 Open access peer-reviewed chapter
Open access peer-reviewed chapter

Abstract
Single nucleotide polymorphisms (SNPs) are heritable variations at defined regions and occur in at least 1% of the population. SNPs are mostly bi-allelic, and their inheritance pattern can be typed in a representative population of few unrelated individuals. Contrary to the STRs (Short tandem repeats), SNPs can be typed relatively easily using next generation sequencing methods. Thus, SNPs have attracted a lot of scientists for application in forensic analysis for cases such as establishing biogeographical ancestry, evolutionary timescale relatedness, immediate family relationships. SNPs are arguably more useful than STRs in certain forensic scenarios. For instance, when the obtained DNA sample from crime scene is a multi-origin mixture or when the DNA is degraded, SNPs offer better utility. SNPs are also valuable in cases where DNA extraction from challenging forensic samples, such as bones or meat, poses technical difficulties. Due to their characteristics, SNPs provide enhanced capabilities for forensic analysis in these specific situation. A plethora of novel techniques and algorithms have been developed to use the available SNP databases for forensic analysis. The developed technologies include hybridization assay, primer extension assay, multiplex polymerase chain reaction (PCR), denaturing high performance liquid chromatography, matrix-assisted laser desorption/ionization. These are just a few examples of the developed technologies utilized in molecular biology and genetic analysis, each with its unique advantages and disadvantages. We discuss the accuracy, sensitivity, specificity, advantages and disadvantages of some of these techniques in detail here.
Keywords
- SNP
- forensic
- SNP typing
- molecular beacons
- oligonucleotide ligation assay
1. Introduction
Single nucleotide polymorphisms (SNPs) refer to genomic positions at which different nucleotides can occur in different individuals [1, 2, 3]. Any nucleotide variation at a genomic position can be considered as an SNP, only if the same variation occurs at a frequency of 1%, in a sampled population. SNPs are found abundantly in the human genome, on an average of 1 in every 1000 nucleotide [1, 2, 3]. Multiple databases have been developed over years to keep up with the ever-increasing number of reported SNPs. Some of these databases are: The SNP consortium (1998), dbSNP [4, 5], 1000 genomes project (2008) [6], the GWAS catalog (2008) [7], ALlele FREquency Database (ALFRED) [8]. Interestingly, these databases also store information on SNPs constituting the same haplotype, SNPs related to distinct disorders and distinct phenotypes.
Mutations often lead to formation of SNPs. The mutation can either be transition mutations (for example: A to G or G to A) or transversion mutations (for example: C to A or G to C). Transition mutation led SNPs account for bi-allelic loci and transversion mutation led SNPs account for bi-allelic, tri-allelic or tetra-allelic loci (Figure 1). Bi-allelic SNPs are found more abundantly in the human genome [1, 9, 10]. It is interesting to note that a few thousands of the reported SNPs existed in the human genome before humans migrated out of South Africa. These SNPs account for the common SNPs found across different human populations on the earth. In addition to these common SNPs, there are thousands of SNPs reported as population-specific SNPs which may have arisen due to distinct biogeographical location, epigenetic factors such as food, lifestyle preferences, local environmental conditions. SNPs have been used for a range of purposes, including human identity verification, differentiation amongst distinct biogeographic human populations, analysis of human immigration patterns, exploration of ancestral history, determination of sibling relationships and identification of criminals across the border [11, 12, 13].

Figure 1.
Single nucleotide polymorphisms (SNPs) can be bi-allelic, tri-allelic or tetra-allelic. (A) Schematic representation of a bi-allelic SNP, presence of C in sample 3 and G at position 7 in sample 4. (B) Schematic representation of a bi-allelic SNP, presence of C in sample 2, T at position 7 in sample 3 and G at position 7 in sample 4.
SNPs provide a genetic tool to be used in defining human populations, distinguishing one individual from the other. However, Short Tandem Repeats (STRs) have been a conventional choice for similar applications. STRs are short DNA sequences (about 2–6 nucleotide long) which make up about 3% of the total human genome [14, 15, 16]. Lately, SNPs have gained popularity as an alternative to STRs due to characteristic properties such as: bi-allelic nature of SNPs, low mutation rates as compared to STRs, inheritance of SNPs in the form of haplotypes, relation to distinct phenotypes, easy identification from smaller amplicons (often obtained from degraded forensic samples), lack of stutter artifacts, amenable to multiplexing assay and automation [1, 2, 3]. However, the use of SNPs has faced certain limitations. These include the need for a large number of SNPs compared to STR markers to achieve discrimination between human and non-human samples. Careful selection of population-specific SNPs is necessary, which can be challenging. SNPs have limited utility in mixture interpretations due to their bi-allelic nature. Additionally, available criminal offender databases such as The Combined DNA Index System (CODIS) [17] predominantly contain STR typing datasets, necessitating the retyping of these databases for SNPs. Developing multiplexing assays for SNPs can be costly and time-consuming, and the resulting information may be less informative compared to STRs.
Multiple SNP typing methods have been developed for their use in basic research and forensic applications. With the advent of technological advancement, many of these methods can be adapted for multiplexing assays. SNP typing methods are based on four different reaction principles: (1) Hybridization, (2) Oligonucleotide ligation, (3) Primer extension and (4) Enzymatic cleavage [2, 3]. The reaction principles have been combined with different combinations of assay formats and detection platforms for use of SNPs in user-specific requirements. Below we describe various methods based on reaction principles, advancements in the methods, and combinations of methods with different assay formats and detection platforms.
2. SNP typing methods based on the principle of hybridization
2.1 Allele-specific hybridization
DNA–DNA hybridization (DDH) involves identification of a target DNA using a complementary DNA probe (short strand of DNA) [18]. DDH has been conventionally used to differentiate between alleles using allele-specific DNA probes [19]. Allele-specific hybridization attempts to identify DNA targets at a polymorphic locus using Allele-Specific Oligonucleotide probes (ASO). The probes are designed such that a central polymorphic nucleotide is flanked by gene specific-DNA sequence. The complementarity between target DNA and the probe determines the stability of hybridization [20]. Two probes, one complementary to each allele, are used in each hybridization reaction. One of the two probes will be completely complementary to the target DNA, thus forming a stable hybrid (Figure 2). Optimized reaction conditions favor the formation of stable hybrid. Detection of the stable hybrid has been attempted with various techniques. However, fluorescence resonance energy transfer (FRET) has remained the detection technique of choice in combination with allele-specific hybridization.

Figure 2.
Allele specific hybridization. The allele specific hybridization depends on the design of probes. Allele specific oligonucleotide probes (ASO) have a polymorphic site at the Centre of the probe. (A) Schematic representation of hybridization of ASO to the target DNA (B) schematic representation of no hybridization between ASO and target DNA due to mismatch at the polymorphic site.
2.2 Fluorescence resonance energy transfer (FRET)
FRET works on the principle of energy transfer from a donor fluorophore to an acceptor fluorophore. Excitation of the donor fluorophore at a wavelength (within its excitation spectrum) leads to emission of energy. This emitted energy serves as a source of excitation of the acceptor fluorophore. The energy transfer is possible because the emission spectrum of the donor overlaps with the excitation spectrum of the acceptor. FRET is a distance sensitive technique, i.e., the donor fluorophore and the acceptor fluorophores have to be in close proximity for a successful energy transfer [21].
2.3 Recent advancements in the method
ASO and FRET were restricted by the amount of DNA available for studies. Retrieval of very small quantities of DNA is often faced by forensic scientists. This affects the sensitivity, efficiency and the accuracy of SNP typing. To overcome this, ASO and FRET were combined with polymerase chain reaction (PCR). PCR uses a target DNA and amplifies it using primers (short DNA sequences; complementary to target DNA), DiDeoxy nucleotide tri phosphates (dNTPs), DNA polymerase. Primers provide the 3′ OH group, as a substrate for addition of dNTPs complementary to target DNA, by DNA polymerase. Below we described the three technical advancements depending on the design of the probe, using exonuclease activity of the DNA polymerase. Each of these three advanced methods attempts to enhance sensitivity, accuracy and efficiency of the conventional hybridization method.
2.4 LightCycler®
LightCycler® (

Figure 3.
Recent methods of allele specific hybridization which use FRET for detection. (A) Light cycler uses two ASO, each labeled with a fluorophore at its end. Generation of FRET signal determines the adjacent hybridization of both ASO to the target DNA. (B) TaqMan uses a single ASO labeled with a fluorophore and a quencher. FRET signal determines cleavage of ASO from its 5′ end by Taq polymerase.
Earlier, the assay was designed to use different probe sequences for typing different SNPs. The size of the probes may be similar for distinct target DNAs, however, the polymorphic nucleotide in the probe would vary depending on the target DNA. The difference in the polymorphic nucleotide would lead to different melting temperature of the probe. In case of unknown target DNA, melting curve analysis post PCR reactions determined the SNP in the target DNA. With time, LightCycler® has been updated for use in real-time quantification (LightCycler® 480 Instrument II), diagnostics by multiplexing and endpoint genotyping applications (LightCycler® 96 Instrument) [23].
2.5 TaqMan™ SNP genotyping assay
TaqMan™ SNP genotyping assay (
2.6 Molecular beacons
Molecular Beacons mark the latest advancements in ASO probes. The probes are designed such that they have DNA sequence complementary to the target DNA as well as DNA sequence with intra-molecular complementarity. The intra-molecular complementarity is the characteristic feature of the molecular beacon probes. Intra-molecular complementarity leads to formation of a hair pin loop structure of the probe until it anneals to a target DNA (Figure 4) [28, 29, 30]. Like the TaqMan™ SNP genotyping assay, molecular beacon probes have a fluorophore at the 5′ end and quencher at the 3′ end of the probe. The absence of fluorescence signal signifies that probe did not anneal to the target DNA. The fluorescence signal signifies that the probe has annealed to the target DNA because the annealing of probe to the target DNA arranges the quencher and the fluorophore distant from each other. Like the LightCycler® and the TaqMan™ SNP genotyping assay, the amplification of DNA can be monitored in real time using the intensity of the fluorescent signal [1, 2]. Conventionally, two different molecular beacon probes are designed for two different alleles and/or SNPs. Multiplexing reactions have been conducted using molecular beacon probes with different fluorophores for each allele or SNP in the target DNA and a wavelength shifting molecular beacons. Wavelength shifting molecular beacons are probes attached to different fluorophores. Each of these fluorophores can be excited using a monochromatic light source or a laser but each fluorophore emits a distinct wavelength (distinct color) [31].

Figure 4.
Design of molecular beacon probes used for advanced allele specific hybridization. The molecular beacon probe has intra-molecular complementary sequence leading to formation of a hair pin structure, flanked by a fluorophore at the 5′ end and quencher at the 3′ end. Upon hybridization to the target DNA, the fluorophore and the quencher are placed distant to each other emitting fluorescence signal.
2.6.1 Array hybridization
All three of the above technologies had restricted multiplexing capacity due to: availability of distinct, non-overlapping fluorophores emitting in selected wavelength spectrum, similar reaction conditions (annealing temperatures, melting temperatures, product lengths) for multiple target DNAs to be amplified simultaneously [1, 2]. This was overcome upon selecting a microarray assay format to screen multiple SNPs simultaneously. Microarrays have multiple probes attached to the solid surface. These probes are used to hybridize with different fluorescently labeled PCR products. Each PCR product represents a distinct SNP (Figure 5). However, the hybridization depends on the length of the sequences flanking the polymorphic site [32]. GeneChip® system used multiple ASO probes for the same SNP, spanning all the possible combinations of the polymorphic site and the flanking sequence (Tiling strategy) [33, 34, 35]. Tiling strategy could overcome the ambiguity in the microarray results arising due to the difference in lengths of the sequences flanking the polymorphic site. The multiplexing capacity of the GeneChip® system is beyond the actual need of SNP typing in forensic studies lately. However, it may prove to be useful for future applications.

Figure 5.
Array hybridization. A microarray format for typing multiple SNPs by attaching multiple probes to the solid surface. Probes are used to hybridize with the PCR products. Each PCR product will represent a distinct SNP. Using an array of probes aids identification of many SNPs simultaneously.
3. SNP typing methods based on the principle of primer extension
Primer extension works on the principle of addition of nucleotides complementary to the target DNA using 3′ end of the primer as a starting point. The ability of DNA polymerase to accurately add nucleotide at the 3′ end of a primer sequence forms the basis of the primer extension reaction (Figure 6) [36]. The choice of length extended DNA, use of allele-specific primers has offered adaptations of primer extension reaction. These are: (A) Minisequencing, (B) Pyrosequencing and (C) Allele-specific primer extension [1, 2]. Irrespective of the length of extended DNA or distinct primers, the extended DNA was conventionally separated using electrophoresis technique for a length-based identification of polymorphic sites in the target DNA. However, with technological advancements, fluorescence-based detection and mass spectrometry-based detection have been combined with primer extension reactions for accurate polymorphism identification.
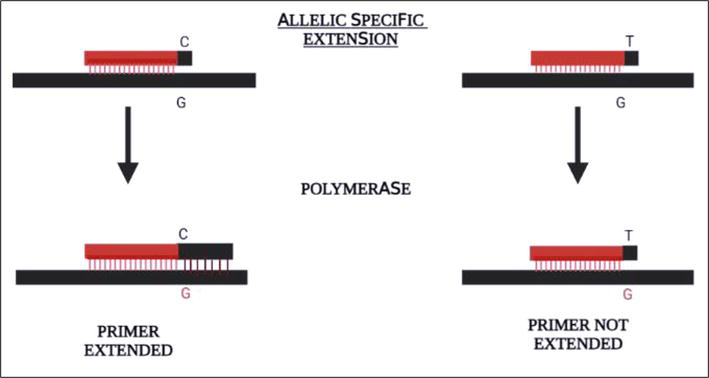
Figure 6.
Primer extension method of SNP typing. A recent method of SNP typing using principle of primer extension. Primers are designed to be allele specific such that primer extension happens only if the polymorphic site on the primer and the target DNA are complementary.
3.1 Minisequencing
It refers to (i) sequencing few nucleotides using labeled DiDeoxy Nucleotide Tri Phosphates (ddNTPs) to detect polymorphic sites (Figure 6) or (ii) identifying the sequence of the target DNA using polymorphic site-specific primers [37, 38]. Case (i) is a conventional PCR except for the use of ddNTPs instead of dNTPs. Primers are designed such that they anneal the target DNA till immediately upstream of the polymorphic nucleotide (Figure 7). The addition of first ddNTP to the 3′ end of the primer identified the sequence at the polymorphic site. Labelling each ddNTP with distinct fluorophores provides real-time detection of the polymorphic nucleotide identification using fluorescence detectors, thus facilitating automation of the entire process. Extended DNA length may overlap between two independent primer extension reactions. Electrophoretic separation of overlapping lengths of DNA proved a challenge for accurate identification of polymorphic nucleotide. Addition of non-human DNA sequences at the 5′ end of the primers solved the problem of overlapping DNA lengths [1, 2]. SNaPshot™ was one of the earliest SNP typing methods that combined minisequencing with detecting a fluorescently labeled ddNTP [39, 40]. Currently the commercially available SNaPshot™ Multiplex Kit offers multiplexing of ten SNPs [41]. These have been used for forensic investigations and applications. Case (ii) is a conventional PCR as well. However, the primers are designed complementary to the polymorphic site of the target DNA. The primer extension happened only if the primer annealed to the target DNA. Thus, the polymorphic site can be identified by the primer sequence of the extended DNA. Use of mass spectrometry for detection of polymorphic site along with primer extension reaction has been used lately [42]. Mass spectrometer identifies the polymorphic site by differentiation in the mass of extended DNA. The difference in the mass of extended DNA is contributed by distinct mass of each ddNTP. Matrix Assisted Laser Desorption Ionization – Time of Flight (MALDI-TOF) is a preferred ionization source and detector combination used for SNP typing of minisequenced products [42]. PROBE assay [43], PinPoint assay [44], GOOD assay [45] are few of the SNP typing assays that use the minisequencing reaction principle coupled with mass spectrometry detection platform. Each of these assays attempts to distinguish extended DNA more accurately than the other. The PROBE assay utilizes both ddNTPs and dNTPs to increase the mass difference between the extended DNA. The PinPoint assay utilizes dNTPs only. The GOOD assay uses modified primer that enhances the purification of the primer extended DNA for detection using mass spectrometry. Like the SNaPshot™ assay, non-overlapping masses of extended DNA are achieved upon addition of non-human DNA sequences at the 5′ end of the primers.

Figure 7.
Minisequencing method of SNP typing. SNP typing using sequencing the polymorphic nucleotide immediately downstream of the primer using distinctly labeled ddNTPs. The label on the annealed ddNTP complementary to the target will determine its sequence.
3.2 Pyrosequencing
It was developed as a method to sequence longer DNA as compared to minisequencing. It works on the principle of sequencing by synthesis. Like a conventional PCR, the reaction mixture contains dNTPs, DNA polymerase, primers. In addition to these, the reaction mixture also contains ATP sulfurylase, luciferase, apyrase. Upon addition of every nucleotide, light is released. The light released is due to the pyrophosphate released upon activity of DNA polymerase. This pyrophosphate acts as a substrate of ATP sulfurylase, which converts it into ATP. Thus formed ATP is used by luciferase to generate light. Excess nucleotides are degraded by apyrase. Nucleotides are added to the sequencing reaction mixture in a pre-defined order. The release of light signifies incorporation of the added nucleotide. Thus, the sequence of the synthesized strand is determined based on the pre-defined order of addition of nucleotides. About 20–30 nucleotides can be sequenced using pyrosequencing [46, 47]. The advantage of this is being able to read the sequences around the polymorphic sites in the target DNA. However, limited multiplexing possibilities and lengthy target preparation procedures make pyrosequencing applicable to limited studies [1, 2].
3.3 Allele-specific primer extension
It involves the use of allele-specific primers and the target DNA. Only those primers which anneal stably (without-mismatch) to the target DNA will be extended by the DNA polymerase whereas the primers with polymorphic site other than the target DNA (mismatched primers) do not get extended [48, 49]. The detection platforms coupled with allele-specific primer extension vary from the conventional electrophoresis, fluorescence signal detection. Multiplexing with allele-specific primer extension has been possible by using microarray technology. Microarray allows detection of multiple polymorphic sites simultaneously or use of FRET primer pairs for real-time SNP typing (Figure 4) [50].
4. SNP typing methods based on the principle of oligonucleotide ligation
4.1 Oligonucleotide ligation assay (OLA)
DNA ligases are a class of enzymes which catalyze the formation of a phosphodiester bond between two subsequently placed nucleotides. The reaction which uses DNA ligase for sealing a nick between two consecutive nucleotides is known as oligonucleotide ligation. OLA uses DNA ligase to ligate two probes positioned one phosphodiester bond apart on a target DNA [51]. Of the two required probes, one probe is designed complementary to the polymorphic site on the target DNA at its 3′ end. The second probe, also known as the common probe, is complementary to the target DNA sequence immediately downstream of the polymorphic site. Ligation of the two probes happens only if both the probes have annealed to their respective target DNA (Figure 8). Thus, upon ligation, the probe sequence can be used to identify the polymorphic nucleotide in the target DNA [1, 2, 51].

Figure 8.
Oligonucleotide ligation assay (OLA) for SNP typing. The OLA uses two probes. Both of them hybridize with the target DNA adjacent to each other. One of the probes are designed specific to the allele in the target DNA. Ligation of the two probes happens only if the probe hybridizes completely to the target DNA.
4.2 Recent advancements in the method
Ligase chain reaction (LCR) combines the OLA and PCR. LCR is designed such that includes a thermostable DNA ligase as a part of the conventional PCR mixture [1, 2, 52]. The ligated probes in the early steps of the reaction form a target for attachment of probes in the next ligation reaction. Use of two strands of the target DNA, one representing each allele, in an LCR helps to identify allele-specific polymorphic sites. Although, it would need two distinct probes (in addition to the common probe), one complementary to each allele at the polymorphic site of the target DNA [1, 2, 52].
Detection of ligated probes in the OLA and LCR has been attempted using biotin-based detection method. The common probes are biotinylated, and the allele-specific probes have a reporter dye attached to them. Ligation of the two probes will lead to capture of the ligated product using streptavidin [1, 2, 52]. Advancements in the detection technologies have replaced the biotin with mobility modifiers and replaced reporter dyes with fluorescent dyes. Mobility modifiers are chemical molecules, often oligomeric which bind to the ligated probes and define its mobility. Thus, distinguishing the ligated probes from the unligated probes. Use of the mobility modifiers served for higher resolution separation of ligated products upon electrophoresis. Importantly, the mobility modifiers regulate the mobility of the ligated products independent of the length of the ligated product. Availability of varied fluorescent dyes provides the possibility of multiplexing the assay. Detection of multiple SNPs is possible after electrophoretic separation of the ligation products and detection of their respective reporter dyes [1, 2, 52].
Coupled Amplification and Oligonucleotide Ligation (CAL) offers a one-step amplification and ligation reaction [53]. Using primers with high melting temperatures and probes with lower temperatures was the key to this. The reagents for both DNA amplification and oligonucleotide ligation are added to the same tube. Firstly, the primers anneal to the target DNA and amplifies the target DNA. The annealing temperature should be higher than the melting temperature of the oligonucleotide probes. In the second step, the ligation happens at a lower temperature, allowing hybridization of the probes which is a pre-requisite of the OLA. Advanced CAL was developed with fluorescently labeled probes for real-time SNP typing. Currently, SNPlex (ThermoFisher Scientific) genotyping system is a commercially available SNP typing method based on OLA [54]. It is used to perform SNP typing taking genomic DNA as the target DNA. More importantly, at times it has been used with fragmented genomic DNA. Multiple unlabelled oligonucleotide probes are used for multiplexing the assay, followed by amplification of the identified target DNA using universal primers. Thus, the recognized polymorphic sites are read using ZipChute probes. These probes hybridize to the target DNA, they are purified and sequenced using capillary electrophoresis.
5. SNP typing methods based on the principle of enzymatic cleavage
5.1 Enzymatic cleavage
Structure-specific Flap endonucleases (FEN) are enzymes which recognize overlapping nucleotides (fork-like structure) and cleave the DNA which hangs loosely (not bound to any DNA strand) (Figure 9) [55, 56]. Archaeal FENs were earlier studied to recognize overlapping oligonucleotides which are hybridized to a target DNA strand and cleave the loosely hanging oligonucleotides. Interestingly, this cleavage was found to be sequence dependent as well [55, 56]. Thus, making the FENs a perfect fit for the enzymes which could be used for detection of polymorphic sites in a given target DNA using overlapping probes (complementary to the target DNA).

Figure 9.
Invader assay for SNP typing. The assay uses two probes. Allele specific probe and invader probe. Both of these probes have an overlap of one nucleotide. If the allele specific probe is complementary to the target DNA, a fork-like structure is formed between the invader probe and the allele specific probe. This structure is recognized by the FEN endonucleases mediating cleavage at the junction between probes.
5.2 Invader assay
Invader assay was one of the first SNP typing methods used in forensic studies [57]. It works on the principle of cleavage of probe annealed to the target DNA at the polymorphic site. Each assay utilizes two probes: An allele-specific probe and an invader oligo. The invader oligo is designed such that it is complementary to the polymorphic site of the target DNA at the extreme 3′ end. The allele-specific probe is designed such that it is complementary to the polymorphic site on the target DNA towards it’s 5′ end with some non-complementary nucleotides following the polymorphic site. The design of the probes leads to formation of an invader structure upon probes annealing to the target DNA. FENs recognize the invader structure which is like a bifurcated fork-like structure and cleave at the 3′ end of the overlapping invader structure (Figure 9). The cleavage can be detected in real time using FRET probes which have a fluorophore at the 5′ end of the probe and a quencher at the 3′ end of the probe [55, 56, 57]. Cleavage separates the quencher and the fluorophore generating a measurable fluorescent signal. In case of mismatch at the polymorphic site, the invader oligo will not anneal to the target DNA, hence no invader structure is formed, and no FEN cleavage is observed.
Despite the simple chemistry and one step method for SNP detection, the assay could not be adapted for forensic studies due to requirement of large amount of target DNA and minimized possibility of multiplexing the reactions [1, 2].
Acknowledgments
The authors thank Mr. Niranjan Vyas for helping create the figures used in this book chapter. The authors express heartfelt gratitude towards the School of forensic science, National Forensic Sciences University, for encouraging to initiate a book chapter on the recent advanced techniques and providing conducive environment for critical thinking and analysis of the subject matter.
References
- 1.
Novroski NM, Cihlar JC. Evolution of single-nucleotide polymorphism use in forensic genetics. WIREs Forensic Science. 2022; 4 (6):e1459 - 2.
Sobrino B, Carracedo A. SNP typing in forensic genetics: A review. Methods in Molecular Biology. 2005; 297 :107-126. DOI: 10.1385/1-59259-867-6:107 - 3.
Syvänen AC. Accessing genetic variation: Genotyping single nucleotide polymorphisms. Nature Reviews. Genetics. 2001; 2 :930-942 - 4.
Smigielski EM, Sirotkin K, Ward M, Sherry ST. dbSNP: A database of single nucleotide polymorphisms. Nucleic Acids Research. 2000; 28 (1):352-355. DOI: 10.1093/nar/28.1.352 - 5.
Sherry ST, Ward M-H, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: The NCBI database of genetic variation. Nucleic Acids Research. Jan 2001; 29 (1):308-311 - 6.
Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, et al. A global reference for human genetic variation. Nature. 1 Oct 2015; 526 :68-74 - 7.
Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Research. 2019; 47 (D1):D1005-D1012. DOI: 10.1093/nar/gky1120 - 8.
Rajeevan H, Osier MV, Cheung KH, Deng H, Druskin L, Heinzen R, et al. ALFRED: The ALelle FREquency database. Update. Nucleic Acids Research. 2003; 31 (1):270-271. DOI: 10.1093/nar/gkg043 - 9.
Brookes AJ. The essence of SNPs. Gene. 1999; 234 (2):177-186. DOI: 10.1016/s0378-1119(99)00219-x - 10.
Phillips C, Amigo J, Carracedo Á, Lareu MV. Tetra-allelic SNPs: Informative forensic markers compiled from public whole-genome sequence data. Forensic Science International. Genetics. 2015; 19 :100-106. DOI: 10.1016/j.fsigen.2015.06.011 - 11.
Mcevoy BP, Powell JE, Goddard M, Visscher PM. Human population dispersal "out of Africa" estimated from linkage disequilibrium and allele frequencies of SNPs. Genome Research. 2011; 21 (6):821-829 - 12.
Stoneking M, Krause J. Learning about human population history from ancient and modern genomes. Nature Reviews. Genetics. 2011; 12 (9):603-614. DOI: 10.1038/nrg3029 - 13.
Huisman J. Pedigree reconstruction from SNP data: Parentage assignment, sibship clustering and beyond. Molecular Ecology Resources. 2017; 17 (5):1009-1024. DOI: 10.1111/1755-0998.12665 - 14.
Wyner N, Barash M, McNevin D. Forensic autosomal short tandem repeats and their potential association with phenotype. Frontiers in Genetics. 2020; 11 :884. DOI: 10.3389/fgene.2020.00884 - 15.
Kayser M. Forensic use of Y-chromosome DNA: A general overview. Human Genetics. 2017; 136 (5):621-635. DOI: 10.1007/s00439-017-1776-9 - 16.
Haddrill PR. Developments in forensic DNA analysis. Emerging Topics in Life Sciences. 2021; 5 (3):381-393. DOI: 10.1042/ETLS20200304 - 17.
Bruce B, Moretti Tamyra R, Niezgoda Stephen J., Brown Barry L. CODIS and PCR-Based Short Tandem Repeat Loci: Law Enforcement Tools. Promega [Retrieved October 8, 2016] - 18.
Kennell DE. Principles and practices of nucleic acid hybridization. Progress in Nucleic Acid Research and Molecular Biology. 1971; 11 :259-301. DOI: 10.1016/s0079-6603(08)60330-x - 19.
Howell W, Jobs M, Gyllensten U, et al. Dynamic allele-specific hybridization. Nature Biotechnology. 1999; 17 :87-88 - 20.
Tortajada-Genaro LA. Design of Oligonucleotides for allele-specific amplification based on PCR and isothermal techniques. Methods in Molecular Biology. 2022; 2392 :35-51. DOI: 10.1007/978-1-0716-1799-1_3 - 21.
Kaur A, Kaur P, Ahuja S. Förster resonance energy transfer (FRET) and applications thereof. Analytical Methods. 2020; 12 (46):5532-5550. DOI: 10.1039/d0ay01961e Erratum in: Anal Methods. 2021;13(5):730 - 22.
Lareu M, Puente J, Sobrino B, Quintáns B, Brión M, Carracedo A. The use of the LightCycler for the detection of Y chromosome SNPs. Forensic Science International. 2001; 118 (2-3):163-168. DOI: 10.1016/s0379-0738(01)00386-3 - 23.
https://lifescience.roche.com/global/en/products/others/lightcycler-480-multiwell-plate-96-clear-381435.html - 24.
Glaab WE, Skopek TR. A novel assay for allelic discrimination that combines the fluorogenic 5′ nuclease polymerase chain reaction (TaqMan) and mismatch amplification mutation assay. Mutation Research. 1999; 430 (1):1-12. DOI: 10.1016/s0027-5107(99)00147-5 - 25.
Holland PM, Abramson RD, Watson R, Gelfand DH. Detection of specific polymerase chain reaction product by utilizing the 5′----3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proceedings of the National Academy of Sciences of the United States of America. 1991; 88 (16):7276-7280. DOI: 10.1073/pnas.88.16.7276 - 26.
Livak KJ, Flood SJ, Marmaro J, Giusti W, Deetz K. Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods and Applications. 1995; 4 (6):357-362. DOI: 10.1101/gr.4.6.357 - 27.
https://www.thermofisher.com/in/en/home/life-science/pcr/real-time-pcr/real-time-pcr-assays/snp-genotyping-taqman-assays/taqman-snp-genotyping-assay-sets.html - 28.
Tyagi S, Kramer FR. Molecular beacons: Probes that fluoresce upon hybridization. Nature Biotechnology. 1996; 14 (3):303-308. DOI: 10.1038/nbt0396-303 - 29.
Tyagi S, Bratu DP, Kramer FR. Multicolor molecular beacons for allele discrimination. Nature Biotechnology. 1998; 16 (1):49-53. DOI: 10.1038/nbt0198-49 - 30.
Kostrikis LG, Tyagi S, Mhlanga MM, Ho DD, Kramer FR. Spectral genotyping of human alleles. Science. 1998; 279 (5354):1228-1229. DOI: 10.1126/science.279.5354.1228 - 31.
Tyagi S, Marras SA, Kramer FR. Wavelength-shifting molecular beacons. Nature Biotechnology. 2000; 18 (11):1191-1196. DOI: 10.1038/81192 - 32.
Budowle B, Planz JV, Campbell RS, Eisenberg AJ. Single nucleotide polymorphisms and microarray Technology in Forensic Genetics - development and application to mitochondrial DNA. Forensic Science Review. 2004; 16 (1):21-36 - 33.
Pease AC, Solas D, Sullivan EJ, Cronin MT, Holmes CP, Fodor SP. Light-generated oligonucleotide arrays for rapid DNA sequence analysis. Proceedings of the National Academy of Sciences of the United States of America. 1994; 91 (11):5022-5026. DOI: 10.1073/pnas.91.11.5022 - 34.
Dalma-Weiszhausz DD, Warrington J, Tanimoto EY, Miyada CG. The affymetrix GeneChip platform: An overview. Methods in Enzymology. 2006; 410 :3-28. DOI: 10.1016/S0076-6879(06)10001-4 - 35.
Wang DG, Fan JB, Siao CJ, Berno A, Young P, Sapolsky R, et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 1998; 280 (5366):1077-1082. DOI: 10.1126/science.280.5366.1077 - 36.
Wu R. Development of the primer-extension approach: A key role in DNA sequencing. Trends in Biochemical Sciences. 1994; 19 (10):429-433. DOI: 10.1016/0968-0004(94)90094-9 - 37.
Syvänen AC. From gels to chips: "minisequencing" primer extension for analysis of point mutations and single nucleotide polymorphisms. Human Mutation. 1999; 13 (1):1-10. DOI: 10.1002/(SICI)1098-1004(1999)13:1 - 38.
Babol-Pokora K, Berent J. SNP-minisequencing as an excellent tool for analysing degraded DNA recovered from archival tissues. Acta Biochimica Polonica. 2008; 55 (4):815-819 - 39.
Geppert M, Roewer L. SNaPshot® minisequencing analysis of multiple ancestry-informative Y-SNPs using capillary electrophoresis. Methods in Molecular Biology. 2012; 830 :127-140. DOI: 10.1007/978-1-61779-461-2_9 - 40.
Schlebusch CM, Naidoo T, Soodyall H. SNaPshot minisequencing to resolve mitochondrial macro-haplogroups found in Africa. Electrophoresis. 2009; 30 (21):3657-3664. DOI: 10.1002/elps.200900197 - 41.
https://www.thermofisher.com/order/catalog/product/4323151 - 42.
Haff LA, Smirnov IP. Single-nucleotide polymorphism identification assays using a thermostable DNA polymerase and delayed extraction MALDI-TOF mass spectrometry. Genome Research. 1997; 7 (4):378-388. DOI: 10.1101/gr.7.4.378 - 43.
Braun A, Little DP, Köster H. Detecting CFTR gene mutations by using primer oligo base extension and mass spectrometry. Clinical Chemistry. 1997; 43 (7):1151-1158 - 44.
Paracchini S, Arredi B, Chalk R, Tyler-Smith C. Hierarchical high-throughput SNP genotyping of the human Y chromosome using MALDI-TOF mass spectrometry. Nucleic Acids Research. 2002; 30 (6):e27. DOI: 10.1093/nar/30.6.e27 - 45.
Sauer S, Lechner D, Berlin K, Lehrach H, Escary JL, Fox N, et al. A novel procedure for efficient genotyping of single nucleotide polymorphisms. Nucleic Acids Research. 2000; 28 (5):E13 - 46.
Lavebratt C, Sengul S, Jansson M, Schalling M. Pyrosequencing-based SNP allele frequency estimation in DNA pools. Human Mutation. 2004; 23 (1):92-97. DOI: 10.1002/humu.10292 - 47.
Ahmadian A, Gharizadeh B, Gustafsson AC, Sterky F, Nyrén P, Uhlén M, et al. Single-nucleotide polymorphism analysis by pyrosequencing. Analytical Biochemistry. 2000; 280 (1):103-110. DOI: 10.1006/abio.2000.4493 - 48.
Russom A, Tooke N, Andersson H, Stemme G. Single nucleotide polymorphism analysis by allele-specific primer extension with real-time bioluminescence detection in a microfluidic device. Journal of Chromatography. A. 2003; 1014 (1-2):37-45. DOI: 10.1016/s0021-9673(03)01033-1 - 49.
Rejali NA, Moric E, Wittwer CT. The effect of single mismatches on primer extension. Clinical Chemistry. 2018; 64 (5):801-809. DOI: 10.1373/clinchem.2017.282285 - 50.
Vallone PM, Just RS, Coble MD, Butler JM, Parsons TJ. A multiplex allele-specific primer extension assay for forensically informative SNPs distributed throughout the mitochondrial genome. International Journal of Legal Medicine. 2004; 118 (3):147-157. DOI: 10.1007/s00414-004-0428-5 - 51.
Landegren U, Kaiser R, Sanders J, Hood L. A ligase-mediated gene detection technique. Science. 1988; 241 (4869):1077-1080. DOI: 10.1126/science.3413476 - 52.
Grossman PD, Bloch W, Brinson E, Chang CC, Eggerding FA, Fung S, et al. High-density multiplex detection of nucleic acid sequences: Oligonucleotide ligation assay and sequence-coded separation. Nucleic Acids Research. 1994; 22 (21):4527-4534 - 53.
Eggerding FA. A one-step coupled amplification and oligonucleotide ligation procedure for multiplex genetic typing. PCR Methods and Applications. 1995; 4 (6):337-345. DOI: 10.1101/gr.4.6.337 PMID: 7580927 - 54.
https://assets.thermofisher.com/TFS-Assets/LSG/Specification-Sheets/cms_059523.pdf - 55.
Lyamichev V, Mast A, Hall J, et al. Polymorphism identification and quantitative detection of genomic DNA by invasive cleavage of oligonucleotide probes. Nature Biotechnology. 1999; 17 :292-296. DOI: 10.1038/7044 - 56.
Olivier M. The invader assay for SNP genotyping. Mutation Research. 2005; 573 (1-2):103-110. DOI: 10.1016/j.mrfmmm.2004.08.016 - 57.
Nakahara H, Sekiguchi K, Hosono N, Kubo M, Takahashi A, Nakamura Y, et al. Criterion values for multiplex SNP genotyping by the invader assay. Forensic Science International. Genetics. 2010; 4 (2):130-136. DOI: 10.1016/j.fsigen.2009.07.005