Alcoholism, Clinical and Experimental Research. 2006; 30 (4):665-672 - 106.
Hogan HA, Argueta F, Moe L, Nguyen LP, Sampson HW. Adult-onset alcohol consumption induces osteopenia in female rats. Alcoholism, Clinical and Experimental Research. 2001; 25 (5):746-754 - 107.
Turner RT, Kidder LS, Kennedy A, Evans GL, Sibonga JD. Moderate alcohol consumption suppresses bone turnover in adult female rats. Journal of Bone and Mineral Research. 2001; 16 (3):589-594 - 108.
Díez-Ruiz A, García-Saura PL, García-Ruiz P, González-Calvin JL, Gallego-Rojo F, Fuchs D. Bone mineral density, bone turnover markers and cytokines in alcohol-induced cirrhosis. Alcohol and Alcoholism. 2010; 45 (5):427-430 - 109.
Luo Z, Liu Y, Liu Y, Chen H, Shi S, Liu Y. Cellular and molecular mechanisms of alcohol-induced osteopenia. Cellular and Molecular Life Sciences. 2017; 74 (24):4443-4453 - 110.
Eby JM, Sharieh F, Callaci JJ. Impact of alcohol on bone health, homeostasis and fracture repair. Current Pathobiological Reports. 2020; 8 (3):75-86 - 111.
Shah VN, Joshee P, Sippl R, Pyle L, Vigers T, Carpenter RD, et al. Type 1 diabetes onset at young age is associated with compromised bone quality. Bone. 2019; 123 :260-264 - 112.
Xu L, Yu J, Wang O, Hou Y, Li W, Zhang H, et al. Comparison of differences in bone microarchitecture in adult- versus juvenile-onset type 1 diabetes Asian males versus non-diabetes males: An observational cross-sectional pilot study. Endocrine. 2021; 71 (1):87-95 - 113.
Halper-Stromberg E, Gallo T, Champakanath A, Taki I, Rewers M, Snell-Bergeon J, et al. Bone mineral density across the lifespan in patients with type 1 diabetes. The Journal of Clinical Endocrinology and Metabolism. 2020; 105 (3):746-753 - 114.
Cortet B, Lucas S, Legroux-Gerot I, Penel G, Chauveau C, Paccou J. Bone disorders associated with diabetes mellitus and its treatments. Joint, Bone, Spine. 2019; 86 (3):315-320 - 115.
Farr JN, Drake MT, Amin S, Melton LJ, McCready LK, Khosla S. In vivo assessment of bone quality in postmenopausal women with type 2 diabetes. Journal of Bone and Mineral Research. 2014; 29 (4):787-795 - 116.
de Araújo IM, Moreira MLM, de Paula FJA. Diabetes and bone. Archives in Endocrinological Metabolism. 2022; 66 (5):633-641 - 117.
Corrado A, Cici D, Rotondo C, Maruotti N, Cantatore FP. Molecular basis of bone aging. International Journal of Molecular Sciences. 2020; 21 (10):3679 - 118.
Kim HN, Chang J, Shao L, Han L, Iyer S, Manolagas SC, et al. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell. 2017; 16 (4):693-703 - 119.
Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, et al. Identification of senescent cells in the bone microenvironment. Journal of Bone and Mineral Research. 2016; 31 (11):1920-1929 - 120.
Müller KH, Hayward R, Rajan R, Whitehead M, Cobb AM, Ahmad S, et al. Poly(ADP-ribose) links the DNA damage response and biomineralization. Cell Reports. 2019; 27 (11):3124-3138 - 121.
Rasheed N, Wang X, Niu QT, Yeh J, Li B. Atm-deficient mice: An osteoporosis model with defective osteoblast differentiation and increased osteoclastogenesis. Human Molecular Genetics. 2006; 15 (12):1938-1948 - 122.
de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, et al. Premature aging in mice deficient in DNA repair and transcription. Science. 2002; 296 (5571):1276-1279 - 123.
Dobson PF, Dennis EP, Hipps D, Reeve A, Laude A, Bradshaw C, et al. Mitochondrial dysfunction impairs osteogenesis, increases osteoclast activity, and accelerates age related bone loss. Scientific Reports. 2020; 10 (1):11643 - 124.
De la Garza-Ramos R, Bydon M, Abt NB, Sciubba DM, Wolinsky JP, Bydon A, et al. The impact of obesity on short- and long-term outcomes after lumbar fusion. Spine (Phila Pa 1976). 2015; 40 (1):56-61 - 125.
Forte YS, Renovato-Martins M, Barja-Fidalgo C. Cellular and molecular mechanisms associating obesity to bone loss. Cell. 2023; 12 (4):521 - 126.
Shapses SA, Pop LC, Wang Y. Obesity is a concern for bone health with aging. Nutrition Research. 2017; 39 :1-13 - 127.
Katsevman GA, Daffner SD, Brandmeir NJ, Emery SE, France JC, Sedney CL. Complexities of spine surgery in obese patient populations: A narrative review. The Spine Journal. 2020; 20 (4):501-511 - 128.
Nakamura A, Akahane M, Shigematsu H, Tadokoro M, Morita Y, Ohgushi H, et al. Cell sheet transplantation of cultured mesenchymal stem cells enhances bone formation in a rat nonunion model. Bone. 2010; 46 (2):418-424 - 129.
Hao C, Wang Y, Shao L, Liu J, Chen L, Zhao Z. Local injection of bone mesenchymal stem cells and fibrin glue promotes the repair of bone atrophic nonunion In vivo. Advances in Therapy. 2016; 33 (5):824-833 - 130.
Kim SJ, Shin YW, Yang KH, Kim SB, Yoo MJ, Han SK, et al. A multi-center, randomized, clinical study to compare the effect and safety of autologous cultured osteoblast(Ossron) injection to treat fractures. BMC Musculoskeletal Disorders. 2009; 10 :20 - 131.
Xu X, Shi D, Liu Y, Yao Y, Dai J, Xu Z, et al. In vivo repair of full-thickness cartilage defect with human iPSC-derived mesenchymal progenitor cells in a rabbit model. Experimental and Therapeutic Medicine. 2017; 14 (1):239-245 - 132.
Rana D, Kumar S, Webster TJ, Ramalingam M. Impact of induced pluripotent stem cells in bone repair and regeneration. Current Osteoporosis Reports. 2019; 17 (4):226-234 - 133.
Jeffery EC, Mann TLA, Pool JA, Zhao Z, Morrison SJ. Bone marrow and periosteal skeletal stem/progenitor cells make distinct contributions to bone maintenance and repair. Cell Stem Cell. 2022; 29 (11):1547-1561.e6 - 134.
Lee CCI, Christensen JE, Yoder MC, Tarantal AF. Clonal analysis and hierarchy of human bone marrow mesenchymal stem and progenitor cells. Experimental Hematology. 2010; 38 (1):46-54 - 135.
Lee CC, Hirasawa N, Garcia KG, Ramanathan D, Kim KD. Stem and progenitor cell microenvironment for bone regeneration and repair. Regenerative Medicine. 2019; 14 (7):693-702 - 136.
Huang YC, Leung VYL, Lu WW, Luk KDK. The effects of microenvironment in mesenchymal stem cell-based regeneration of intervertebral disc. The Spine Journal. 2013; 13 (3):352-362 - 137.
Dozza B, Lesci IG, Duchi S, Della Bella E, Martini L, Salamanna F, et al. When size matters: Differences in demineralized bone matrix particles affect collagen structure, mesenchymal stem cell behavior, and osteogenic potential. Journal of Biomedical Materials Research. Part A. 2017; 105 (4):1019-1033 - 138.
Vlaikou AM, Kouroupis D, Sgourou A, Markopoulos GS, Bagli E, Markou M, et al. Mechanical stress affects methylation pattern of GNAS isoforms and osteogenic differentiation of hAT-MSCs. Biochimica Biophysica Acta Molecular Cell Research. 2017; 1864 (8):1371-1381 - 139.
Denry I, Kuhn LT. Design and characterization of calcium phosphate ceramic scaffolds for bone tissue engineering. Dental Materials. 2016; 32 (1):43-53 - 140.
Chen W, Zhou H, Weir MD, Bao C, Xu HHK. Umbilical cord stem cells released from alginate-fibrin microbeads inside macroporous and biofunctionalized calcium phosphate cement for bone regeneration. Acta Biomaterialia. 2012; 8 (6):2297-2306 - 141.
Holderegger C, Schmidlin PR, Weber FE, Mohn D. Preclinical in vivo performance of novel biodegradable, electrospun poly(lactic acid) and poly(lactic-co-glycolic acid) nanocomposites: A review. Materials (Basel). 2015; 8 (8):4912-4931 - 142.
Wu DT, Munguia-Lopez JG, Cho YW, Ma X, Song V, Zhu Z, et al. Polymeric scaffolds for dental, Oral, and craniofacial regenerative medicine. Molecules. 2021; 26 (22):7043 - 143.
Park S, Park J, Jo I, Cho SP, Sung D, Ryu S, et al. In situ hybridization of carbon nanotubes with bacterial cellulose for three-dimensional hybrid bioscaffolds. Biomaterials. 2015; 58 :93-102 - 144.
Hung BP, Naved BA, Nyberg EL, Dias M, Holmes CA, Elisseeff JH, et al. Three-dimensional printing of bone extracellular matrix for craniofacial regeneration. ACS Biomaterials Science & Engineering. 2016; 2 (10):1806-1816 - 145.
Levengood SL, Zhang M. Chitosan-based scaffolds for bone tissue engineering. Journal of Materials Chemistry B. 2014; 2 (21):3161-3184 - 146.
Mathews S, Mathew SA, Gupta PK, Bhonde R, Totey S. Glycosaminoglycans enhance osteoblast differentiation of bone marrow derived human mesenchymal stem cells. Journal of Tissue Engineering and Regenerative Medicine. 2014; 8 (2):143-152 - 147.
Lee SS, Fyrner T, Chen F, Álvarez Z, Sleep E, Chun DS, et al. Sulfated glycopeptide nanostructures for multipotent protein activation. Nature Nanotechnology. 2017; 12 (8):821-829 - 148.
Koleva PM, Keefer JH, Ayala AM, Lorenzo I, Han CE, Pham K, et al. Hyper-Crosslinked carbohydrate polymer for repair of critical-sized bone defects. Bioresearch Open Access. 2019; 8 (1):111-120 - 149.
Scott RT, Hyer CF. Role of cellular allograft containing mesenchymal stem cells in high-risk foot and ankle reconstructions. The Journal of Foot and Ankle Surgery. 2013; 52 (1):32-35 - 150.
Camargo WA, de Vries R, van Luijk J, Hoekstra JW, Bronkhorst EM, Jansen JA, et al. Diabetes mellitus and bone regeneration: A systematic review and Meta-analysis of animal studies. Tissue Engineering. Part B, Reviews. 2017; 23 (5):471-479 - 151.
Daniels TR, Younger ASE, Penner MJ, Wing KJ, Le ILD, Russell IS, et al. Prospective randomized controlled trial of Hindfoot and ankle fusions treated with rhPDGF-BB in combination with a β-TCP-collagen matrix. Foot & Ankle International. 2015; 36 (7):739-748 - 152.
Krell ES, DiGiovanni CW. The efficacy of platelet-derived growth factor as a bone-stimulating agent. Foot and Ankle Clinics. 2016; 21 (4):763-770 - 153.
Gadomski BC, Labus KM, Puttlitz CM, McGilvray KC, Regan DP, Nelson B, et al. Evaluation of lumbar spinal fusion utilizing recombinant human platelet derived growth factor-B chain homodimer (rhPDGF-BB) combined with a bovine collagen/β-tricalcium phosphate (β-TCP) matrix in an ovine model. JOR Spine. 2021; 4 (3):e1166