 Open access peer-reviewed chapter
Open access peer-reviewed chapter
Abstract
Dental pulp stem cells (DPSCs) are a special mesenchymal stem cell (MSC) type. These cells can be isolated from the dental pulp (DP) of deciduous, adult, and wisdom teeth. Stem cells from milk/baby teeth fall naturally, representing an advantageous source of young stem cells. These cells are less studied than MSCs from bone marrow, adipose tissue, and umbilical cord. MSCs from these sources are currently widely used in clinical studies. However, obtaining significant quantities of DPSCs from one donor is still challenging, thus limiting their systemic application in patients, which requires doses starting from 5 × 105 per kg of weight and higher. In this chapter, we would like to share our experience of more than 20 years in the isolation and scaling up of DPSC from deciduous teeth. We will also provide information about their in vitro growth, differentiation, and therapeutic potential observed in animal models that mimic human diseases or injuries in preclinical studies. Finally, we will discuss our experience of DPSC production under good manufacturing practice conditions and their use in regulated clinical studies in Brazil for Huntington’s disease.
Keywords
- dental pulp stem cells
- deciduous teeth
- scaling up
- preclinical studies
- therapeutic potential
1. Introduction
The term “stem cells” was first proposed by Russian histologist Alexander Maksimov in 1908 to explain the ability of specific tissues, such as blood, to self-renew for the lifetime of an organism. Maksimov’s unitary theory of hematopoiesis was based on stem cells and substantiated the concept of hematopoietic stem cells in various animal models [1]. Another Russian scientist, Alexander Friedenstein, studied the interactions between bone tissue and the blood system and proposed the idea of a hematopoietic microenvironment formed by populations of non-hematopoietic stromal stem cells or bone marrow-derived osteogenic precursors. He showed that these cells are adhesive fibroblast-like clonogenic cells and also showed high replicative ability and the possibility of differentiation into osteoblasts, chondrocytes, adipocytes, and stromal cells that support hematopoiesis [2, 3, 4, 5, 6].
Arnold Caplan, Professor of Biology and Director of the Skeletal Research Center at Case Western Reserve University, who was also the first to demonstrate the biotechnological and therapeutic potential of these cells, first suggested the term “mesenchymal stem cells” (MSCs). MSC means that the cells were isolated from mesoderm-bone marrow (BM) and can undergo
Mesenchymal stem cells were isolated from different tissues besides BM, such as adipose tissue, umbilical blood and cord, menstrual blood, placenta, DP, and other tissue sources. Moreover, these MSCs beyond mesoderm (BM-MSC) originated from extraembryonic tissue, such as the umbilical cord and placenta, and ectoderm as DP [10, 11, 12, 13, 14]. As demonstrated by Friedenstein and Caplan, all these MSCs are fibroblastic, adherent, and colony forming. Fibroblastic colony-forming units (CFU-F) were developed for plastic-adherent clonogenic BM-MSC, which grow in a monolayer and are self-renewing, and are used to characterize MSC from other sources. These cells can also differentiate from mesoderm derivatives
Caplan and his colleagues were the first to discover cell-surface antigens such as CD105 (SH2) and CD73 (SH3/4) on human BM-MSC, which are detected by monoclonal antibodies [15]. Currently, these antibodies are recognized as principal markers for MSC identification and are part of the minimum criteria for defining MSC, along with CFU-F and differentiation assays [16].
Dental pulp stem cells (DPSCs) can be isolated from deciduous, adult, and wisdom teeth. Their discovery is more recent compared with BM-MSC. DPSC from adult DP for the first time was isolated in 2000, and 3 years later, these cells were isolated from human exfoliated deciduous teeth (SHED) [17]. These cells and their potential therapeutic application have been evaluated mainly in dentistry for a long time. Only recently, these cells and their potential use have attracted attention from other medical areas, for instance, for treating spinal cord injury, neurological and neurodegenerative diseases [18, 19].
Whether DPSC or MSC is still unknown, although they fit in the MSC definition proposed by the International Society for Cellular Therapy [16]. In this chapter, we would like to provide comprehensive, critical, and concise information about our group’s experience with the isolation, characterization, nonclinical, clinical application, and scaling-up expansion to obtain significant quantities of DPSC from deciduous teeth, which can allow further possible commercialization of these cells.
2. Origin and function of MSC and DPSC
Figure 1 demonstrates the embryonic origin of tissues used for MSC isolation. This figure shows that BM-MSCs are originated from paraxial mesoderm [20], which gives rise to the axial skeleton. Adipose tissue-derived MSCs (AT-MSCs) [10] are derived from lateral plate mesoderm that gives rise to the appendicular skeleton and adipose tissue. DP is originated from neural crest (that gives rise to the craniofacial skeleton) and head mesenchyme (that gives rise to the bones, cartilages, muscles, tongue, craniofacial nerves, and teeth, and dental ectomesenchymal stem cells and connective tissues in the craniofacial complex) [14, 21, 22]. Umbilical cord and placenta-derived MSC are originated from extraembryonic mesoderm [23].
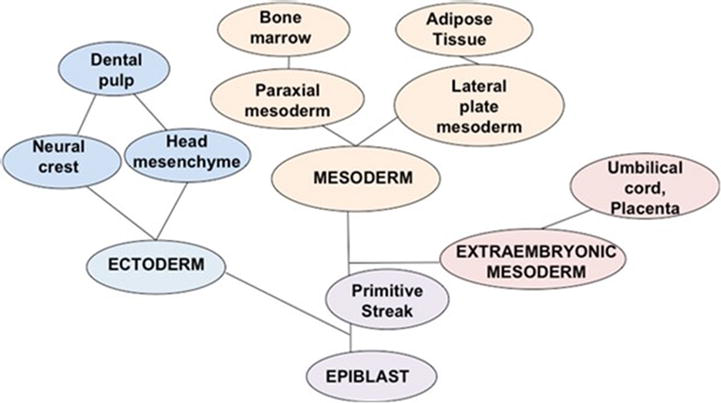
Figure 1.
Embryonic development ontology tree. This figure demonstrates the different embryonic origins of tissues, which can originate MSCs. MSC in the adult organism can be isolated from DP (ectoderm), bone marrow and adipose tissue (mesoderm), and umbilical cord and placenta (extraembryonic mesoderm).
Mesenchymal stem cells, independently of origin, exert similar but not identical functions. Accumulation of scientific data about MSC showed that old ideas about replacing injured tissues with MSC have no basis anymore.
In recent years, the research has been directed toward a better understanding of the mechanisms of MSCs function. This includes the rescue and repair of injured organs and tissues. It has been discovered that MSCs, when transplanted into injured tissue, can be used mainly for paracrine activity involving the secretion of proteins/peptides and hormones by these cells [27, 28, 29, 30, 31].
2.1 Function of MSC and pericytes
Crisan et al. [32] demonstrated the link between MSC and perivascular cells, also called pericytes. His group compared the MSC and the pericytes’
Because of these findings, endogenous pericytes were widely associated with MSC, even though the cell fate plasticity of endogenous pericytes
The fact that MSC/pericytes secreted bioactive molecules expects their involvement in a regenerative microenvironment for a variety of injured adult tissues. These cells can limit the area of damage and induce a self-regulated endogenous regenerative response. In this context, the regenerative microenvironment was referred to as trophic, and MSCs play the role of valuable mediators for tissue repair and regeneration [38]. Therefore, the differentiation capacity of MSC was relegated to the background because the
2.2 Dental pulp from deciduous teeth
Dental pulp is a soft, gelatinous, and non-mineralized oral tissue. A jelly-like core is composed of soft, loose connective tissue and vascular, lymphatic, and nervous cells found in each tooth’s dental pulp cavity. DP is made up of Type I and Type III collagens; however, it lacks elastin fibers.
Dental pulp from deciduous teeth, discarded after exfoliation, represents a valuable source of young stem cells. The fact that the cells from deciduous teeth are young is significant for their therapeutic use. Evidence shows that MSCs function declines with age, thus limiting their therapeutic potential [38, 40, 41, 42].
The main difference in the biology of the DP isolated from deciduous and permanent teeth is that the lifetime of deciduous teeth is rather limited. Different from adult teeth, they start to grow during embryonic life, and their loss begins at the age of 6 years, starting with the central incisors and followed about a year later by the lateral incisors [43]. However, from a tiny piece of DP of deciduous teeth, significant quantities of young cells can be isolated [44].
3. DPSC isolation and characterization
As mentioned, one of the characteristics of MSCs is their adherence to plastic surfaces. Therefore, the principal method of MSC isolation is based on the ability of the MSCs to selectively adhere to plastic surfaces. The method of isolation and culturing of MSC is challenging and depends on the MSC method of preparations, which should account for variability in tissue sources, and donor-related issues, for instance, age, disease, gender, etc., culture media, and population doublings.
3.1 Explant culture
For the first time, we isolated human immature DP stem cells (hIDPSCs) from the DP of deciduous teeth using the explant method [44, 45]. This method is also denominated as organ culture and used for the
However, only recently, eleven years after our pioneering work [44], the explant method was recognized as more advantageous for MSCs isolation compared to enzymatic one. This method is gainful because first, it excludes the proteolysis step during cell isolation. Second, it provides the presence of small tissue pieces in the primary cell culture. Third, it removes lytic stress on cells and reduces
3.2 Isolation of human dental pulp stem cells (hDPSCs)
Using an explant method, the cells like MSC from other tissues can be isolated from DP, which is a very small organ; its length is approximately 4.04 mm, while the width is 1.0 mm. In deciduous teeth, a greater cellular density was observed in the coronal region, approximately 47.30 ± 14.71 cells/mm2 [47]. However, this size allows an easy
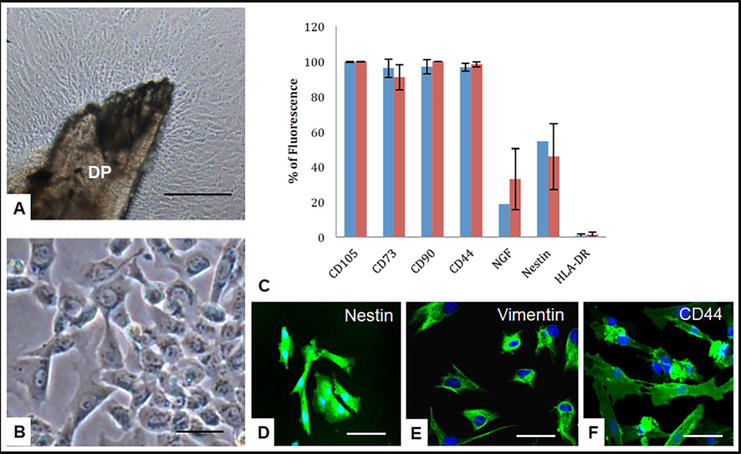
Figure 2.
Isolation and characterization of hIDPSC. A. The cells outgrowing from DP, passage 0. B. HIDPSC after passing (passage 1, P1). C. Flow cytometry demonstrates the expression of CD105, CD73, CD90, CD44, neurotrophic growth factor (NGF), nestin, and human leukocyte antigen HLA-DR isotype (major histocompatibility complex (MHC) class II cell-surface receptor). Immunofluorescence shows positive staining of hIDPSC to nestin (D), vimentin (E), and CD44 (cell-surface glycoprotein) (F). Azul—expression of studied markers after the first DP adhesion (P5), red—expression of studied markers after the first DP transfer (P5). Scale bar: (A, B) = 100 μm, (D–F) = 10 μm. A, B—light microscopy, phase contrast, D–F—epifluorescence.
3.3 Characterization of the population of hIDPSC
After isolation, the cells present a fibroblast-like phenotype and adherent to the plastic. The term “immature” is due to the discovery that human immature DP stem cells (hIDPSCs) express embryonic stem cell markers, such as octamer-binding transcription factor 4 (Oct-4), Nanog, stage-specific embryo antigen 3 (SSEA-3), stage-specific embryo antigen 4 (SSEA-4), tumor resistance antigen 1–60 (TRA-1-60), and tumor resistance antigen 1–81 (TRA-1-81), at early passages and mainly in outgrowth culture (before enzymatic treatment) and under conditions described in Kerkis et al. [44]. These cells also express MSC markers, such as CD105, CD73 [16], and CD13, which is not a typical MSC marker, and they were negative for CD34, CD43, HLA-DR (Figure 2C), and CD45 ruling out the absence of contamination with hematopoietic and endothelial cells. The cells grow during at least 25 passages while maintaining the normal karyotype [44, 48]. Further investigation of the molecular phenotype of the cells demonstrated that they also express such markers as Nestin, vimentin, CD44, neurotrophic growth factor (NGF) (Figure 2C–F), CD29, CD146, CD31, alpha-fetoprotein (AFP), STRO-1, CD146, CD117 (c-KIT), and other markers [45].
4. In vitro differentiation and priming of hIDPSC
We also showed that hIDPSC,
4.1 Osteogenic and chondrogenic hIDPSC potential
The priming hIDPSC demonstrated the osteogenic and chondrogenic potential
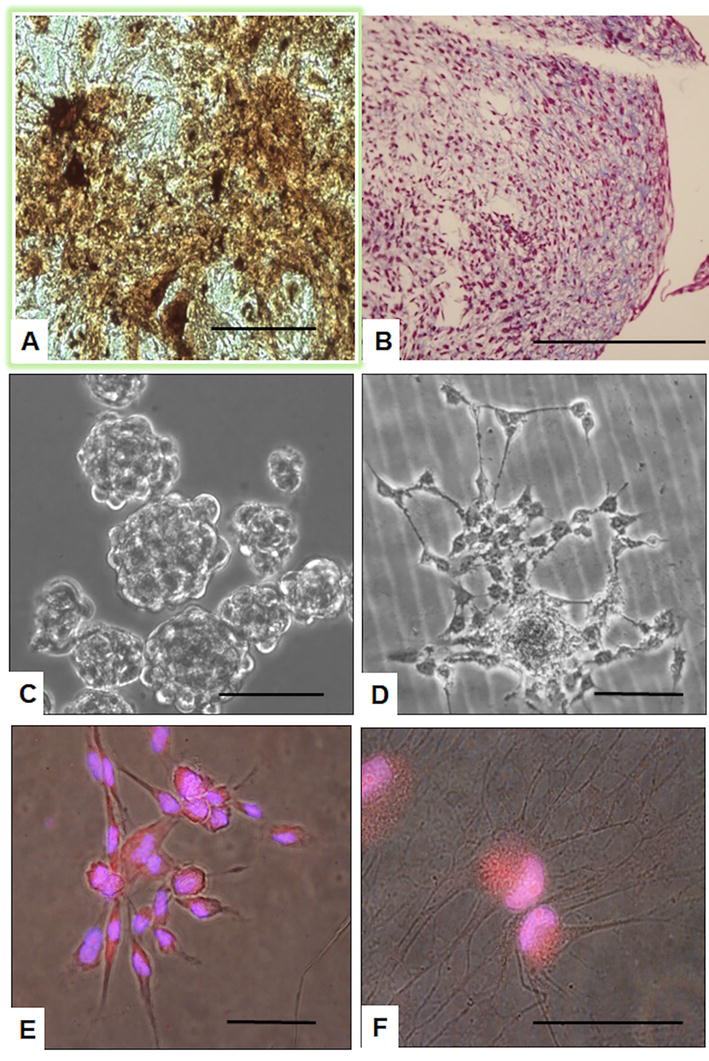
Figure 3.
Induced
4.2 Neuronal in vitro differentiation
When treated with retinoic acid (RA)—a potential inductor of neuronal differentiation—hIDPSC exhibited morphology like that of neural cells. In addition to expressing neural proteins (Nestin, Beta-Tubulin, and anti-glial fibrillary acidic protein (GFAP)), hIDPSC presented an electrophysiological response for sodium and potassium and can trigger an action potential (Figure 3C–F). Curiously, the cells without RA treatment also express these markers and present an electrophysiological response. In addition, hIDPSCs were able to direct neural differentiation of embryonic stem cells in coculture assays [51, 52].
5. The immunomodulatory capacity of hIDPSC
One of the most essential characteristics of MSCs and their mechanism of action are immunomodulatory properties, which have critical clinical applications. Therefore, the hIDPSC’s immunomodulatory potential has also been studied.
The immunomodulatory effects of hIDPSC on differentiation, maturation of dendritic cells (DCs) derived from monocytes (mo-DC), and their ability to activate T cells and check soluble factors released in coculture cells were analyzed. Peripheral blood mononuclear cells (PBMCs), T cells, and hIDPSCs were obtained from unrelated donors (n = 4). Monocytes were obtained by adhering and removing non-adherent cells from the PBMC cultivation. They were induced to differentiate into mo-DC by culture in the presence of interleukin 4 (IL-4) and granulocyte macrophage colony-stimulating factor (GM-CSF) for seven days. Lipopolysaccharide (LPS), added after five days of culture, was used to induce mo-DC maturation. Effects of hIDPSC were analyzed by flow cytometry after its addition to the cultures from day zero or after five days of culture at a ratio of 1:10. Monocytes derived from DC exposed to hIDPSC from day zero showed a reduction in mean fluorescence intensity (MFI) of the markers, such as blood dendritic cell antigen 1 (BDCA-1) (70%) and CD11c (32%), compared to the control. After activation by LPS, there was a decrease in MFI of CD40 levels (52%), CD80 (35%), CD83 (67%), and CD86 (50%) compared to the control. Mo-DC exposed to hIDPSC from day five showed no changes in the expression of markers. To assess the ability of mo-DC exposed to hIDPSC from day zero in activating T cell responses, mo-DC (HLA-DR +) were separated from hIDPSC (HLA-DR-) by magnetic beads and were cocultured (ratio 1:10) with T cells (carboxyfluorescein diacetate (CFSE)-labeled). After five days, cell proliferation was assessed by CFSE dilution. The proliferation of CD4+ T cells induced by mo-DC (exposed to hIDPSC) decreased by 63% for immature mo-DC (IDC) and 50% for mo-DC activated by LPS (mDC), compared to control non-cultivated with hIDPSC. Similarly, the proliferation of CD8+ T cells decreased by 40 and 26% by culture with immature dendritic cells (IDCs) and mature dendritic cells (mDCs), respectively, when cocultured with hIDPSC. There was an increase in the proportion of CD4 + FoxP3 + IL-10+ T cells and CD4 + FoxP3 + IFN-γ + T cells after cocultured with mDC previously cultivated with hIDPSC. The levels of the released soluble factors in cocultures demonstrate the immunomodulation of the studied cells. The levels of pro-inflammatory factors, such as interleukin 2 (IL-2), tumor necrosis factor alpha (TNF-α), and interferon gamma (IFN-γ), reduced while that of the anti-inflammatory factor IL-10 increased. These data showed that hIDPSC affects the differentiation of mo-DC, a phenomenon reflected in the reduction of markers of mo-DC maturation and a decreased ability of mo-DC to induce T cell proliferation. The anti-inflammatory balance of factors released to the medium supports observations [51, 52, 53].
6. In vivo biodiodistribution of hIDPSC in adult, embryonic, and fetal environment
Mesenchymal stem cells are one of the principal products of advanced cell therapies, which are of great interest due to their paracrine effect that provides a microenvironment improvement, and regenerative activity of endogenous cells in the injury site. The biodistribution of MSCs is an essential step in their characterization that is used to identify addressed migration and engraftment of MSC in injured sites. These properties are very important for defining MSC safety and efficacy after transplantation to human organisms. To understand MSC’s therapeutic potential and differentiation capacities, biodistribution studies should be performed at different development stages.
6.1 Biodistribution in nude mice
After
6.2 Biodistribution and fate of hIDPSC in developing mouse embryo
We injected the cells in mouse compacted morulae or early blastocysts to verify the biodistribution of hIDPSC in developing mouse embryos and their possible differentiation in the embryonic or fetal microenvironment [54]. Production of human/animal preterm chimeras is widely used to analyze mammalian cells’ developmental potency in biomedical research [55, 56, 57].
We showed that hIDPSC presented biological compatibility with the mouse host embryo environment and could survive, proliferate, and contribute to the inner cell mass after introduction into early mouse embryos (n = 28), which achieved the hatching stage following 24 and 48 h in culture. This result demonstrates that hIDPSCs were not toxic for early stage embryo development. When transferred to foster mice (n = 5), these blastocysts with hIDPSC (n = 57) yielded embryos (n = 3) and fetuses (n = 6) (Figure 4A–D). The hIDPSC demonstrated robust engraftment in 11 d.p.c. (days post coitum) embryos in the nervous system in primary vesicles, telencephalon, mesencephalon, and rhombencephalon. Additionally, the hIDPSC engrafted in the ocular region on 18-d.p.c. hIDPSC showed comprehensive individual, organ, and tissue biodistribution in mouse fetuses. The cells were observed in different organs of the chimeras, such as the brain, liver, intestine, and muscles (Figure 4E and F). However, as expected, while fetal age increased the number of grafted hIDPSCs decreased. We verified whether hIDPSC would also be able to differentiate into tissue-specific cell types in the mouse environment. The contribution of hIDPSC in at least two types of tissues, muscles, and epithelial tissue, was confirmed. We showed that hIDPSC survived, proliferated, and differentiated in mice developing preterm chimeras [54].
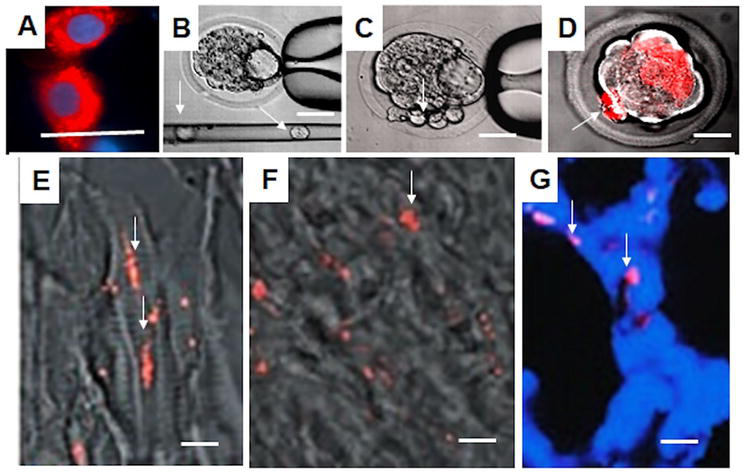
Figure 4.
Human immature dental pulp stem cell’s biological compatibility with the mouse embryo environment. A. hIDPSC stained with Vybrant—cytoplasm, nucleus stained with 4′,6-diamidino-2-phenylindole (DAPI) (DNA stain). B. Mouse embryo and glass capillary with hIDPSC inside (white arrow). C. hIDPSC observed inside mouse embryo (white arrow). D. Epifluorescence + phase contrast showing Vybrant-stained hIDPSC (red, white arrow) aggregated with mouse blastocyst cells. Fluorescence
Overall, these findings were significant for further exploration of the therapeutic potential of hIDPSC since the cells grafted into the growing fetus’s brain and other tissues that form the animal head. Thus, this study suggested hIDPSC similarity with neural crest cells, which produce diverse cell lineages, such as melanocytes, craniofacial cartilage and bone, smooth muscle, peripheral and enteric neurons, and glia. Another important observation is that the cells could express human proteins in a tissue-specific manner under the environment clues provided by the fetuses.
6.3 Biodistribution of hIDPSC following in utero transplantation in canine model (Canis lupus familiaris )
Intrauterine stem cell transplantation (IUSCT) treats genetic, congenital, hematological, and immunological diseases. Basic research provides a model for studying the dynamics of migration, graft, and functional status of different types of stem cells. The cells can be transplanted in different moments of the gestational period, which can be divided into equivalent quarters. The choice of the cells and the quarter where the stem cells will be applied can influence cell behavior and transplantation results. Fetal and adult hematopoietic or bone marrow-derived MSCs were mainly used for IUSCT [58, 59, 60]. Our study aimed to evaluate the migration capacity, proliferation, and homing of IDPSCs after IUSCT during the third fetal period in dogs. All experimental procedures were approved by the Ethical Committee of the School of Veterinary Medicine and Animal Science of San Paulo University and were performed under appropriate anesthesia. Up to 1 × 106 undifferentiated green fluorescent protein (GFP)-positive human IDPSCs (GFP-hIDPSCs) were transplanted into five fetuses at 45 days of gestation. This transplantation occurred through laparotomy and intraperitoneal injection, guided by intra-operative ultrasound control. Five fetuses, which did not receive IDPSCs, were used as a control. Ultrasound analyses were performed daily before the collection of the fetuses. After seven days of ovarian hysterectomy, fetuses were collected; organs and tissues were isolated and fixed or cryopreserved. The biodistribution of GFP-IDPSCs within the organs and tissues was analyzed on cryosections (5 μm) under a confocal microscope. Homing of GFP-IDPSCs was observed in organs derived from three germ lines, endoderm, ectoderm, and mesoderm. GFP-IDPSCs were found in the intraglandular space and muscular mucosae in the stomach and the intestine. In the liver, these cells were observed in the hepatic parenchyma, in the heart within the myocardium, and in the brain within blood vessels in the cerebellum, specifically within Purkinje cells. Among the different organs, expressive homing was observed in the heart, spleen, and liver myocardium. The hIDPSCs were also found in the canine placenta, especially in blood vessels. These data were confirmed using anti-human nucleus antibody (immunohistochemistry), GFP-hIDPSC, and FISH analysis for the human chromosomes. Human IDPSC showed high migration and proliferation potential after IUSCT in dog fetuses. Therefore, hIDPSC demonstrated homing in fetal hematopoietic (placenta), epithelial (gastric glands), and perivascular stem cell niches [61].
7. hIDPSC local application and in vivo differentiation
Most
7.1 hIDPSC sheet transplantation in the damaged ocular surface in rabbits: unilateral limbal stem cell deficiency
The ability of hIDPSC to differentiate
Two distinct types of epithelial cells, conjunctival and corneal epithelia, compose the ocular surface. Corneal epithelial stem cells reside at the corneoscleral limbus, whose microenvironment is essential in maintaining their stemness. However, the limbal stem cells may be partially or depleted after limbus damage. Such stem cell deficiency results in corneal surface abnormalities that lead to “conjunctivalization” and corneal vascularization, producing an irregular and unstable epithelium and vision deficiency. Limbal stem cell transplantation is commonly used in patients with either uni- or bilateral total limbal stem cell deficiency (LSCD), which can be reproduced in rabbits. Commonly, the chemical burn in one eye of rabbits induces LSCD [62]. We verified hIDPSC
First, we verified whether hIDPSC expresses markers in common with LSCD, such as ABCG2 (adenosine triphosphate (ATP)-binding cassette subfamily G member 2), integrin beta 1, vimentin, p63, connexin 43, and cytokeratin 3/12 (K3/12). The expression of these markers was confirmed in undifferentiated hIDPSC, excluding K3/12, a marker of the differentiated corneal epithelium [63]. Next, we induced a rabbit model of unilateral burn eye—LSCD. We demonstrated that hIDPSC could reconstruct the eye surface after induction of LSCD in rabbits and transplantation of the priming hIDPSC sheet growing on the scaffold directly onto the exposed stromal bed. Morphological and immunohistochemical analysis using human-specific antibodies against limbal and corneal epithelium demonstrated corneal epithelium reconstruction, presenting an expression of human antibodies such as integrin beta 1 (ITGB1), cytokeratin 18 (CK18), and K3 [63].
Further investigation with a higher number of animals confirms that this study provided new histological electron microscopy and demonstrated the expression of corneal epithelium human’s protein (ABCG2, CK18, p63, and K3 expression in a rabbit LSCD model) [64].
7.2 Reconstruction of significant cranial defects in rats using hIDPSC
Mesenchymal stem cells are critical in bone fracture repair by differentiating between bone-formation osteoblasts and cartilage-forming chondrocytes [65]. Kerkis and coworkers [44] showed remarkable
8. hIDPSC and genetic disease animal model
It is known that adult stem cells can be used in treating genetic diseases. For instance, osteogenesis imperfecta (OI) is not a curable chronic disease. Off-the-shelf MSC appears as a candidate in a clinical therapy for OI. This is due to MSC’s high osteogenic potency. Recently, preclinical and initial clinical data supported the use of MSC in treating OI [67]. We present here our preclinical study using DPSC to treat Duchenne muscular dystrophy (DMD) in dogs [68].
8.1 hIDPSC for muscle regeneration in golden retriever muscular dystrophy (GRMD) dogs: intramuscular and intra-arterial applications
Our previous study [44] also suggests using hIDPSC for muscle regeneration. Duchenne muscular dystrophy (DMD) is a lethal X-linked disease affecting newborn males. It is caused by mutations in a large gene located at Xp21 that encodes the muscle protein dystrophin. The closest model for human DMD is the golden retriever muscular dystrophy (GRMD) dog, which causes analogous skeletal and cardiac muscle disease and shows the absence of dystrophin. GRMD dogs are typically succumbing to the disease by around age 2.
In Kerkis and coauthors [68], transplantation of hIDPSC (2n = 46, XY) of four affected littermate GRMD (2 males and 2 females) aged 28–40 days by either arterial or muscular injections (6 × 107 cells per animal), and without using immunosuppression has been carried out. The females received one unique injection, whereas the males were treated with monthly injections, and one male received six intramuscular (biceps femoris) injections. In contrast, another subject received nine systemic injections of arterial (femoral artery) (Figure 5A–C). No signs of immune rejection were observed, and these results suggested that hIDPSC transplantation might be done without immunosuppression. Indeed, white blood cell counting did not present any critical changes in response to cell transplantation, and no lymphocyte infiltration was observed in muscle cells. A 1-year-old dog, which received nine systemic injections, showed a good performance with moderate scores, mainly in postural tone, standing up, crossing barriers, and hoping. At 26 months old, this dog still showed no decline. Our data suggested systemic multiple deliveries seemed more effective than local injections. Biopsies from the dog’s muscles were obtained and checked by immunohistochemistry (dystrophin markers) and FISH using human antibodies and X and Y DNA probes. We showed that hIDPSC presented significant engraftment in GRMD dog muscles, although human dystrophin expression was modest and limited to several muscle fibers [68] (Figure 5D–G). Two years after we published our study on GRMD dogs, the dog that received nine systemic transplantations of hIDPSC was still alive (data not published).
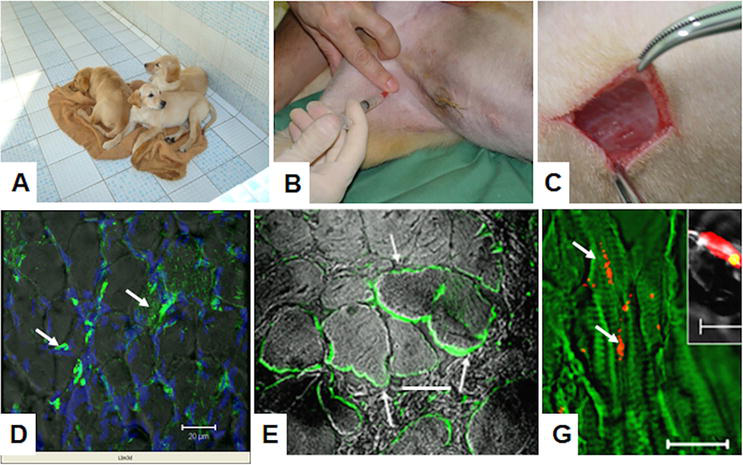
Figure 5.
Transplantation of hIDPSC to golden retriever muscular dystrophy (GRMD) dogs. A. Rare litter of GRMD dogs. B. Intra-arterial hIDPSC injection. C. Biceps femoralis biopsy. D. Chimeric human/canine muscle fibers present positive green, fluorescent immunostaining with anti-human nucleus antibody (white arrows). E. Positive immunostaining with anti-human dystrophin antibody, clone 2C6 (MANDYS106) in large dystrophic fibers (white arrows). G. FISH analysis of dystrophic male’s muscles using specific human probe for chromosome: Y (red) and in inset X. Yellow, because merged images of propidium iodide (PI) (red)-stained nucleus and probe of chromosome X (green) are presented. D-G confocal microscopy: Epifluorescence + DIC. G—green artificial color. (D) Nucleus stained with DAPI (blue). Scale bars: A, E = 50 μm G = 10 μm.
Further studies confirmed our finding demonstrating that DMD mice and dog’s phenotypes, such as pathological inflammation and motor dysfunction, can be significantly improved by repeated systemic injections of hIDPSC [69].
9. hIDPSC and neuroregenerative potential
Currently, the neuroregenerative potential of hIDPSC derived from young and adult teeth is widely explored and discussed in scientific literature. This is because hIDPSCs are a population affluent in cell proliferation and multipotency. After transplantation in animal models that mimic neurodegenerative disease, hIDPSCs showed neuron-protective effects. These protective effects are related to trophic factors released from hIDPSCs, which are neurotrophic and were effective for models of neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [70, 71, 72].
9.1 hIDPSC delivery precision in a mouse model of compressive spinal cord injury: localized administration approach
A spinal cord injury (SCI) is damage to any part of the spinal cord or nerves at the end of the spinal canal. This injury causes temporary or permanent changes in spinal cord function. In the USA, 17,000 new SCIs occur yearly, while in Brazil, approximately 150,000 cases occur yearly. Among the factors that contribute to limited SCI recovery are the reduced ability of remyelination spared demyelinated axons, failure of axons to overcome the local expression of myelin-associated inhibitory molecules, a lack of neurotrophic factors to support axonal growth, and other factors [73, 74, 75].
We aimed to investigate the efficacy of hIDPSC after local transplantation in a compression lesion of the spinal cord induced in mice. Therefore, SCI was induced in C57/BL6 mice females via laminectomy at T9 and spinal cord compression with a vascular clip for 1 min. The cells were transplanted 7 days or 28 days after the lesion. The recovery in a subacute or chronic phase of SCI was evaluated after cell therapy. We demonstrated that transplantation of hIDPSC into the site of the spinal cord lesion improved tissue organization in the injured site significantly. Larger areas of white-matter (WM) preservation were also evidenced only in groups treated with hIDPSC but not in control. In addition, in treated groups, the morphological ultrastructural analysis demonstrated large numbers of standard fibers, many axons being remyelinated by either Schwann cells or oligodendrocytes, and preserved neurons exhibiting intact synapses on their cell bodies. These effects were attributed to the statistically significant release of trophic factors observed in CSI animals treated with hIDPSC. This is because we showed in the present study that hIDPSCs express neurotrophic factors, such as brain-derived neurotrophic factor (BDNF), glial cell-derived neurotrophic factor (GDNF), nerve growth factor, beta polypeptide (NGF-b), neurotrophin 3 (NT-3), and neurotrophin 4/5 (NT-4/NT-5), as detected by reverse transcriptase-polymerase chain reaction (RT-PCR). Statistically significant differences in BDNF, NGFb, NT-3, and NT-4 expression in brain tissues between treated (increased) and control animals were observed. Before transplantation, the hIDPSCs labeled with cell tracer (red) were observed undestroyed and distributed in the white matter and demonstrated co-localization with anti-GFAP and anti-S-100 antibodies. Furthermore, we used the basic mobility scale (BMS) and the global mobility test to assess motor performance in animals with CSI treated with hIDPSC. Our data suggest that hIDPSC-treated groups showed better locomotor performance in both functional tests and exhibited higher speeds than the control groups, especially in the subacute animals that received cell transplantation seven days after injury. Therefore, we demonstrated the hIDPSC therapeutic potential in both subacute and chronic stages of a mouse compressive SCI, which suggests possible use of hIDPSC in human trials shortly [76].
9.2 hIDPSC in multiple sclerosis: clinical cases of canine distemper (hardpad disease), intravenous injection
Multiple sclerosis (MS) is a progressive disease affecting 2.8 million people worldwide without a definitive cure. The immune system attacks the brain and spinal cord tissues (central nervous system) and nerves by the own organism, causing MS. The etiology of MS is unclear. However, it may be a combination of genetic and environmental factors. Recently, it has been proposed that infection with the Epstein-Barr virus (EBV) may cause this disease [77].
In MS, the immune system attacks the protective sheath (myelin) that covers nerve fibers and causes communication problems between the brain and the rest of the body, thus causing permanent damage or deterioration of the nerve fibers. Canine distemper is an animal model for MS. Canine distemper is a viral disease of dogs, and this virus belongs to the paramyxovirus group. In approximately half of the cases, it is fatal. Impossible to cure, canine distemper is a severe viral illness that attacks a dog’s body on all fronts. It can cause persistent infection of the dog’s central nervous system, resulting in a progressive, multifocal demyelinating disease. Once an animal develops neurological symptoms of the disease, such as seizures or paralysis, its chances of surviving are slim, its quality of life is bound to worsen, and the animal dies [78].
Therefore, we conducted the study using hIDPSC in canine distemper following international animal care guidelines. The animals were enrolled in an experimental procedure after obtaining informed consent signed by the animals’ owners. The treatment was also realized without cost to the owners, and animals received long-term follow-up. Eight dogs of variable breeds aged between 4 and 6 years and weighing 8–16 kg in canine distemper were diagnosed. They presented symptoms such as vomiting, diarrhea, and fever; others were enrolled. Before cell transplantation, these dogs received antibiotics to fight the cough and pneumonia, which did not provide any clinical amelioration. Following the dog’s ability to fend off the effects of the virus, they acquired the symptoms of a neural form of this disease, such as paralysis of the hind paws or both hind and front paws.
We performed transplantation of previously cryopreserved hIDPSC (P3–P5) only after the effect of the virus was ended. The cells were thawed before transplantation and washed twice in prewarmed (37°C) sterile phosphate-buffered saline (PBS) following centrifugation for 5 min at 800 × g. At that moment, the viability of the cells was tested using trypan blue staining and was approximately 98% of live cells. The cell number used in each application was established following the dog weights and varied between 2 × 106 (8 kg) and 4 × 106 (16 kg). The cells were suspended in a mean volume of ±0.5 mL sterile physiological solution for subsequent intravenous injection. The dog received an injectable anesthetic, which dose was computed according to the weight of the dog and the sensitivity of certain breeds to the anesthetic. The dogs received single or multiple (no more than three) applications according to disease severity. Human IDPSCs were transplanted without any immunosuppressive protocol. None of the animals showed signs of immune rejection following single or numerous hIDPSC transplantations. Soon after the first hIDPSC application, all animals showed significant amelioration of symptoms of the neurological form of canine distemper. The animals, which were able only to crawl, started to half-rise, while those that were able to half-rise started to get up and stroll. Each animal had its healing dynamics, and those with stronger musculature demonstrated accelerated dynamics of amelioration. The dogs that presented more advanced clinical symptoms and could only crawl demonstrated significant amelioration after the third IDPSC application and even with difficulty, but these animals could walk. At a follow-up 4 months later, all animals recuperated their movement and even running capacities. At a follow-up 2 years later, the animals demonstrated that hIDPSC transplantation was safe and could maintain the physical performance of dogs, which were healthy. None of the animals demonstrated tumor formation, which supposedly can be caused by stem cell transplantation [79, 80].
9.3 hIDPSC in rat model of Huntington’s disease
Huntington’s disease (HD) is an inherited neurodegenerative disorder. It is caused by genetic mutation expansion of cytosine-adenine-guanine (CAG) repeats in the huntingtin (Htt) gene localized on chromosome 4. This rare disease is found in 0.4–5.7 cases per 100,000 people. The clinical symptoms appear in middle-aged or older people and present motor, cognitive, and behavioral changes. HD causes neuronal loss in the striatum and cortex. Mutant Htt protein forms neuronal intranuclear aggregates in medium-spiny neurons (MSNs). HD causes neuronal loss in the striatum and cortex. Mutant Htt protein forms neuronal intranuclear aggregates in medium-spiny neurons. This neuronal population of HD-vulnerable γ-aminobutyric acid (GABA) neurons in the striatum constitutes a hallmark of degeneration in HD because these neurons die first.
Cumulative evidence has demonstrated that BDNF-expressing MSCs can confer neuroprotection, promoting functional recovery in rodent Huntington’s disease (HD) models. To investigate the capability of BDNF-expressing hIDPSC to restore the BDNF, dopamine- and cAMP-regulated phosphoprotein, 32 kDa (DARPP32), and dopamine D2 receptor (D2R) expression, Wistar rats subjected to a subacute treatment with 3-nitropropionic acid (3-NP), a toxin that irreversibly inhibits the mitochondrial succinate dehydrogenase (SDH), leading to mitochondrial dysfunction and neurodegeneration of striatal cells, mimic the characteristics of HD. Although it is impossible to fully mimic the HD pathology since no genetically modified or chemically induced animal models exhibit the primary clinical signal of the disease, the chorea, the 3-NP has been successfully used to promote selective brain damage that inevitably leads to striatal neurons’ (MSN) loss. Two doses of hIDPSC (single versus three consecutive injections) were injected intravenously. The cells demonstrated homing in the striatum, cortex, and subventricular zone. Thirty days after hIDPSC administration, the cells found in the brain still express hallmarks of undifferentiated MSC (Figure 6). Immunohistochemistry quantitative analysis revealed a significant increase in BDNF, DARPP32, and D2R positively stained cells in the striatum and cortex in the hIDPSC groups. The differences were more expressive in animals that received only one administration of hIDPSC. These data suggest that intravenous hIDPSC can restore the BDNF, DARPP32, and D2R expression, promoting neuroprotection and neurogenesis [81].
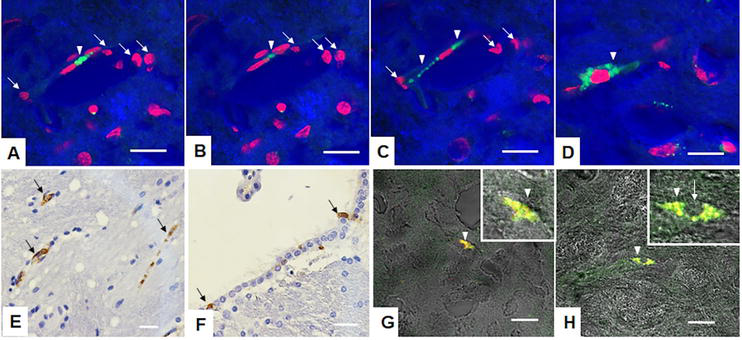
Figure 6.
Human immature dental pulp stem cell engraftment in mice brain after the treatment with 3-nitropropionic acid. A–C hIDPSCs stained with Vybrant (green) optical cuts demonstrate the polarization of hIDPSC, suggesting their migration within brain capillaries. White arrows indicate hIDPSC green cytoplasm is shown by white arrowheads, the same cells on A–C. D. hIDPSC fibroblastic morphology in brain parenchyma. hIDPSC engraftment observed in the striatum E and subventricular zone in F. Overlapping (yellow) of Vybrant-stained hIDPSC (red) with immunopositive staining for mesenchymal stem cell markers—CD105 (G) and CD73 (H) both in green. A–D, G, H—confocal microscopy: Epifluorescence + DIC. G—blue artificial color. (D) Nucleus stained with PI (red).
9.4 hIDPSC in rat facial nerve regeneration, local application
Facial nerve paralysis compromises muscles controlling smiling, blinking capacity, and other facial movements. It is also accompanied by disorders in speech and mastication, as well as esthetics. The successful facial nerve lesion regeneration depends on the Schwann cells’ (SCs’) support, which participates in the phagocytosis of axon and myelin debris and later in the myelination of newly formed axonal fibers, besides the production of neurotrophic factors that contribute to regularization and modulation of neuronal survival. We induced unilateral facial nerve crush in 70 Wistar rats (CG—control not injured group, G1—crushed group, G2—crushed + hIDPSC). The functional recovery was evaluated after 3, 7, 14, 21, and 42 postoperative days. The cells (5 × 105) were transplanted immediately after the crush injury. G2 exhibited statistically more significant values (p < 0.05) in nerve growth factor expression compared with CG and G1 at 7 days and showed complete functional recovery at 14 days, while G1 recovered after 42 days. Additionally, G2 presented histological improvement, evidencing better axonal and structural organization of the myelin sheath, and exhibited statistically higher values for the outer and inner perimeters and g-ratio (degree of myelination). At 42 days, both groups were close to the levels observed in the control group. Therefore, a single injection of hIDPSC accelerates facial nerve trunk regeneration [82].
10. Osteoarthritis and HIDPSC
Osteoarthritis (OA) affects millions of people worldwide. This common disease occurs because the protective cartilage cushions the ends of the bones and is gradually destroyed along the life, and it frequently affects hands, joints, knees, hips, and spine. These irreversible damages and diseased people suffer from joint pain and movement limitations. MSC can be injected into a joint helping to relieve pain, swelling, and loss of movement. They can also be used to make artificial cartilage in a laboratory, which can be transplanted into a joint helping to repair damaged bone, ligaments, and cartilage [83, 84, 85].
10.1 Cartilage regeneration observed after intra-articular administration of hIDPSC in experimental osteoarthritis rat model
Osteoarthritis is an incurable condition mainly affecting joints in your hands, knees, hips, and spine. Due to the complexity of the disease, we investigated the molecular and morphological effects after intra-articular injection of one single dose of hIDPSC (8 × 105) in the OA rat model. We also compared the effect of this treatment with the effect of diacerein and glucosamine-chondroitin drugs, which are known to relieve osteoarthritis symptoms. The cell therapy-treated group received the cells on day 14 after osteoarthritis induction, and euthanasia was performed after 60 days. The drug-treated groups were given 50 mg/kg of diacerein and 400/500 mg/kg of glucosamine-chondroitin starting on day 14 for 60 days. The morphological analysis and expression of SRY-Box Transcription Factor 5 (SOX-5), Indian hedgehog (IHH), matrix metalloproteinase-8 (MMP-8), matrix metalloproteinase-13 (MMP-13), and Type II collagen antibodies were statistically analyzed in lateral femoral condyle cartilage. We showed that after hIDPSC transplantation, structural reorganization of the tissues of lateral femoral condyles was observed, while the glucosamine-chondroitin sulfate provided anti-inflammatory modulation. The diacerein treatment preserves the primordial cartilage of femoral condyles. In conclusion, we demonstrated that a single hIDPSC injection significantly improved cartilage regeneration in an OA rat model compared with the positive therapeutic effect of daily administered conventional drugs [86].
11. Aplastic anemia and hIDPSC
Aplastic anemia and myelodysplastic syndromes are rare disorders characterized by bone marrow failure, which lead to a significant reduction in the hematopoietic stem/progenitor cells (HSPCs), which results in defective mature blood cell production and peripheral pancytopenia [87, 88].
11.1 Hematoprotective role of hIDPSC for aplastic anemia and potentially for other hematopoietic failures
To induce the acquired aplastic anemia (AA), a mouse model subjected to total body irradiation (TBI) was used. After three consecutive hIDPSC (1 × 106 cells/animal) transplantations, we observed that the irradiated mice showed high BM cellularity, recovering the normal BM histology 62 days after cell transplantation (short-term treatment) when compared to irradiated mice treated with saline (placebo). Furthermore, we showed that at D182, the irradiated mice treated with hIDPSC demonstrated stable BM tissue improvement, as evidenced by histological studies. In contrast, BM of the irradiated and placebo-treated mice still presented a significant fat deposit. These data suggest that hIDPSC can stimulate BM tissue recovery and long-term hematoprotection. They (hIDPSCs) maintained fibroblast-like morphology and influence positively the expression of endogen Nestin, which is a marker of vascular structures in BM [44] and a selective marker of BM perivascular MSC that was downregulated after irradiation. In addition, the endogenous CD44+ cell increased in cell-treated mouse BM 180 days after hIDPSC transplantation. CD44 is essential for human hematopoietic regulation, including lymphocyte migration and activation, progenitor cell proliferation, and BM environment restoration. This result suggests that although hIDPSCs are of ectomesenchymal origin and differ from bone marrow MSCs, they accurately respond to BM microenvironment control, thus providing BM recovery [89].
12. Tumorigenic potenial of hIDPSC
Only the cell, which presents a modified genome leading to loss of control of cell growth, can produce a malignant mass tumor. Analyzing factors (proliferation rate, karyotype integrity, and mutations’ presence) that may influence the tumorigenicity of hIDPSC to generate malignant tumors discards such a possibility. Moreover, no scientific evidence supports malignant tumor generation by the MSC [90].
Teratoma formation is essential in determining the pluripotency of any pluripotent cells, such as embryonic or induced pluripotent stem cells (ES and iPS cells). A consistent protocol for assessing the cells’ teratoma-forming ability was established and used in studies [91]. This method is helpful for the biosafety analysis of other adult/mesenchymal stem cells (MSCs), such as those derived from DP of deciduous teeth, umbilical cord adipose tissue, and others. To form teratomas, the normal pluripotent stem cells should have epithelial-like morphology and present gap junction, which helps to keep these cells connected. Functionally, gap junctions maintain cellular homeostasis by allowing communication between adjacent cells [92]. However, gap junctions also play an essential role in teratoma formation because they do not allow the migration of pluripotent cells from the local injection of the cells aiming to form teratoma. Furthermore, the enzymatically dissociated cells are used for teratoma formation. Therefore, the cells are co-injected with Matrigel, a solubilized basement membrane matrix used for pluripotent cell adherence to ensure cell mass formation. Co-injection of the pluripotent cells with Matrigel increased subcutaneous teratoma formation efficiency from 25 to 40% to 80–100% [93]. In early development, MSCs originated from pluripotent embryonic cells. Furthermore, they underwent epithelial-mesenchymal transition and became migratory cells. This argument discards the possibility of forming a teratoma by MSC because the cells should stay together to form a teratoma or tumor.
12.1 Experimental evidence demonstrates that hIDPSC does not produce teratomas
In Kerkis et al. [44], we reported that a few cells from the hIDPSC population express two of three pluripotent stem cell markers, such as octamer-binding transcription factor (Oct) 3/4 and Nanog. Therefore, hIDPSC can supposedly produce a teratoma. This capacity of hIDPSC was tested. For this purpose, BALB/c nude mice—females (Institute of Biomedical Sciences (ICB)/University of São Paulo (USP), Isogenic Mouse Vivarium) received an intraperitoneal injection of 1 × 106 hIDPSC. Animals (n = 15) were sacrificed 1, 2, and 3 months after injection by approved methods, and tissues of interest were freshly frozen to verify the presence of human cells. No teratoma or other type of tumor formation was observed. The human DNA sequence encoding the glucocerebrosidase (GBA) gene was detected by PCR in the sites of hIDPSC engraftment and found in the liver, spleen, brain, kidney, and other organs [44].
Furthermore, the capacity of hIDPSC to form teratomas was verified when hIDPSC was reprogrammed to induce pluripotent hIDPSCs using four Yamanaka factors [95]. Reprogrammed hIDPSC-induced pluripotent stem cells (iPSCs) acquired embryonic stem cell (ESC)-like morphology, expressed pluripotent markers, possessed stable, normal karyotypes, and demonstrated the ability to differentiate
13. hIDPSCs display unique transcriptional signature
Since 2016, the hIDPSC isolation and cultivation technology [44, 48, 79], whose development started in 2000 by a research group led by Dr. Irina Kerkis within the Brazilian Butantan Institute, was licensed to Cellavita Pesquisas Científicas Ltda. Brazilian Startup Company in the middle of 2016. Based on this technology, an advanced cell therapy investigational product was developed for treating neurodegenerative diseases, particularly Huntington’s disease (HD). Currently, it comprises a hIDPSC suspension in sterile saline solution (0.9% NaCl), which expresses Nestin, a protein known for axon growth, and secretes high levels of brain-derived neurotrophic factor (BDNF). This revolutionary product is composed of cryopreserved hIDPSC, which after thawing corresponds to the active component of the NestaCell® product. Following intravenous (IV) administration, NestaCell® can migrate and home in the brain tissues. Because of the low expression of HLA-DR antigens, NestaCell® is safe for heterologous use without immune suppression. To provide further characterization and to demonstrate stability of three different batches (donors) of NestaCell® product, we performed the transcriptome analysis of these cells produced on a large scale using RNA-Seq. We used bioinformatics tools to obtain the list of differentially expressed genes (DEGs), which were next subjected to functional enrichment analysis. One of the most important findings is that the hIDPSC samples showed clustering with a less dimensional distance compared with other analyzed MSC samples. Although the hIDPSC shares at least 72% of transcripts with the other 137 MSC samples from different donors of adipocyte-derived (AD-MSC), bone marrow (BM-MSC), hepatocyte-derived (HD-MSC), menstrual blood (MB-MSC), umbilical cord (UC-MSC), and vertebral tissue (vMSC), they did not present overlapping with these samples. Additionally, we showed that NestaCell® uniquely expressed 375 genes that represent 4.61% (375/8128 genes) of the whole transcriptome of the product, and these genes mainly promote axon growth and guidance [96].
14. NestaCell® use in clinical regulatory studies in Huntington’s disease and COVID-19
The neurogenic potential of hIDPSC was demonstrated in various animal models: spinal cord compressive injury model in mice, in Wistar rats that suffered unilateral injury due to crushing of the facial nerve, in dogs with neurological sequelae of distemper, and in a pharmaceutical model of HD [76, 80, 81, 82]. All these studies, to some extent, are related to HD pathologies. For instance, spinal cord compression injury induces brain white-matter (WM) impairment. HD also leads to WM degeneration, possibly due to an early breakdown in axon myelination [97]. The facial nerve regeneration model is also essential since uncontrollable movements of the face are between later symptoms of HD [98]. Therefore, all these studies justify the clinical investigation of NestaCell® product in HD.
Mesenchymal stem cell use in COVID-19 treatment appears helpful because of these cells’ immunomodulatory, anti-inflammatory, and regenerative properties. It has been suggested that MSC can improve the survival rate of critically ill COVID-19 patients via inflammation control [99]. NestaCell also showed immunomodulatory potential
14.1 Clinical trials on intravenous administration of NestaCell® product in patients with Huntington’s disease and COVID-19
NestaCell product was used in three clinical studies. The first-in-human, non-randomized, Phase I study entitled “SAVE” aimed to evaluate the safety and tolerability and preliminary evidence of the effectiveness of the NestaCell® after intravenous administration in HD patients (ClinicalTrials.gov ID NCT02728115). This study started on October 16, 2017 and enrolled six patients who received three intravenous injections, two doses 1 × 106 and 2 × 106 cells/kg of body weight and followed for 5 years. The study showed that participants tolerated NestaCell® and that no treatment-related serious adverse events were observed at the end of 2 years and up to the present. Five of six patients presented improvement in the Unified Huntington’s Disease Rating Scale (UHDRS) observed for six to nine months after the last administration [100]. The second is a Phase II study entitled “ADORE” (ClinicalTrials.gov ID NCT03252535), aimed at identifying the optimal NestaCell® dose for HD treatment. This is a prospective, Phase II, single-center, randomized, triple-blind, placebo-controlled study with two test doses of NestaCell® (= Cellavita HD) product. This study enrolled 35 patients who started on January 15, 2018. The patients were randomized in a 2:2:1 ratio for the groups G1: lower dose (1 × 106 cells/weight range), G2: higher dose (2 × 106 cells/weight range), or G3: placebo. The study confirmed that NestaCell® is safe for humans with no treatment-related adverse effects. The Phase II ADORE study was extended, and the patients from the control group started to receive the product since the patients from the experimental group demonstrated improvements in UHDRS (motor score). Thus, the extension study evaluates the long-term safety and efficacy of NestaCell® in patients who concluded the ADORE trial. The product NestaCell is also being evaluated to proceed with the Phase III study.
Clinical trial entitled “HOPE” (ClinicalTrials.gov ID NCT04315987) was conducted on COVID-19 hospitalized patients to evaluate the NestaCell® safety and preliminary efficacy. This Phase I/II prospective, randomized, double-blind, multicenter, placebo-controlled clinical trial enrolled the patients hospitalized with COVID-19 but not requiring mechanical ventilation. A total of 90 male and female patients received up to four intravenous injections of 20 million cells per injection (45 patients) or saline (45 patients) every other day (a maximum of 80 million cells per patient). NestaCell® was safe with no relevant treatment-related adverse effects [101]. However, there was no clinical or laboratory benefit in comparison with placebo.
14.2 NestaCell® do not engraft in preexisting malignant neoplasm
Although it is well stated that MSCs, including hIDPSCs, are safe in terms of tumorigenicity, there is a constant concern with the use of cell therapy in patients with preexisting malignancies. This is because several studies have reported that cancer naturally recruits stem cells (particularly bone marrow MSC), which can engraft within the tumor microenvironment (TME), contributing to cancer progression and metastasis, as previously revised by us in Costa et al. [102]. This concern is still higher for lung cancer. Due to the pulmonary first-passage effect associated with intravenous infusion (the most used route of MSC administration), there is a rising concern that these cells could be entrapped in the lungs and grafted in preexisting lung cancer [103]. Despite this concern, until 2021, no clinical report describes whether manufactured stem cells, when transplanted in patients, could engraft within TME, contributing to the oncogenic process.
In this regard, during the clinical trial of the NestaCell® product, a patient with Huntington’s disease who attended all roll-in criteria established by the study protocol was subjected to the intravenous treatment with two cycles (one per year) of three doses of 1 × 106 cells/kg each, receiving a dose per month. However, a preexisting pulmonary nodule was identified during the trial screening. The patient was referred to a pulmonologist who considered the nodule to be a benign tumor and authorized enrollment. One month after the last dose, it was observed as a nodule growth. For this reason, the nodule was biopsied through a bronchoscopy biopsy, which revealed a lung adenocarcinoma. The neoplasm was surgically excised, and the tumor was sectioned into six fragments (representing different areas). These fragments were subjected to RNA-Seq analysis. The transcriptome of each tumor section was compared with the transcriptomes of transplanted hIDPSCs (which correspond to the active component of the NestaCell® product). Using reduction dimension analysis (PCA or principal component analysis), we demonstrated no evidence of the hIDPSCs within the lung adenocarcinoma. This result suggests that transplanted manufactured hIDPSCs neither home nor graft within the pulmonary nodule [104], reinforcing that the cell therapy with the NestaCell® is safe even for patients with preexisting lung cancer.
15. Conclusions
The hIDPSC differential is the original method of isolation and culturing, which allows for obtaining significant quantities of cells from one DP. This method is based on DP explant culture and multiple DP transference to a new culture flask, avoiding excessive passages requiring enzymatic DP and cell treatment. This method drastically decreases the number of donors used for cell isolation and, consequently, genetic variability observed between donors during clinical studies. These cells were used in multiple nonclinical studies demonstrating safety and efficiency in animal models. As a result of technology transfer from academic to commercial areas through licensing, a new advanced cell therapy investigational product was created, which is currently under clinical investigation. Therefore, NestaCell® is an innovative, cryopreserved, off-the-shelf, allogenic cell therapy product, whose active ingredient is based on young, undifferentiated, early passage, hIDPSC, and release of high levels of brain-derived neurotrophic factor (BDNF)
Acknowledgments
This work was supported by FAPESP (São Paulo Research Foundation), CNPq (National Council for Scientific and Technological Development), Cellavita Pesquisas Científicas Ltda, and Butantan Foundation, São Paulo, SP, Brazil. I am very grateful to my scientific collaborators, laboratory technicians, laboratory staff, and postgraduate students who participated in this research.
Conflict of interest
Dr. Prof. Irina Kerkis from Butantan Institute receives financial support from Cellavita Pesquisas Científicas Ltda, São Paulo, SP, Brazil.
References
- 1.
Konstantinov IE. In search of Alexander A. Maximow: The man behind the unitarian theory of Hematopoiesis. Perspectives of Biology and Medicine. 2000; 43 :269-276 - 2.
Friedenstein AJ. Osteogenic stem cells in bone marrow. In: Heersche JNM, Kanis JA, editors. Bone and Mineral Research. Amsterdam: Elsevier; 1990. pp. 243-272 - 3.
Bianco P, Robey PG, Simmons PJ. Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell. 2008; 2 (4):313-319 - 4.
Friedenstein AJ, Chailakhjan RK, Lalykina KS. The development of fibroblast colonies in monolayer cultures of Guinea-pig bone marrow and spleen cells. Cell and Tissue Kinetics. 1970; 3 (4):393-403 - 5.
Owen M, Friedenstein AJ. Stromal stem cells: Marrow-derived osteogenic precursors. Ciba Foundation Symposium. 1988; 136 :42-60 - 6.
Friedenstein AJ, Chailakhyan RK, Gerasimov UV. Bone marrow osteogenic stem cells: In vitro cultivation and transplantation in diffusion chambers. Cell and Tissue Kinetics. 1987; 20 (3):263-272 - 7.
Caplan AI. Mesenchymal stem cells. Journal of Orthopaedic Research. 1991; 9 (5):641-650 - 8.
Bruder SP, Fink DJ, Caplan AI. Mesenchymal stem cells in bone development, bone repair, and skeletal regeneration therapy. Journal of Cellular Biochemistry. 1994; 56 (3):283-294 - 9.
Caplan AI. The mesengenic process. Clinics in Plastic Surgery. 1994; 21 (3):429-435 - 10.
Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H, et al. Human adipose tissue is a source of multipotent stem cells. Molecular Biology of the Cell. 2002; 13 (12):4279-4295 - 11.
Lee OK, Kuo TK, Chen WM, Lee KD, Hsieh SL, Chen TH. Isolation of multipotent mesenchymal stem cells from umbilical cord blood. Blood. 2004; 103 (5):1669-1675 - 12.
Beeravolu N, McKee C, Alamri A, Mikhael S, Brown C, Perez-Cruet M, et al. Isolation and characterization of mesenchymal stromal cells from human umbilical cord and fetal placenta. Journal of Visualized Experiments. 2017; 3 (122):55224 - 13.
Wagner W, Wein F, Seckinger A, Frankhauser M, Wirkner U, Krause U, et al. Comparative characteristics of mesenchymal stem cells from human bone marrow, adipose tissue, and umbilical cord blood. Experimental Hematology. 2005; 33 (11):1402-1416 - 14.
Gronthos S, Mankani M, Brahim J, Robey PG, Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2000; 97 (25):13625-13630 - 15.
Haynesworth SE, Baber MA, Caplan AI. Cell surface antigens on human marrow-derived mesenchymal cells are detected by monoclonal antibodies. Bone. 1992; 13 (1):69-80 - 16.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006; 8 (4):315-317 - 17.
Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG, et al. SHED: Stem cells from human exfoliated deciduous teeth. Proceedings of the National Academy of Sciences. 2003; 100 (10):5807-5812 - 18.
Ueda T, Inden M, Ito T, Kurita H, Hozumi I. Characteristics and therapeutic potential of dental pulp stem cells on neurodegenerative diseases. Frontiers in Neuroscience. 2020; 14 :532435 - 19.
Candelise N, Santilli F, Fabrizi J, Caissutti D, Spinello Z, Moliterni C, et al. The importance of stem cells isolated from human dental pulp and exfoliated deciduous teeth as therapeutic approach in nervous system pathologies. Cell. 2023; 12 (13):1686 - 20.
Caplan AI. Mesenchymal stem cells: The past, the present, the future. Cartilage. 2010; 1 (1):6-9 - 21.
Chai Y, Jiang X, Ito Y, Bringas P Jr, Han J, Rowitch DH, et al. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 2000; 127 (8):1671-1679 - 22.
Solis-Castro OO, Rivolta MN, Boissonade FM. Neural crest-derived stem cells (NCSCs) obtained from dental-related stem cells (DRSCs): A literature review on current knowledge and directions toward translational applications. International Journal of Molecular Sciences. 2022; 23 (5):2714. DOI: 10.3390/ijms23052714 - 23.
Nagamura-Inoue T, He H. Umbilical cord-derived mesenchymal stem cells: Their advantages and potential clinical utility. World Journal of Stem Cells. 2014; 6 (2):195-202. DOI: 10.4252/wjsc. v6.i2.195 - 24.
Costa LA, Eiro N, Fraile M, Gonzalez LO, Saá J, Garcia-Portabella P, et al. Functional heterogeneity of mesenchymal stem cells from natural niches to culture conditions: Implications for further clinical uses. Cellular and Molecular Life Sciences. 2021; 78 (2):447-467. DOI: 10.1007/s00018-020-03600-0 - 25.
Czapla J, Matuszczak S, Kulik K, et al. The effect of culture media on large-scale expansion and characteristic of adipose tissue-derived mesenchymal stromal cells. Stem Cell Research & Therapy. 2019; 10 :235 - 26.
Quinn C, Flake AW. In vivo differentiation potential of mesenchymal stem cells: Prenatal and postnatal model systems. Transfusion Medicine and Hemotherapy. 2008; 35 (3):239-247. DOI: 10.1159/000129129 - 27.
Alvites R, Branquinho M, Sousa AC, Lopes B, Sousa P, Maurício AC. Mesenchymal stem/stromal cells and their paracrine activity-immunomodulation mechanisms and how to influence the therapeutic potential. Pharmaceutics. 2022; 14 (2):381. DOI: 10.3390/pharmaceutics14020381 - 28.
Merimi M, El-Majzoub R, Lagneaux L, Moussa Agha D, Bouhtit F, Meuleman N, et al. The therapeutic potential of mesenchymal stromal cells for regenerative medicine: Current knowledge and future understandings. Frontiers in Cell and Development Biology. 2021; 9 :661532. DOI: 10.3389/fcell.2021.661532 - 29.
Rasmusson I, Ringden O, Sundberg B, Le Blanc K. Mesenchymal stem cells inhibit the formation of cytotoxic T lymphocytes, but not activated cytotoxic T lymphocytes or natural killer cells. Transplantation. 2003; 76 :1208-1213. DOI: 10.1097/01.TP.0000082540.43730.80 - 30.
Oliva AA, McClain-Moss L, Pena A, Drouillard A, Hare JM. Allogeneic mesenchymal stem cell therapy: A regenerative medicine approach to geroscience. Aging Medicine (Milton). 2019; 2 (3):142-146. DOI: 10.1002/agm2.12079 - 31.
Durand N, Zubair AC. Autologous versus allogeneic mesenchymal stem cell therapy: The pros and cons. Surgery. 2022; 171 (5):1440-1442. DOI: 10.1016/j.surg.2021.10.057 - 32.
Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008; 3 (3):301-313. DOI: 10.1016/j.stem.2008.07.003 - 33.
Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nature Neuroscience. 2011; 14 (11):1398 - 34.
Girolamo F, Errede M, Bizzoca A, Virgintino D, Ribatti D. Central nervous system pericytes contribute to health and disease. Cell. 2022; 11 (10):1707 - 35.
Guimarães-Camboa N, Cattaneo P, Sun Y, Moore-Morris T, Gu Y, Dalton ND, et al. Pericytes of multiple organs do not behave as mesenchymal stem cells In vivo. Cell Stem Cell. 2017; 20 (3):345-359.e5. DOI: 10.1016/j.stem.2016.12.006 - 36.
Caplan AI. All MSCs are pericytes? Cell Stem Cell. 2008; 3 (3):229-230. DOI: 10.1016/j.stem.2008.08.008 - 37.
Caplan AI. New MSC: MSCs as pericytes are sentinels and gatekeepers. Journal of Orthopaedic Research. 2017; 35 (6):1151-1159. DOI: 10.1002/jor.23560 - 38.
Caplan AI. Adult mesenchymal stem cells for tissue engineering versus regenerative medicine. Journal of Cellular Physiology. 2007; 213 (2):341-347 - 39.
Caplan AI. Mesenchymal stem cells: Time to change the name! Stem Cells Translational Medicine. 2017; 6 (6):1445-1451. DOI: 10.1002/sctm.17-0051 - 40.
Hosseiniyan Khatibi SM, Kheyrolahzadeh K, Barzegari A, Rahbar Saadat Y, Zununi VS. Medicinal signaling cells: A potential antimicrobial drug store. Journal of Cellular Physiology. 2020; 235 (11):7731-7746. DOI: 10.1002/jcp.29728 - 41.
Zhang Y, Guo L, Han S, Chen L, Li C, Zhang Z, et al. Adult mesenchymal stem cell ageing interplays with depressed mitochondrial Ndufs6. Cell Death & Disease. 2020; 11 (12):1075 - 42.
Hung G, Ashvetiya T, Leszczynska A, Yang W, Hwang CW, Gerstenblith G, et al. Paracrine-mediated rejuvenation of aged mesenchymal stem cells is associated with downregulation of the autophagy-lysosomal pathway. NPJ Aging. 2022; 8 (1):10 - 43.
Arnold WH, Naumova LA, Goulioumis V. Pulp biology of deciduous and permanent teeth. In: Neuhaus KW, Lussi A, editors. Management of Dental Emergencies in Children and Adolescents. Wilay Online Library. Hoboken, New Jersey, United States: Wiley-Blackwell; 2019. Ch 1,2. pp. 12-13 - 44.
Kerkis I, Kerkis A, Dozortsev D, Stukart-Parsons GC, Gomes Massironi SM, Pereira LV, et al. Isolation and characterization of a population of immature dental pulp stem cells expressing OCT-4 and other embryonic stem cell markers. Cells, Tissues, Organs. 2006; 184 (3-4):105-116 - 45.
Kerkis I, Caplan AI. Stem cells in dental pulp of deciduous teeth. Tissue Engineering. Part B, Reviews. 2012; 18 (2):129-138 - 46.
Hendijani F. Explant culture: An advantageous method for isolation of mesenchymal stem cells from human tissues. Cell Proliferation. 2017; 50 (2):e12334 - 47.
Guerrero-Jiménez M, Nic-Can GI, Castro-Linares N, Aguilar-Ayala FJ, Canul-Chan M, Rojas-Herrera RA, et al. In vitro histomorphometric comparison of dental pulp tissue in different teeth. PeerJ. 2019; 7 :e8212. DOI: 10.7717/peerj.8212 - 48.
Lizier NF, Kerkis A, Gomes CM, Hebling J, Oliveira CF, Caplan AI, et al. Scaling-up of dental pulp stem cells isolated from multiple niches. PLoS One. 2012; 7 (6):e39885 - 49.
Noronha NC, Mizukami A, Caliári-Oliveira C, Cominal JG, Rocha JLM, Covas DT, et al. Correction to: Priming approaches to improve the efficacy of mesenchymal stromal cell-based therapies. Stem Cell Research & Therapy. 2019; 10 (1):132 - 50.
Lobo SE, Glickman R, da Silva WN, Arinzeh TL, Kerkis I. Response of stem cells from different origins to biphasic calcium phosphate bioceramics. Cell and Tissue Research. 2015; 361 (2):477-495 - 51.
Pelegrino KO. Caracterização e diferenciação neural in vitro de células-tronco de polpa de dente decíduo humano [Dissertação de Mestrado]. São Paulo: Instituto de Biociências, Universidade de São Paulo; 2009. DOI: 10.11606/D.41.2009.tde-18092009-141614. Available from: www.teses.usp.br [Accessed: January 06, 2023] - 52.
de Sá Silva F. Investigação da capacidade imunomoduladora de células-tronco imaturas de polpa dentária humana [tese]. São Paulo: Biotecnologia; 2013. DOI: 10.11606/T.87.2013.tde-05022013-090052 - 53.
Silva Fde S, Ramos RN, de Almeida DC, Bassi EJ, Gonzales RP, Miyagi SP, et al. Mesenchymal stem cells derived from human exfoliated deciduous teeth (SHEDs) induce immune modulatory profile in monocyte-derived dendritic cells. PLoS One. 2014; 9 (5):e98050 - 54.
Siqueira da Fonseca SA, Abdelmassih S, de Mello Cintra Lavagnolli T, Serafim RC, Clemente Santos EJ, Mota Mendes C, et al. Human immature dental pulp stem cells' contribution to developing mouse embryos: Production of human/mouse preterm chimaeras. Cell Proliferation. 2009; 42 (2):132-140 - 55.
Lensch MW, Schlaeger TM, Zon LI, Daley GQ. Teratoma formation assays with human embryonic stem cells: A rationale for one type of human-animal chimaera. Cell Stem Cell. 2007; 1 :253-258 - 56.
Yokoo T, Ohashi T, Shen JS, Sakurai K, Miyazaki Y, Utsunomiya Y, et al. Human mesenchymal stem cells in rodent whole-embryo culture are reprogrammed to contribute to kidney tissues. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102 :3296-3300 - 57.
Muotri AR, Nakashima K, Toni N, Sandçer VM, Gage FH. Development of functional human embryonic stem cell-derived neurons in mouse brain. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102 :18644-18648 - 58.
Guillot PV, Abass O, Bassett JH, Shefelbine SJ, Bou-Gharios G, Chan J, et al. Intrauterine transplantation of human fetal mesenchymal stem cells from first-trimester blood repairs bone and reduces fractures in osteogenesis imperfecta mice. Blood. 2008; 111 (3):1717-1725. DOI: 10.1182/blood-2007-08-105809 - 59.
Chan JK, Götherström C. Prenatal transplantation of mesenchymal stem cells to treat osteogenesis imperfecta. Frontiers in Pharmacology. 2014; 5 - 60.
Huang J, Li Q , Yuan X, Liu Q , Zhang W, Li P. Intrauterine infusion of clinically graded human umbilical cord-derived mesenchymal stem cells for the treatment of poor healing after uterine injury: A phase I clinical trial. Stem Cell Research & Therapy. 2022; 13 (1):85. DOI: 10.1186/s13287-022-02756- - 61.
Reginato AL. Estudo da biodistribuição de células tronco de polpa de dente decíduo humana (CTPDDh) após o transplante intra-uterino no modelo canino (Canis lupus familiares) [tese]. São Paulo: Faculdade de Medicina Veterinária e Zootecnia; 2012. DOI: 10.11606/T.10.2012.tde-07082013 - 62.
Dua HS, Azuara-Blanco A. Limbal stem cells of the corneal epithelium. Survey of Ophthalmology. 2000; 44 (5):415-425 - 63.
Monteiro BG, Serafim RC, Melo GB, Silva MC, Lizier NF, Maranduba CM, et al. Human immature dental pulp stem cells share key characteristic features with limbal stem cells. Cell Proliferation. 2009; 42 (5):587-594 - 64.
Gomes JA, Geraldes Monteiro B, Melo GB, Smith RL, Pereira C, da Silva M, et al. Corneal reconstruction with tissue-engineered cell sheets composed of human immature dental pulp stem cells. Investigative Ophthalmology & Visual Science. 2010; 51 (3):1408-1414 - 65.
Oryan A, Kamali A, Moshiri A, Baghaban EM. Role of mesenchymal stem cells in bone regenerative medicine: What is the evidence? Cells, Tissues, Organs. 2017; 204 (2):59-83 - 66.
de Mendonça CA, Bueno DF, Martins MT, Kerkis I, Kerkis A, Fanganiello RD, et al. Reconstruction of large cranial defects in nonimmunosuppressed experimental design with human dental pulp stem cells. The Journal of Craniofacial Surgery. 2008; 19 (1):204-210 - 67.
Götherström C, Walther-Jallow L. Stem cell therapy as a treatment for osteogenesis imperfecta. Current Osteoporosis Reports. 2020; 18 (4):337-343. DOI: 10.1007/s11914-020-00594-3 - 68.
Kerkis I, Ambrosio CE, Kerkis A, Martins DS, Zucconi E, Fonseca SA, et al. Early transplantation of human immature dental pulp stem cells from baby teeth to golden retriever muscular dystrophy (GRMD) dogs: Local or systemic? Journal of Translational Medicine. 2008; 6 :35. DOI: 10.1186/1479-5876-6-35 - 69.
Nitahara-Kasahara Y, Kuraoka M, Guillermo PH, et al. Dental pulp stem cells can improve muscle dysfunction in animal models of Duchenne muscular dystrophy. Stem Cell Research & Therapy. 2021; 12 :78. DOI: 10.1186/s13287-020-02099-3 - 70.
Ueda T, Inden M, Ito T, Kurita H, Hozumi I. Characteristics and therapeutic potential of dental pulp stem cells on neurodegenerative diseases. Frontiers in Neuroscience. 2020; 14 :407. DOI: 10.3389/fnins.2020.00407 - 71.
Staniowski T. Therapeutic potential of dental pulp stem cells according to different transplant types. Molecules. 2021; 26 (24):7423. DOI: 10.3390/molecules26247423 - 72.
Xiao Z, Lei T, Liu Y, et al. The potential therapy with dental tissue-derived mesenchymal stem cells in Parkinson’s disease. Stem Cell Research & Therapy. 2021; 12 :5. DOI: 10.1186/s13287-020-01957-4 - 73.
Blesch A, Fischer I, Tuszynski MH. Gene therapy, neurotrophic factors and spinal cord regeneration. Handbook of Clinical Neurology. 2012; 109 :563-574. DOI: 10.1016/B978-0-444-52137-8.00035-8 - 74.
Hu X, Xu W, Ren Y, Wang Z, He X, Huang R, et al. Spinal cord injury: Molecular mechanisms and therapeutic interventions. Signal Transduction and Targeted Therapy. 2023; 8 (1):1-28. DOI: 10.1038/s41392-023-01477-6 - 75.
Tian T, Zhang S, Yang M. Recent progress and challenges in the treatment of spinal cord injury. Protein & Cell. 2023; 14 (9):635-652. DOI: 10.1093/procel/pwad003 - 76.
de Almeida FM, Marques SA, Ramalho Bdos S, Rodrigues RF, Cadilhe DV, Furtado D, et al. Human dental pulp cells: A new source of cell therapy in a mouse model of compressive spinal cord injury. Journal of Neurotrauma. 2011; 28 (9):1939-1949 - 77.
Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. 2022; 375 (6578):296-301. DOI: 10.1126/science. abj8222 - 78.
Carvalho OV, Botelho CV, Ferreira CG, Scherer PO, Soares-Martins JA, Almeida MR, et al. Immunopathogenic and neurological mechanisms of canine distemper virus. Advances in Virology. 2012; 2012 :163860. DOI: 10.1155/2012/163860 - 79.
Kerkis I, Glozman S. Multifunctional immature dental pulp stem cells and therapeutic applications (US Patent # 9,790,468). IP USA BOOTH UDALL FULLER. 2014. Available from: https://patents.justia.com/patent/20200339954 ;https://patentcenter.uspto.gov/applications/14214016 - 80.
Barros MAD. Evaluation of allogenic transplantation of mesenchymal stem cells isolated from adipose tissue in dogs with neurological sequela caused by dumpster. 2017. Available from: https://www.oasisbr.ibict.br/vufind/Record/BRCRIS_10dd783a47b1ac25ebb41856df7b26c4 - 81.
Wenceslau CV, de Souza DM, Mambelli-Lisboa NC, Ynoue LH, Araldi RP, da Silva JM, et al. Restoration of BDNF, DARPP32, and D2R expression following intravenous infusion of human immature dental pulp stem cells in Huntington's disease 3-NP rat model. Cell. 2022; 11 (10):1664. DOI: 10.3390/cells11101664 - 82.
Saez DM, Sasaki RT, Martins DO, Chacur M, Kerkis I, da Silva MCP. Rat facial nerve regeneration with human immature dental pulp stem cells. Cell Transplantation. 2019; 28 (12):1573-1584 - 83.
Mancuso P, Raman S, Glynn A, Barry F, Murphy JM. Mesenchymal stem cell therapy for osteoarthritis: The critical role of the cell secretome. Frontiers in Bioengineering and Biotechnology. 2019; 7 :419383. DOI: 10.3389/fbioe.2019.00009 - 84.
Freitag J, Wickham J, Shah K, Tenen A. Real-world evidence of mesenchymal stem cell therapy in knee osteoarthritis: A large prospective two-year case series. Regenerative Medicine. 2022; 17 (6):355-373. DOI: 10.2217/rme-2022-0002 - 85.
Lv Z, Cai X, Bian Y, Wei Z, Zhu W, Zhao X, et al. Advances in mesenchymal stem cell therapy for osteoarthritis: From preclinical and clinical perspectives. Bioengineering (Basel). 2023; 10 (2):195. DOI: 10.3390/bioengineering10020195 - 86.
Correa Maldonado D, Nicoliche T, Faber J, Kerkis I, Martinez Saez D, Tetsuo Sasaki R, et al. Intra-articular human deciduous dental pulp stem cell administration vs. pharmacological therapy in experimental osteoarthritis rat model. European Review for Medical and Pharmacological Sciences. 2021; 25 (9):3546-3556 - 87.
Gonzaga VF, Wenceslau CV, Lisboa GS, Frare EO, Kerkis I. Mesenchymal stem cell benefits observed in bone marrow failure and acquired aplastic anemia. Stem Cells International. 2017; 2017 :8076529. DOI: 10.1155/2017/8076529 - 88.
Gonzaga V, Policiquio B, Wenceslau C, et al. Alternative immune-mediated-based methods in the aplastic anemia treatment. In: Human Blood Group Systems and Haemoglobinopathies. London, UK: IntechOpen; 2021. DOI: 10.5772/intechopen.89090 - 89.
Gonzaga VF, Wenceslau CV, Vieira DP, Policiquio BO, Khalil C, Araldi RP, et al. Therapeutic potential of human immature dental pulp stem cells observed in mouse model for acquired aplastic anemia. Cell. 2022; 11 (14):2252. DOI: 10.3390/cells11142252 - 90.
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999; 284 :143-147 - 91.
Gropp M, Shilo V, Vainer G, Gov M, Gil Y, Khaner H, et al. Standardization of the teratoma assay for analysis of pluripotency of human ES cells and biosafety of their differentiated progeny. PLoS One. 2012; 7 (9):e45532 - 92.
Houghton FD. Role of gap junctions during early embryo development. Reproduction. 2005; 129 (2):129-135 - 93.
Prokhorova TA, Harkness LM, Frandsen U, Ditzel N, Schrøder HD, Burns JS, et al. Teratoma formation by human embryonic stem cells is site dependent and enhanced by the presence of Matrigel. Stem Cells and Development. 2009; 18 (1):47-54 - 94.
Cooke MJ, Stojkovic M, Przyborski SA. Growth of teratomas derived from human pluripotent stem cells is influenced by the graft site. Stem Cells and Development. 2006; 15 (2):254-259 - 95.
Beltrão-Braga PC, Pignatari GC, Maiorka PC, Oliveira NA, Lizier NF, Wenceslau CV, et al. Feeder-free derivation of induced pluripotent stem cells from human immature dental pulp stem cells. Cell Transplantation. 2011; 20 (11-12):1707-1719 - 96.
Araldi RP, Viana M, Colozza-Gama GA, Dias Pinto JR, Ankol L, Wenceslau CV, et al. Unique transcriptional signatures observed in stem cells from the dental pulp of deciduous teeth produced on a large scale. Pharmacologia. 2023; 14 (1):72-95 - 97.
Bourbon-Teles J, Bells S, Jones DK, Coulthard E, Rosser A, Metzler-Baddeley C. Myelin breakdown in human Huntington's disease: Multi-modal evidence from diffusion MRI and quantitative magnetization transfer. Neuroscience. 2019; 403 :79-92. DOI: 10.1016/j.neuroscience.2017.05.042 - 98.
Roos RA. Huntington's disease: A clinical review. Orphanet Journal of Rare Diseases. 2010; 5 :40. DOI: 10.1186/1750-1172-5-40 - 99.
Abu-El-Rub E, Khasawneh RR, Almahasneh F, Altaany Z, Bataineh N, Zegallai H, et al. Mesenchymal stem cells and COVID-19: What they do and what they can do. World Journal of Stem Cells. 2021; 13 (9):1318-1337. DOI: 10.4252/wjsc.v13.i9.1318 - 100.
Macedo J, Pagani E, Wenceslau C, Ferrara L, Kerkis I. A phase I clinical trial on intravenous administration of immature human dental pulp stem cells (NestaCell HDTM) to Huntington's DISEASE patients. Cytotherapy. 2021; 23 (4):1. DOI: 10.1016/j.jcyt.2021.02.008 - 101.
Araldi RP, Prezoto BC, Gonzaga V, Policiquio B, Mendes TB, D'Amélio F, et al. Advanced cell therapy with low tissue factor loaded product NestaCell® does not confer thrombogenic risk for critically ill COVID-19 heparin-treated patients. Biomedicine & Pharmacotherapy. 2022; 112920 :72-95. DOI: 10.1016/j.biopha.2022.112920 - 102.
Costa VR, Araldi RP, Vigerelli H, D’Amélio F, Mendes TB, Gonzaga V, et al. Exosomes in tumor microenvironment: From biology to clinical applications. Cell. 2021; 10 (10):2617 - 103.
Fischer UM, Harting MT, Jimenez F, Monzon-Posadas WO, Xue H, Savitz SI, et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: The pulmonary first-pass effect. Stem Cells and Development. 2009; 18 (5):683-692. DOI: 10.1089/scd.2008.0253 - 104.
Silva JM, Araldi RP, Colloza-Gama GA, Pagani E, Sid A, Valverde CW, et al. Human immature dental pulp stem cells did not graft into preexisting human lung adenocarcinoma. Case Reports in Oncology. 2021; 15 (1):413-422