Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
Chronic Kidney Disease: Mineral Bone Disorder (CKD-MBD) is a complex syndrome, encompassing biochemical, skeletal, and cardiovascular complications. The heterogeneity of individual manifestations, the multitude of contributory factors, and the absence of biomarkers to identify the complications and intervene early in the course have resulted in the current clinical approach, where clinicians are addressing primarily biochemical abnormalities in the advanced stages of chronic kidney disease. The late intervention and the use of surrogate targets that are far removed from the relevant clinical outcomes of fracture and accelerated cardiovascular disease, causes the poor survival of individuals with CKD. The identification of early events in the development of CKD-MBD and the recognition of novel mediators of CKD-associated skeletal and cardiovascular disease are setting the stage for a revolution in how we approach and treat CKD-MBD. The complexity of CKD-MBD lends itself to an alternative approach to management using an artificial intelligence approach combined with a sophisticated mathematical model of the disease to help clinicians target disease complications in a personalized fashion.
Medical Services, VA North Texas Health Care System, Dallas, TX, United States
Department of Medicine, University of Texas Southwestern Medical Center, Dallas, TX, United States
Michael E. Brier
Department of Medicine, University of Louisville School of Medicine, Louisville, KY, United States
Research Service, Robley Rex VA Medical Center, Louisville, KY, United States
Adam E. Gaweda
Department of Medicine, University of Louisville School of Medicine, Louisville, KY, United States
*Address all correspondence to: eleanor.lederer@utsouthwestern.edu
1. Introduction
The derangements of mineral metabolism that occur as chronic kidney disease (CKD) progresses encompass far more than mere biochemical abnormalities of calcium and phosphorus. Recognition of the full manifestation of the syndrome was acknowledged by the international guideline development organization, Kidney Disease: Improving Global Outcomes in 2006. Named Chronic Kidney Disease: Mineral and Bone Disorder (CKD-MDB), this complex syndrome includes the biochemical abnormalities (hyperphosphatemia, hypocalcemia, secondary hyperparathyroidism), the skeletal abnormalities (renal osteodystrophy), and the development of soft tissue calcification, especially in the vasculature [1].
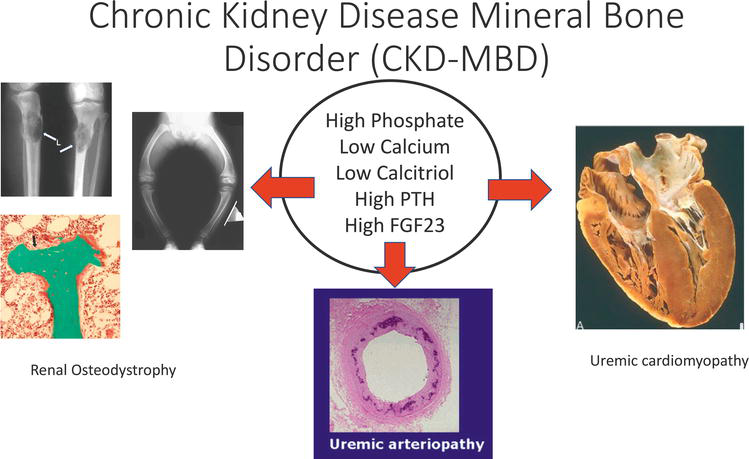
CKD-MBD has effects far beyond the balance of minerals, with the two major clinical sequelae of CKD-MBD being fractures and accelerated cardiovascular disease (Figure 1) [2, 3, 4]. Individuals with CKD develop bone fragility due to decreased mineralization and abnormal bone structure. The incidence of “osteoporosis” in individuals with CKD is not truly known as few large-scale studies have systematically assessed bone density; however, multiple studies have shown that the incidence of fracture is higher in CKD patients when compared to the general population [5, 6, 7, 8, 9, 10, 11]. The other major sequela of CKD-MBD is accelerated cardiovascular disease manifested as cardiomyopathy, cardiac arrhythmias, sudden cardiac death, calcific valvular heart disease, pulmonary hypertension, and vascular smooth muscle calcification, also known as uremic or medical arteriolar calcification [12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22]. These complications contribute heavily to the poor survival of patients with CKD. The increased mortality can be seen as early as stage 1 CKD. While the decrease in survival is modest with stage 1 CKD, this mortality effect is amplified with each increasing stage of CKD with the greatest effect seen in younger patients. For example, a recent study observed that a 30-year-old individual starting dialysis would have an expected life span of only 12 years, markedly decreasing from the expected 45–50 years of a healthy individual [12]. As the stage of CKD progresses the fraction of patients dying of cardiovascular disease increases to as high as 50% of all deaths [12, 13]. Undergoing kidney transplantation, our best therapy for end-stage kidney disease (kidney failure), improves but does not confer a normal life span. In fact, the individual who receives a kidney transplant can expect a survival comparable to an individual with stage 3 CKD [22].
Figure 1.
Definition of chronic kidney disease mineral bone disorder. This figure graphically depicts the major components of CKD-MBD.
Although life-saving, individuals with complete kidney failure treated with either dialysis or transplantation do not enjoy a normal lifespan. Notably, these therapies are for complete kidney failure, suggesting that interventions late in the course of CKD are inadequate to achieve the optimal impact on the morbidity and mortality of CKD. Disappointingly, the application of interventions such as statins, blood pressure control, and weight control in these individuals have failed to change materially the outcome for these individuals with complete kidney failure, confirming the observation that late intervention is ineffective [23, 24, 25, 26]. Although renal osteodystrophy as a sequela of CKD has been recognized for over 60 years, the impact of abnormal mineral metabolism on the cardiovascular system has only been recently appreciated [15, 16]. Recent advances in the field have uncovered previously unknown mediators of mineral metabolism such as klotho, activin A, DKK1 (dickkopf 1), FGF23, and sclerostin, to name just a few, that have an impact on skeleton and cardiovascular systems to produce the chronic complications known as CKD-MBD [27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37]. Alterations in the regulation of many of these mediators occur at the earliest stages of CKD. Within the past few years, several investigators have conducted trials intervening earlier in the course of CKD to ameliorate the skeletal and cardiovascular manifestation of CKD-MBD, generally targeting single discrete targets. These studies are in preliminary stages and the results have been decidedly mixed. For example, one group of investigators tested the hypothesis that limiting phosphate absorption early in the course of kidney disease before serum phosphate was frankly elevated could ameliorate the long-term outcomes [38]. In this relatively short-term trial, the outcomes were changes in urine phosphate and serum levels of FGF23. Unfortunately, the primary outcomes were not achieved. On the other hand, interventions to ameliorate the acidosis associated with CKD by giving either a higher fruit/vegetable diet, supplemental bicarbonate, or an acid-binding polymer suggest slower deterioration of kidney function [39, 40, 41]. The effects on skeletal and cardiovascular outcomes have not been reported as yet. Another interesting study demonstrated that administration of an inhibitor of the inflammatory cytokine, IL-1, decreased biochemical markers of inflammation and improved markers of vascular health [42] but had no discernible effects on markers of mineral metabolism. The long-term effects are not yet available. However, these and other studies justify the ongoing investigation into the identification of earlier mediators of the development of CKD-MBD that could become therapeutic targets to improve outcomes [43, 44, 45, 46, 47].
The recognition of the systemic nature of the pathogenesis of CKD-MBD has led to a greater awareness of the marked heterogeneity in the manifestations of CKD-MBD and difficulty in identifying individual pathology, particularly with regard to bone health [48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63]. At this time, the absence of clear evidence from clinical trials for early interventions has hindered the development of clinical guidelines for evaluation of skeletal and cardiovascular status or risk factors in individuals with CKD. No standardized assessments of skeletal and cardiovascular health have been established. Phenotypic classification to define individual risks has not been accomplished. For the individual patient, a clinician is unable to predict which complications will likely occur. The result is that all patients with CKD are approached similarly with the same tools and the same targets, which are primarily the biochemical targets of CKD-MBD – calcium, phosphorus, and PTH levels. Overall, these considerations have led to the inevitable conclusion that as a medical community, we need to 1) treat patients with CKD earlier in the course to prevent the deadly complications, 2) identify the best biomarkers to predict adverse complications, 3) develop new therapies for CKD-MBD based on pathogenesis, 4) target the clinically significant sequelae of CKD-MBD, and 5) tailor therapies to the individual patient. The complexity of CKD-MBD makes it an ideal condition to apply artificial intelligence techniques to improve the outcomes for individuals afflicted with this deadly disorder.
The initiating event thought to trigger the development of CKD-MBD is the challenge of maintaining phosphate balance in the setting of declining kidney function [64, 65]. Dietary intake and absorption of phosphate are well in excess of daily needs. The kidneys are primarily responsible for maintaining phosphate balance by altering tubular reabsorption of phosphate from glomerular ultrafiltrate [66]. Even at the earliest stage of CKD, an orchestrated series of changes occur to ensure that the kidneys can excrete the daily load of phosphate. These changes include decreased expression of transport proteins in the kidney that reabsorb filtered phosphate through the activation of a hormone called fibroblast growth factor 23 (FGF23). FGF23 is a hormone, made by osteocytes primarily in response to a phosphate load that interacts with FGF receptors on the kidney proximal tubule in conjunction with its essential cofactor, klotho, a glucuronidase made almost exclusively in kidney [32, 33, 34, 35, 36, 67, 68]. The result is the activation of a signaling cascade that reduces the expression of sodium-coupled phosphate transporters on kidney proximal tubule cells and increased the excretion of phosphate by those nephrons. The other major phosphaturic hormone, parathyroid hormone (PTH), also contributes to the enhancement of kidney excretion of phosphate at the single nephron level by reducing the expression of phosphate transporters [66, 69, 70]. While immediate and transient changes in FGF23 and PTH can be elicited in individuals without CKD by providing a phosphate load, the presence of CKD leads to early (stage 1 CKD) sustained increases in FGF23 followed by sustained increases in PTH, generally beginning late stage 3 CKD. The other major effect of FGF23 that influences phosphate homeostasis is the downregulation of the formation of 1,25 dihydroxy vitamin D, the active form of vitamin D that is responsible for enhancing intestinal absorption of both calcium and phosphate, necessary for normal bone mineralization [32, 33, 34, 35, 36, 67, 71]. The final activating step of vitamin D occurs in the kidney. FGF23 suppresses the formation of active vitamin D by decreasing the production of 1 alpha-hydroxylase (CYP27B1), the activating enzyme. Simultaneously, FGF23 promotes inactivation of vitamin D through the increased production of 24 hydroxylase (CYP24A1), one of the vitamin D inactivating enzymes. Together, these two effects of FGF23 to decrease intestinal absorption of phosphate and decrease kidney reabsorption of phosphate serve to maintain phosphate balance through the early stages of CKD. Patients with CKD do not become hyperphosphatemic until the late stages of CKD, while the increase in FGF23 and the decrease in calcitriol occur very early. Importantly, neither FGF23 nor calcitriol is routinely measured parameters in patients with CKD.
In addition to a loss of excretory function, kidney injury leads to the elaboration and release of additional circulating substances that influence mineral metabolism [27, 28, 29, 30, 31]. Two of these substances, DKK1 and activin A, are released from injured kidneys into the circulation, stimulating the release of sclerostin from osteocytes. Sclerostin, in turn, inhibits bone formation. Additional molecules released by injured kidney cells, damage-associated molecular patterns (DAMPs) or alarmins, stimulate the release of inflammatory cytokines that also have a direct effect on bone turnover, indirect effects through their detrimental effects on muscle, and increase FGF23 production [72, 73, 74, 75, 76]. The expression of klotho, the essential cofactor for FGF23 signaling in the kidney proximal tubule, decreases at the onset of CKD, resulting in impaired FGF23 signaling [36]. This consequence then leads to increased stimulation of FGF23 to accomplish the primary task of blocking kidney phosphate reabsorption. Klotho also has anti-inflammatory and anti-oxidant properties; thus, the loss of klotho amplifies the inflammatory response, additionally contributing to FGF23 release. The development of anemia associated with CKD also has an effect on FGF23 release, thought to be mediated by the abnormalities in iron utilization [77, 78]. Anemia increases both the production and the cleavage of FGF23. The current understanding of FGF23 effects is that intact FGF23 is the active entity; however, the cleaved products may also have as yet unknown effects on mineral metabolism.
The consequences of the accelerating pace of FGF23 generation are manifold. Calcitriol production and expression continue to decline. FGF23 at very high levels also appears to interact with FGF receptors on the heart which can be activated in the absence of klotho. The results of FGF23 stimulation of heart FGF receptors is cardiac muscle remodeling leading to left ventricular hypertrophy and eventual heart failure [32, 33, 68, 79, 80]. The markedly increased FGF23 levels have been implicated in the development of vascular calcification. FGF23 inhibition of calcitriol expression coupled with the presence of high levels of sclerostin impairs bone formation and turnover, leading to defects in skeletal structure and mineralization [27, 31]. Active vitamin D has also been implicated in vascular health; thus, the decrease in active vitamin D levels may also contribute to uremic vasculopathy [81, 82].
As CKD progresses, the ability of the body to maintain relatively normal levels of serum phosphate, calcium, and PTH falters. Eventually, serum phosphate rises and serum calcium falls. Secondary hyperparathyroidism develops due to the combination of hyperphosphatemia, hypocalcemia, and low calcitriol levels. Despite attainment of PTH levels well in excess of the upper limits of normal, calcitriol levels remain low. PTH, in part, combats the decrease in bone formation mediated by sclerostin but the alterations in bone turnover and mineralization persist. The interplay between the high bone turnover effects of hyperparathyroidism and the low bone turnover effects of high sclerostin and low calcitriol yields divergent clinical sequelae, which are nearly impossible to distinguish without bone biopsy. Some individuals show predominantly high turnover bone disease, others low turnover and yet others demonstrate a combined picture. Irrespective of bone histology, these individuals are at high risk for fracture. An additional consequence of the prolonged and increasingly deranged mineral metabolism is that the smooth muscle cells of the vasculature undergo a transformation by which they gain properties of osteoblasts, i.e., increased expression of osteoblast genes and proteins such as collagen, osteocalcin, alkaline phosphatase, and the phosphate transporter PiT-1 and decreased expression of the inhibitor of soft tissue calcification, matrix GLA protein [80, 83, 84]. The consequence is the elaboration of matrix vesicles that promote vascular smooth muscle calcification, a consequence that has been implicated in the development of severe vascular complications including peripheral tissue gangrene and calciphylaxis.
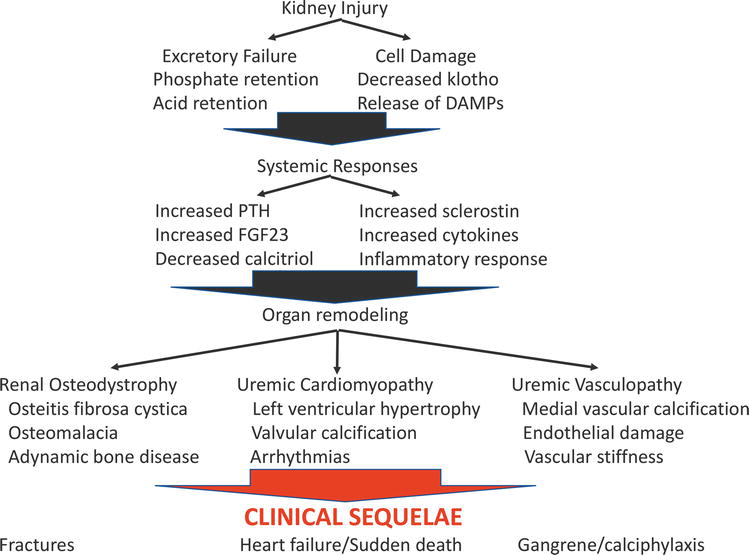
Figure 2 summarizes the pathogenesis of CKD-MBD from the skeletal and cardiovascular perspectives. Two fundamental processes merge to produce the complex picture of CKD-MBD: renal excretory failure involving primarily challenges to maintaining phosphate homeostasis and the kidney cell injury response leading to the release of circulating factors that affect skeletal health, systemic inflammatory status, and oxidative stress. The tissue level results are bone fragility, cardiomyopathy, and vasculopathy. The clinical consequences experienced by the patients are fractures, heart failure, arrhythmias, peripheral gangrene, valvular heart disease, and calciphylaxis. This extensive review of the pathogenesis sets the stage for appreciating the complexity of the pathogenesis and the varied clinical manifestations that have defied effective therapy for decades. Clearly, the current targets for therapy of CKD-MBD, i.e., control of the abnormal biochemical parameters calcium, phosphorus, and PTH, do not address the fundamental pathogenesis of CKD-MBD. Significant effort has been exerted to control serum phosphate and combat phosphate retention through diet, phosphate binders, and dialysis. A variety of active vitamin D analogs to treat secondary hyperparathyroidism have been developed and compared. The advent of the use of calcium-sensing receptor agonists as an alternative means to control PTH without raising serum calcium was met with great enthusiasm. Trials describing the use of and comparing these agents have verified that PTH can be controlled but have not resulted in a meaningful increase in survival for individuals with CKD. These targets for therapy are biochemical markers that become abnormal late in the course of kidney disease and are far removed from the meaningful clinical sequelae of CKD-MBD – renal osteodystrophy, uremic cardiomyopathy, and uremic vasculopathy. It is our contention that the complexity of CKD-MBD mandates a sophisticated approach employing artificial intelligence techniques that will allow clinicians to identify individuals at high risk for specific complications of CKD-MBD and will facilitate the development of effective and personalized therapies early in the course of disease to ameliorate the complications and thus the accelerated mortality associated with CKD-MBD.
Figure 2.
Pathogenesis of CKD-MBD. This figure depicts, in simplified form, the major features outlining the development of CKD-MBD from the initial insult (kidney injury) to the clinical sequelae.
3. Artificial intelligence applications to kidney disease
The preceding section has outlined the complexity of CKD-MBD where the interplay of multiple bowel chemical factors working in concert or counter to other factors makes treatment multidimensional. This multi-dimensionality is naively addressed during drug development without a requirement for an existing drug to be optimized following the introduction of a new therapeutic agent or biochemical factors more directly related to the outcomes of interest. The use of artificial intelligence in combination with advanced mathematical modeling can be applied to solve this dilemma.
The concept of applying Artificial Intelligence (AI) to assist in diagnosing and treating kidney diseases is not a new idea but has been underexplored. Artificial Intelligence methods are merely computational models of cognitive and learning functions inherent to human beings (Table 1). Important in this definition is the assertion that at the current state of the art the AI tools are there to aid, not replace, the clinician and the implication that AI can enhance human capabilities but cannot act autonomously. Of all AI branches, Machine Learning (ML) has been the family of methods most commonly applied in nephrology. The basic underpinning of ML is the principle that a computational system using a human-like learning process can be trained to recognize and predict patterns known (supervised learning) or unknown (unsupervised learning) to human users. However, we wish to move past the thesis of Turing in 1950 where he proposed the question “can machine think?” [85]. It is simply not the goal of these advanced mathematical techniques to “fool” the user and believe that treatment recommendations are coming from a human. To this end, we wish to create new knowledge and use that knowledge represented as a mathematical equation to describe the process we wish to improve and two understand the repercussions of multiple drug therapies on the process we wish to control.
AI methods
Application Area
Supervised Learning: Decision Trees Neural Networks
Diagnostics Predictive Modeling Information Mining
Commonly Used AI Methods in Medicine and Their Applications.
In general terms, Supervised Learning recapitulates and generalizes human knowledge encoded in data. A good example of supervised learning is a diagnostic application where the machine is trained to “read” histology slides of biopsies from human kidneys previously labeled by a human expert. Several publications document the utility of AI methods in this realm. The machine or “agent” is presented with a large number of kidney biopsy slides demonstrating the range of pathology and is trained to recognize specific pathologic findings as would be performed by a human pathologist. With appropriate “training”, a supervised learning agent can provide very rapid and reproducible histopathologic diagnoses. The application of this form of ML can dramatically increase the diagnostic throughput of cases, enabling more rapid reading of slides and transmission of the diagnoses to the referring physicians. Supervised Learning AI methods have been applied to the identification of kidney transplant rejection and multiple forms of glomerular disease. Malcolm Gladwell has famously stated that an individual needs to devote 10,000 hours to become an expert [86]. For a clinician, this goal may require years or even decades to achieve. For the computer, this process can be compressed into hours.
Unsupervised Learning is a technique that can be used to autonomously discover patterns in data. Importantly, these patterns may or may not be previously known to the human operator. Similar to supervised learning, the machine is provided with data from a large number of subjects, encompassing the spectrum of the disease process to be studied. The machine can organize the subjects into different categories using measures of similarity specified by the human operator. For example, a common clinical question that has been addressed with unsupervised ML is the prediction of acute kidney injury in the intensive care unit. While intensivists have attempted to identify patients at high risk for the development of acute kidney injury, the usual parameters that have been tested have demonstrated poor sensitivity and specificity. Being able to determine who is most likely to develop acute kidney injury in this setting could lead to preventive measures such as avoiding nonsteroidal anti-inflammatory drugs or other nephrotoxic drugs. Thus, Unsupervised Learning is useful for clinical questions that have been previously unexplored or unresolved The advantage of applying AI methods to this type of scenario is that the computational capacity allows the processing of much larger amounts of information than can be processed by the human brain, enabling the machine to organize the analyzed variables in various different ways. That being said, it cannot be emphasized enough that although the learning process itself is free of bias, the quality of the output produced is dependent on the data being provided. Clearly, data generated through human intervention contain multiple sources of bias, leaving open the possibility that important factors to address the specific question may not be included in the dataset provided to the computational system. Incorporating additional AI applications into systems with greater computational capacity can address this potential deficiency to some degree. For example, including information from patient records through Natural Language Processing (NLP) can markedly enrich the source information and potentially lead to real breakthroughs in the understanding disease process. For example, inclusion of patient symptomatology from the electronic medical record in addition to easily digitized patient information has led to the discovery that the prevalence of genetic disorders is more common than previously recognized [87, 88]. Several other examples of the use of AI for data fusion to identify new patterns of disease presentation or prevalence include the eMERGE initiative, the Million Veteran Program, and Vanderbilt University’s Synthetic Derivative database.
These examples leverage the vast quantities of data stored in the electronic medical record and the advances in image processing that allow a digital image to be obtained. Most endeavors of this type have demonstrated almost no difference in recognition of disease over the current clinical practice. In areas where the economic cost of humans performing this work is high, an artificial intelligence approach may prove beneficial. However, our goal should be to leverage this data and the associations, whether known or unknown, between observation and outcome to exceed current clinical care.
4. Application of artificial intelligence techniques to CKD-MBD
The arguments for considering the application of AI to CKD-MBD have been touched on. CKD-MBD is an extraordinarily complex clinical syndrome. The pathophysiology is incompletely understood but recent studies have uncovered novel mediators such as FGF23 and sclerostin that present very early during kidney disease. These mediators are not routinely measured, but some are actually targets of new therapies for bone disease. Determining if these new therapies could be used for the treatment of CKD-MBD is a timely question. In fact, the treatment of CKD-MBD has not materially changed in decades. The targets remain control of serum phosphate, control of serum calcium, and control of parathyroid hormone levels. The current guidelines provide little guidance on the evaluation and treatment of CKD-MBD in the early stages CKD primarily because of a dearth of clinical research. Consequently, clinicians have been unable to materially improve the outcomes of individuals with CKD. The survival of patients with CKD remains unacceptably poor.
Several groups of investigators have looked into methods of using large databases to develop a phenotypic characterization of individuals with CKD and predictive models for some specific outcomes such as progression of CKD to ESRD or development of cardiovascular events. The research program in the Nephrology Division at the University of Louisville School of Medicine and the affiliated Department of Veterans Affairs Robley Rex VA Medical Center has focused on the development of a comprehensive Systems Biology approach to the complications of CKD over the past 20 years. The goal of the initial project was to develop a tool that would individualize the therapy of the anemia of kidney disease in dialysis patients. For this project, the investigators employed an Artificial Neural Network (ANN) combined with Model Predictive Control. The ANN is a complex nonlinear regression model, originally created as a way of mimicking the structure of human brain that can be used to replicate complex relationships encoded in large data sets using Supervised Learning. Model Predictive Control is a modern automatic control methodology that, in the context of anemia control, uses a model of the patient to predict the response to erythropoietin therapy. Based on the predicted patient responses to specific drug manipulations, optimal dose adjustments are found that optimize the likelihood of maintaining the relevant blood parameters within defined guideline levels. The model can be capable of “learning” each individual patient’s pattern of response to therapy thus enabling treatment personalization. This approach was highly successful, resulting in avoidance of wide swings in hematocrit and more efficient utilization of erythropoietic therapy. As stated previously, these investigators reached the goal of improving anemia management over current clinical practice through the publication of two double-blind, randomized, clinical trials [89, 90]. Hemoglobin variability was significantly decreased using an artificial intelligence approach. In a follow-up publication on long-term use of the artificial intelligence approach, the researchers also were able to demonstrate a 25% decrease in erythropoietin exposure [91].
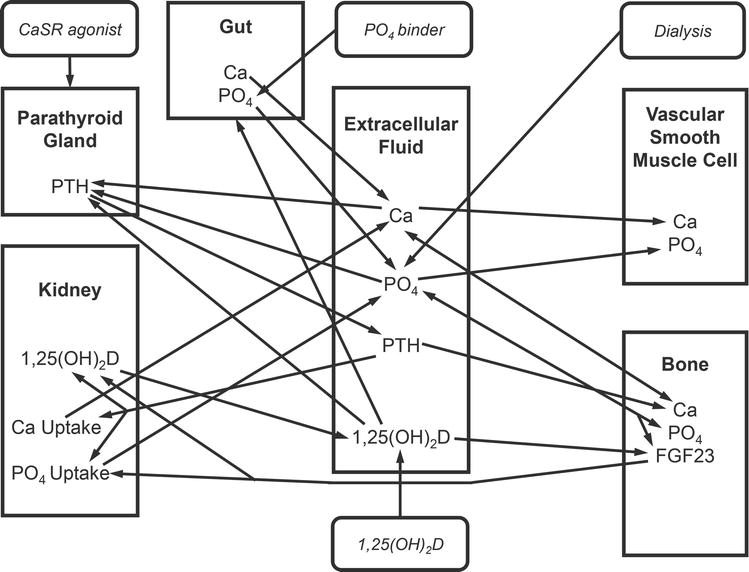
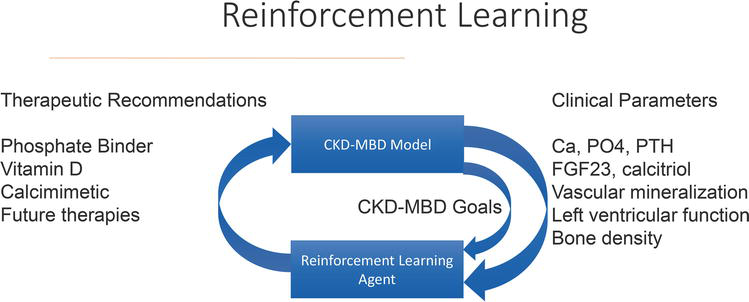
Based on the success of this project, the group next tackled the challenge of individualization of therapy of CKD-MBD, initially focusing on the achievement of published guidelines for calcium, phosphorus, and PTH levels in dialysis patients. This project represented a significant escalation in complexity. First, three separate but interconnected goals of therapy are targeted. Second, multiple therapies are available for use: dialysis prescription, intestinal phosphate binders, active vitamin D analogues, and calcium-sensing receptor agonists. Third, patient behavioral issues significantly influence the outcomes. Adherence to phosphate binder therapy is entirely under patient control. Many of the vitamin D analogs and the calcium-sensing receptor agonists are also oral agents, and thus their efficacy is impacted by patient adherence to the regimen. The solution applied to this project differed somewhat from the previously described AI techniques. A model to describe the evolution of CKD-MBD as kidney failure progresses was developed and validated (Figure 3) [92]. This model, based on a previously published model of mineral metabolism in healthy individuals, was modified to describe the changes in mineral metabolism, bone turnover, and soft tissue calcification that occur through ordinary differential Equations [93]. The challenge of optimal CKD-MBD treatment was addressed using an AI technique called Reinforcement Learning (RL). This ML modality is a hybrid between Supervised and Unsupervised Learning that incorporates a reward system inherent to trial-and-error human learning in order to let an intelligent agent discover the optimal pattern of behavior on its own (Figure 4). Using a systems biology model of a disease, an RL agent can be trained by exposure to a wide variety of simulated patient data that describe the spectrum of disease, in this case, calcium, phosphorus, and PTH in dialysis patients. The agent is provided by the human operator with the goals of therapy for all three biochemical parameters, for example, the targets defined by the published guidelines. By mimicking the human trial-and-error learning, the agent discovers which drugs to use, when, and in what dose amounts, to achieve these goals. One advantage of this approach, especially in the case of complex diseases such as CKD-MBD, is the ability to discover new, previously unknown treatment strategies without exposing human subjects to unnecessary risk. In experiments using simulated subjects, achievement of the guideline goals of therapy occurred more rapidly using the Reinforcement Learning approach as opposed to a standard-of-care approach. The merger of an artificial intelligence approach (reinforcement learning) and a quantitative systems pharmacology (QSP) model of CKD-MBD creates a powerful tool. Although time-consuming to develop, the QSP model allows for extrapolation outside of the collected data that may exist in an electronic medical record. Substitution of the QSP model with another technique like an artificial neural network would significantly constrain the scope of the system, as the artificial neural network which is not capable of performing such extrapolations would likely respond to dose combinations at the edges of the data space with nonsensical results. Adding reinforcement learning allows rigorous probing of the entire data space to discover new unrecognized treatment approaches. Further, this approach is scalable when new biochemical markers are discovered, and new therapeutic agents are developed to target those markers.
Figure 3.
Block diagram model of CKD-MBD. This figure depicts a simplified version of the systems biology model of CKD-MBD encompassing the major organs involved (kidney, intestine, bone, parathyroid), the biochemical features (Ca [calcium], PO4 [phosphate], PTH [parathyroid hormone], 1,25 vitamin D [calcitriol], FGF23 [fibroblast growth factor 23]), and the interventions (CaSR agonist, 1,25 (OH)2, dialysis). The arrows designate relationships between components. The final model consists of over 100 different parameters, the relationships described as ordinary differential equations.
Figure 4.
Schema of reinforcement learning. This figure depicts the iterative and exploratory process of reinforcement learning. The agent is provided with therapies (depicted on the left-hand side), clinical parameters (depicted on the right-hand side), and the goals of therapy (CKD-MBD goal). The agent then chooses a therapy, runs it through the model to assess the outcome, and makes a therapeutic recommendation. If the outcome measured against the goal is favorable, the agent receives a “reward” which reinforces the specific approach. In contrast, if the outcome from the therapeutic recommendation does not come closer to achieving the pre-set goals of therapy, the agent does not receive a “reward” and alters the therapeutic recommendation.
The development of the comprehensive model that recapitulated the progression of CKD-MBD throughout the spectrum of CKD stages yielded additional interesting considerations. The model included changes in the levels of calcitriol (active vitamin D) and FGF23 with the progression of CKD, biochemical parameters that are not routinely measured. The model also included equations that described the movement of calcium and phosphorus in and out of bone and in and out of soft tissue. These features of CKD-MBD are not measurable but represent the processes that result in the clinically relevant outcomes including increased fracture risk and vascular disease. The model predicted changes in calcitriol and FGF23 at the onset of CKD, corresponding to published literature on the subject. More significantly, the model predicted that the loss of bone minerals and the increase in vascular calcification started at the earliest stages of CKD and accelerated throughout the course of CKD. The model also predicted that the application of the Reinforcement Learning agent to achieve the goals of CKD-MBD for calcium, phosphorus, and PTH resulted in improvement in calcitriol and FGF23 levels and a reduction in bone mineral loss that was superior to the results seen with the Supervised agent (i.e., standard of care).
The capabilities of this artificial intelligence approach combining the comprehensive model of CKD-MBD with Reinforcement Learning make it possible to examine different approaches to CKD-MBD through in silico clinical trials, targeting the relevant clinical outcomes of fracture, cardiomyopathy, and peripheral vascular disease. For example, instead of setting specific levels of calcium, phosphorus, and PTH as the goals of therapy, the goals can be minimization of bone mineral loss or minimization of vascular smooth muscle calcification. Setting these goals may yield vastly different recommendations for the use of the calcium-sensing receptor agonists or vitamin D. Additionally, the model has the flexibility to incorporate additional parameters. For example, the effects of sclerostin, activin A, and inflammatory markers can be incorporated into the model as additional mediators of CKD-MBD. Many currently available treatments such as the monoclonal antibodies directed against sclerostin (romosuzamab) used for osteoporosis or FGF23 (burosumab) used for genetic forms of rickets can be tested for their effects on the biochemical parameters, the mineralization abnormalities, and the cardiomyopathy of CKD-MBD in silico. The results from these simulations could then be used to inform the development of clinical trials. One additional advantage of the in-silico approach is that multiple interventions can be tested both simultaneously and singly very rapidly. It is quite difficult to develop clinical trials testing multiple interventions because of the number of subjects needed to achieve adequate power and the difficulties in interpretation in the setting of multiple interventions. The application of this approach is expected to result in potentially paradigm-changing concepts of the pathogenesis as well as the treatment of CKD-MBD. For decades, the cornerstone of therapy of CKD-MBD has been the use of phosphate binders for limiting intestinal absorption. Yet a recent extensive review clarified that no trials have demonstrated a survival benefit with the use of any phosphate binder [94]. On the surface, this would suggest that phosphate binders have no role in the therapy of CKD. On the other hand, another study identified a link between phosphate control and a beneficial effect of statins in advanced CKD, suggesting that examining more complex patterns of disease development and therapy may yield better results [95]. This type of approach can be significantly facilitated by the application of AI.
Artificial intelligence techniques are powerful tools that hold the promise of revolutionizing the delivery of medical care. The complexity, profound clinical impact, and rapidly evolving understanding of CKD-MBD suggest that artificial intelligence could accelerate the development of newer effective therapies for this deadly disorder. Challenges remain. Refinement of the model to ensure the accurate and complete representation of all aspects of CKD-MBD is an ongoing project. Validation of the predictions and recommendations from this computational system will require human trials. Finally, provider and patient acceptance of a computer program contributing to medical care may not be immediate. Despite these challenges, the use of a variety of artificial intelligence applications in multiple clinical settings is increasing and is increasingly accepted. More to come.
References
1.Moe SM, Drueke T, Cunningham J, Goodman W, et al. Definition, evaluation, and classification of renal osteodystrophy: A position statement from kidney disease: Improving global outcomes (KDIGO). Kidney International. 2006;69(11):1945-1953
2.Moe SM, Drueke T, Lameire N, Eknoyan G. Chronic kidney disease-mineral-bone disorder: A new paradigm. Advances in Chronic Kidney Disease. 2007;14(1):3-12
3.Pazianas M, Miller PD. Current understanding of mineral and bone disorders of chronic kidney disease and the scientific grounds on the use of exogenous parathyroid hormone in its management. Journal of Bone Metabolism. 2020;27(1):1-13
4.de Albuquerque Suassuna PG, Sanders-Pinheiro H, de Paula RB. Uremic cardiomyopathy: A new piece in the chronic kidney disease-mineral and bone disorder puzzle. Frontiers in medicine (Lausanne). 2018;5:206
5.Allen MR, Swallow EA, Metzger CE. Kidney disease and bone: Changing the way we look at skeletal health. Current Osteoporosis Reports. 2020;18(3):242-246
6.Gordon PL, Frassetto LA. Management of osteoporosis in CKD stages 3 to 5. American Journal of Kidney Diseases. 2010;55(5):941-956
7.Ketteler M, Block GA, Evenapoel P, Fukagawa M, et al. Executive summary of the 2017 KDIGO chronic kidney disease-mineral bone disorder (CKD-MBD) guideline update: what’s changed and why it matters. Kidney International. 2017;92(1):26-36
8.Pimentel A, Urena-Torres P, Bover J, Luis Fernandez-Martin J, et al. Bone fragility fractures in CKD patients. Calcified Tissue International. 2021;108(4):539-550
9.Jadoul M, Albert JM, Akiba T, et al. Incidence and risk factors for hip or other bone fractures among hemodialysis patients in the dialysis outcomes and practice patterns study. Kidney International. 2006;70:1358-1366
10.Tentori F, McCullough K, Kilpatrick RD, et al. High rates of death and hospitalization follow bone fracture among hemodialysis patients. Kidney International. 2014;85:166-173
11.Iseri K, Carrero JJ, Evans M, Felländer-Tsai L, Berg HE, Runesson B, et al. Fractures after kidney transplantation: Incidence, predictors, and association with mortality. Bone. 2020;140:115554
12.Gansevoort RT, Correa-Rotter R, Hemmelgarn BR, Jafar TH, Heerspink HJL, et al. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. The Lancet. 2013;382(9889):339-352
13.Thompson S, James M, Wiebe N, Hemmelgarn B, Manns B, et al. Cause of death in patients with reduced kidney function. Journal of the American Society of Nephrology. 2015;26:2504-2511
14.Rroji M, Figurek A, Spasovski G. Should we consider the cardiovascular system while evaluating CKD-MBD? Toxins (Basel). 2020;12(3):140
15.Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, et al. Kidney disease as a risk factor for development of cardiovascular disease. Hypertension. 2003;42(5):1050-1065
16.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu C-y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. The New England Journal of Medicine. 2004;350(13):1296-1305
17.Budoff MJ, Rader DJ, Reilly MP, Mohler ER, Lash J, et al. Relationship of estimated GFR and coronary artery calcification in the CRIC (chronic renal insufficiency cohort) study. American Journal of Kidney Diseases. 2011;58(4):519-526
18.Go AS, Tan TC, Chertow GM, Ordonez JD, Fan d, et al. Primary nephrotic syndrome and risks of ESKD, Cardovascular events, and death: The Kaiser Permanente nephrotic syndrome study. Journal of the American Society of Nephrology 2021; 32(9):2303-2314.
19.Mace ML, Egstrand S, Morevati M, Olgaard K, Lewin E. New insights to the crosstalk between vascular and bone tissue in chronic kidney disease-mineral and bone disorder. Metabolites. 2021;11(12):849
20.Wang AA, Cai X, Srivastava A, Prasad PV, Sprague SM, et al. Abnormalities in cardiac structure and function among individuals with CKD: The COMBINE trial. Kidney. 2021;3(2):258-268
21.Ginsberg C, Jouben AJHM, Malhotra R, Berendschot TTJM, Dagnelie PC, et al. Serum phosphate and microvascular function in a population-based cohort. CJASN. 2019;14:1626-1633
24.Dasgupta I, Zoccali C. Is the KDIGO systolic blood pressure target <120mm Hg for chronic kidney disease appropriate in routine clinical practice? Hypertension. 2022;79(1):4-11
25.Zewinger S, Speer T, Kleber ME, Scharnagl H, Woitas R, et al. HDL cholesterol is not associated with lower mortality in patients with kidney dysfunction. Journal of the American Society of Nephrology. 2014;25:1073-1082
26.Kotsis V, Martinez F, Trakatelli C, Redon J. Impact of obesity in kidney diseases. Nutrients. 2021;13:4482
27.Carrillo-López N, Martínez-Arias L, Fernández-Villabrille S, Ruiz-Torres MP, Dusso A, et al. European renal osteodystrophy (EUROD) workgroup. Role of the RANK/RANKL/OPG and Wnt/beta-catenin systems in CKD bone and cardiovascular disorders. Calcified Tissue International. 2021;108:439-451
28.Cianciolo G, La Manna G, Capelli I, Gasperoni L, Galassi A, Ciceri P, et al. The role of activin: The other side of chronic kidney disease-mineral bone disorder? Nephrology, Dialysis, Transplantation. 2020;36(6):966-974
29.Sugatani T. Systemic activation of Activin a signaling causes chronic kidney disease-mineral bone disorder. International Journal of Molecular Sciences. 2018;19(9):2490
30.Pereira L, Frazao JM. The bone-vessel axis in chronic kidney disease: An update on biochemical players and its future role in laboratory medicine. Clinica Chimica Acta. 2020;508:221-227
31.Figurek A, Rroji M, Spasovski G. Sclerostin: A new biomarker of CKD-MBD. International Urology and Nephrology. 2020;52(1):107-113
32.Law JP, Price AM, Pickup L, Radhakrishnan A, Weston C, et al. Clinical potential of targeting fibroblast growth Factor-23 and αklotho in the treatment of uremic cardiomyopathy. Journal of the American Heart Association. 2020;9(7):e016041
33.Memmos E, Papagianni A. New insights into the role of FGF-23 and klotho in cardiovascular disease in chronic kidney disease patients. Current Vascular Pharmacology. 2021;19(1):55-62
34.Neyra JA, Hu MC, Moe OW. Fibroblast growth factor 23 and αklotho in acute kidney injury: Current status in diagnostic and therapeutic applications. Nephron. 2020;144(12):665-672
35.Neyra JA, Hu MC, Moe OW. Klotho in clinical nephrology: Diagnostic and therapeutic implications. Clinical Journal of the American Society of Nephrology. 2020;16(1):162-176
36.Kuro-O M, Moe OW. FGF23-αklotho as a paradigm for a kidney-bone network. Bone. 2017;100:4-18
37.Kreiner FF, Kraaijenhof JM, von Herrath M, Hovingh GKK, von Scholten BJ. Interleukin 6 in diabetes, chronic kidney disease, and cardiovascular disease: Mechanisms and therapeutic perspectives. Expert Review of Clinical Immunology. 2022;18(4):377-389
38.Ix JH, Isakova T, Larive B, Raphael KL, Raj DS, et al. Effects of nicotinamide and lanthanum carbonate on serum phosphate and fibroblast growth Factor-23 in CKD: The COMBINE trial. Journal of the American Society of Nephrology. 2019;30:1096-1108
39.Melamed ML, Raphael KL. Metabolic acidosis in CKD: A review of recent findings. Kidney Medicine. 2021;3(2):267-277
40.Goraya N, Munoz-Maldonado Y, Simoni J, Wesson DE. Treatment of chronic kidney disease-related metabolic acidosis with fruits and vegetables compared to NaHCO3 yields more and better health outcomes and at comparable 5-year cost. Journal of Renal Nutrition. 2021;31(3):239-247
41.Raphael KL, Isakova T, Ix JH, Raj DS, Wolf M, et al. A randomized trial comparing the safety, adherence, and pharmacodynamics profiles of two doses of sodium bicarbonate in CKD: The BASE pilot trial. Journal of the American Society of Nephrology. 2020;31(1):161-174
42.Nowak KL, Chonchol M, Ikizler TA, Farmer-Bailey H, Salas N, et al. IL-1 inhibition and vascular function in CKD. Journal of the American Society of Nephrology. 2017;28:971-980
43.Nowak KL, Hung A, Ikizler TA, Farmer-Bailey H, Salas-Cruz N, et al. Interleukin-1 inhibition, chronic kidney disease-mineral and bone disorder, and physical function. Clinical Nephrology. 2017;88(3):132-139
44.Vongpatanasin W, Peri-Okonny P, Velasco A, Arbique D, Wang Z, Ravikumar P, et al. Effects of potassium magnesium citrate supplementation on 24-hour ambulatory blood pressure and oxidative stress marker in Prehypertensive and hypertensive subjects. The American Journal of Cardiology. 2016;118:849-853
45.Quiñones H, Hamdi T, Sakhaee K, Pasch A, Moe OW, Pak CYC. Control of metabolic predisposition to cardiovascular complications of chronic kidney disease by effervescent calcium magnesium citrate: A feasibility study. Journal of Nephrology. 2019;32(1):93-100
46.Toto RD et al. Correction of hypomagnesemia by dapagliflozin in patients with type 2 diabetes: A post hoc analysis of 10 randomized, placebo-controlled trials. Journal of Diabetes and its Complications. 2019;33(10):107402
47.Shlipak MG, Tummalapalli SL, Boulware LE, Grams ME, Ix JH, et al. The case for early identification and intervention of chronic kidney disease: Conclusions from a kidney disease: Improving global outcomes (KDIGO) controversies conference. Kidney Internat. 2021;99:34-47
48.Machuca-Gayet I, Quinaux T, Bertholet-Thomas A, et al. Bone disease in nephropathic Cystinosis: Beyond renal osteodystrophy. International Journal of Molecular Sciences. 2020;21(9):3109
49.Komaba H, Ketteler M, Cunningham J, Fukagawa M. Old and new drugs for the Management of Bone Disorders in CKD. Calcified Tissue International. 2021;108(4):486-495
50.Ferreira AC, Cohen-Solal M, D'Haese PC, Ferreira A. European renal osteodystrophy (EUROD), an initiative of the CKD-MBD working group of the ERA-EDTA: The role of bone biopsy in the management of CKD-MBD. Calcified Tissue International. 2021;108:528-538
51.Bover J, Ureña-Torres P, Cozzolino M, Rodríguez-García M, Gómez-Alonso C. The non-invasive diagnosis of bone disorders in CKD. Calcified Tissue International. 2021;108:512-527
52.Evenepoel P, Cunningham J, Ferrari S, Haarhaus M, Javaid MK, Lafage-Proust MH, et al. European renal osteodystrophy (EUROD) workgroup, an initiative of the CKD-MBD working group of the ERA-EDTA, and the committee of scientific advisors and National Societies of the IOF. European consensus statement on the diagnosis and management of osteoporosis in chronic kidney disease stages G4-G5D. Nephrology, Dialysis, Transplantation. 2021;36:42-59
53.Bover J, Ureña-Torres P, Torregrosa JV, Rodríguez-García M, Castro-Alonso C. Osteoporosis, bone mineral density and CKD-MBD complex(I): Diagnostic considerations. Nefrología. 2018;38:476-490
54.Jørgensen HS, David K, Salam S, Evenepoel P. European renal osteodystrophy (EUROD) workgroup, an initiative of the CKD-MBD working group of the ERA-EDTA. Traditional and non-traditional risk factors for osteoporosis in CKD. Calcified Tissue International. 2021;108:496-511
55.Doumouchtsis KK, Perrea DN, Doumouchtsis SK. The impact of sex hormone changes on bone mineral deficit in chronic renal failure. Endocrine Research. 2009;34:90-99
56.Mazzaferro S, Cianciolo G, De Pascalis A, Guglielmo C, Urena Torres PA, et al. Bone, inflammation and the bone marrow niche in chronic kidney disease: What do we know? Nephrology, Dialysis, Transplantation. 2018;33:2092-2100
57.Melamed ML, Buttar RS, Coco M. CKD-mineral bone disorder in stage 4 and 5 CKD: What we know today? Advances in Chronic Kidney Disease. 2016;23:262-269
58.Adamczyk P, Szczepanska M, Pluskiewicz W. Skeletal status assessment by quantitative ultrasound and bone densitometry in children with different renal conditions. Osteoporosis International. 2018;29:2667-2675
59.Glenn DA, Denburg MR. Bone health in glomerular kidney disease. Current Osteoporosis Reports. 2019;17:570-579
60.Bover J, Ureña-Torres P, Mateu S, DaSilva I, Gràcia S, et al. Evidence in chronic kidney disease-mineral and bone disorder guidelines: Is it time to treat or time to wait? Clinical Kidney Journal. 2020;13:513-521
61.Bover J, Ureña-Torres P, Laiz Alonso AM, Torregrosa JV, Rodríguez-García M, et al. Osteoporosis, bone mineral density and CKD-MBD (II): Therapeutic implications. Nefrología. 2019;39:227-242
62.Damasiewicz MJ, Nickolas TL. Bisphosphonate therapy in CKD: The current state of affairs. Current Opinion in Nephrology and Hypertension. 2020;29:221-226
63.Zerwekh J, Glass K, Jowsey J, Pak CYC. An unique form of osteomalacia associated with endorgan refractoriness to 1,25-(OH)2-vitamin D and with apparent defective synthesis of 25-OH-vitamin D. The Journal of Clinical Endocrinology & Metabolism. 1979;49:171-175
64.Bricker NS, Fine LG, Kaplan M, Epstein M, Bourgoignie JJ, et al. “Magnification phenomenon” in chronic renal disease. The New England Journal of Medicine. 1978;299(23):1287-1293
65.Martin KJ, Gonzalez EA. Prevention and control of phosphate retention/hyperphosphatemia in CKD-MBD: What is normal, when to start, and how to treat? Clinical Journal of the American Society of Nephrology. 2011;6(2):440-446
66.Hernando N, Gagnon K, Lederer E. Phosphate transport in epithelial and nonepithelial tissue. Physiological Reviews. 2021;101(1):1-35
67.Hu MC, Shizaki K, Kuro-O M, Moe OW. Fibroblast growth factor 23 and klotho: Physiology and pathophysiology of an endocrine network of mineral metabolism. Annual Review of Physiology. 2013;75:503-533
68.Hu MC, Shi M, Cho HJ, Adam-Huet B, Paek J, et al. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. Journal of the American Society of Nephrology. 2015;26:1290-1302
69.Hassan A, Khalaily N, Kilav-Levin R, Nechama M, Volovelsky O, et al. Molecular mechanisms of parathyroid disorders in chronic kidney disease. Metabolites. 2022;12(2):111
70.Duque EJ, Elias RM, Moyses RMA. Parathyroid hormone: A uremic toxin. Toxins (Basel). 2020;12(3):189
71.Meyer MB, Pike JW. Mechanistic homeostasis of vitamin D metabolism in the kidney. The Journal of Steroid Biochemistry and Molecular Biology. 2020;196:105500
72.Canata-Andia JB, Martin-Carro B, Martin-Virgala J, Rodriguez-Carrio J, Bande-Fernandez JJ, et al. Chronic kidney disease-mineral and bone disorders: Pathogenesis and management. Calcified Tissue International. 2021;108(4):410-422
73.Jaramillo-Morales J, Korucu B, Pike MM, Lipworth L, Stewart T, et al. Effects of caloric restriction and aerobic exercise on circulating cell-free mitochondrial DNA in patients with moderate to severe chronic kidney disease. American Journal of Physiology. Renal Physiology. 2022;322(1):F68-F75
74.Tammaro A, Kers J, Scantlebery AML, Florquin S. Metabolic flexibility and innate immunity in renal ischemia reperfusion injury: The Fine balance between adaptive repair and tissue degeneration. Frontiers in Immunology. 2020;11:1346
75.Li L, Tang W, Yi F. Role of Inflammasome in chronic kidney disease. Advances in Experimental Medicine and Biology. 2019;1165:407-421
76.Turner CM, Arulkumaran N, Singer M, Unwin RJ, Tam FW. Is the Inflammasome a potential therapeutic target in renal disease? BMC Nephrology. 2014;15:21
77.Simic P, Babitt JL. Regulation of FGF23: Beyond bone. Current Osteoporosis Reports. 2021;19(6):563-573
78.Edmonston D, Wolf M. FGF23 at the crossroads of phosphate, iron economy and erythropoiesis. Nature Reviews. Nephrology. 2020;16(1):7-19
79.Faul C. Cardiac actions of fibroblast growth factor 23. Bone. 2017;100:69-79
80.Leifheit-Nestler M, Haffner D. How FGF23 shapes multiple organs in chronic kidney disease. Molecular and Cellular Pediatrics. 2021;8(1):12
81.Dusso AS, Bauerle KT, Bernal-Mizrachi C. Non-classical vitamin D actions for renal protection. Frontiers in Medicine (Lausanne). 2021;8:790513
82.De la Guia-Galipienso F, Martinez-Ferran M, Vallecilo N, Lavie CJ, Sanchis-Gomar F, et al. Vitamin D and cardiovascular health. Clinical Nutrition. 2021;40(5):2946-2957
83.Villa-Bellosta R. Vascular calcification: Key roles of phosphate and pyrophosphate. International Journal of Molecular Sciences. 2021;22(24):13536
84.Lanzer P, Hannan FM, Lanzer JD, Janzen J, Raggi P, et al. Medial arterial calcification: JACC state-of-the-art review. Journal of the American College of Cardiology. 2021;78(11):1145-1165
85.Turing AM. Computing machinery and intelligence. Mind. 1950;49:433-460
86.Gladwell M. Outliers: The Story of Success. Boston, MA, USA: Little, Brown and Company; 2008
87.Kho AN, Rasmussen LV, Connolly JJ, Peissig PL, Starren J, et al. Practical challenges in integrating genomic data into the electronic health record. Genetics in Medicine. 2013;15(10):772-778
88.Kho AN, Pacheco JA, Peissig PL, Rasmussen L, Newton KM, et al. Electronic medical records for genetic research: Results of the eMERGE consortium. Science Translational Medicine. 2011;3(79):79re1
89.Brier ME, Gaweda AE, Dailey A, Aronoff GR, Jacobs AA. Randomized trial of model predictive control for improved anemia management. Clinical Journal of the American Society of Nephrology. 2010;5(5):814-820
90.Gaweda AE, Aronoff GR, Jacobs AA, Rai SN, Brier ME. Individualized anemia management reduces hemoglobin variability in hemodialysis patients. Journal of the American Society of Nephrology: JASN. 2014;25(1):159-166
91.Gaweda AE, Jacobs AA, Aronoff GR, Brier ME. Individualized anemia management in a dialysis facility - long-term utility as a single-center quality improvement experience. Clinical Nephrology. 2018;90(4):276-285
92.Gaweda AE, McBride DE, Lederer ED, Brier ME. Development of a quantitative systems pharmacology model of chronic kidney disease: Metabolic bone disorder. American Journal of Physiology. Renal Physiology. 2021;320(2):F203-F211
93.Peterson MC, Riggs MM. A physiologically based mathematical model of integrated calcium homeostasis and bone remodeling. Bone. 2010;46:49-63
94.Lioufas NM, Pascoe EM, Hawley C, Elder GJ, Bdve SV, et al. Systematic review and meta-analyses of the effects of phosphate-lowering agents in nondialysis CKD. Journal of the American Society of Nephrology. 2022;33(1):59-76
95.Massy ZA, Merkling T, Wagner S, Girerd N, Essig M, et al. Association of Serum Phosphate with efficacy of statin therapy in hemodialysis patients. CJASN. 2022;17(4):546-554
Written By
Eleanor Lederer, Michael E. Brier and Adam E. Gaweda
Submitted: 03 May 2022Reviewed: 13 May 2022Published: 31 May 2023
 Open access peer-reviewed chapter
Open access peer-reviewed chapter