Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
Epstein-Barr virus (EBV) is a lymphotropic herpesvirus termed human herpesvirus 4 (HHV4). It was initially identified in biopsies of Burkitt’s lymphoma, arising in the jaw and other site of the body in childhood or early adolescent individuals in sub-Saharan region. Subsequently, its tight association with other type of lymphomas has been described, and the tightest association has been seen in nasopharyngeal carcinoma (NPC), endemic with southeast Asia and southern part of China. The malignant transforming potential of EBV has been identified in immune compromised individuals; in the context all viral genomic products are expressed among which oncogenic proteins or non-coding RNAs are expressed. The interactions between cellular and viral oncoprotein as well as host gene expression regulation by the viral genetic products have been investigated in human tumors. The switch from latent form of infection to lytic phase has been studied in EBV-associated human tumors, and the modulation by intracellular signaling pathways has been known to be of importance in EBV-mediated carcinogenesis.
Department of Pathophysiology, Guangdong Medical University, Dongguan, Guangdong, China
Zhe Zhang
Department of Ear, Nose and Throat (ENT) and Head and Neck Surgery, The First Affiliated Hospital, Guangxi Medical University, Nanning, Guangxi, China
Pankaj Trivedi
Department of Experimental Medicine, La Sapienza University of Rome, Rome, Italy
*Address all correspondence to: zhangxn_2006@126.com
1. Introduction
Epstein-Barr virus (EBV) is a lymphotropic DNA virus belonging to herpesviridae family. The human herpesviruses fall into a family with eight members; systematically, EBV is termed human herpesvirus type 4 (HHV-4); it was initially identified more than 60 years from the biopsies of Burkitt’s lymphoma (BL), a well-differentiated tumor arising in the jaw of individuals at childhood and early adolescence in sub-Saharan region of Africa [1]. It was the first virus documented to be of direct association with human cancers, leading to nasopharyngeal carcinoma (NPC) [2], endemic of southern China, Southeast Asia, North Africa, and Greenland. Biologically, EBV adopts two distinct phases in its life cycle when replicating on entering the human host; that is a lytic form of infection during which the virus replicates, and mature particles are assembled with the production of new virions with infection potentially to enter new host cells on releasing from the previous host cell having established parasitism.
EBV also established a latent form of infection when the full-length viral DNA is integrated into the genome in the invaded cells persisting in dormancy throughout the lifetime of the host [for a review, see Ref. [3]]. The infection of EBV in B cells has been intensively studied. It has been shown that the viral is capable of entering a cell type suitable for long-term latency and periodic reactivation, as manifested by replication ending with rupture of the host cells and release of mature viral particles [4].
EBV assesses to the site in the host B cell to initiate its life cycle of latent infection mimics the natural differentiation triggered by antigen exposure in the same type of cells. The presence of EBV at different developmental stages of B cells, and its ability to infect a range of cells of epithelial origin, also contributes to the pathogenesis, including the genesis of diverse lymphomas and carcinomas [5, 6, 7], exemplified by non-Hodgkin’s lymphoma, lymphomas and lymphoproliferative diseases in the immunocompromised, post-transplantation lymphoma, and NPC- and EBV-associated gastric cancer.
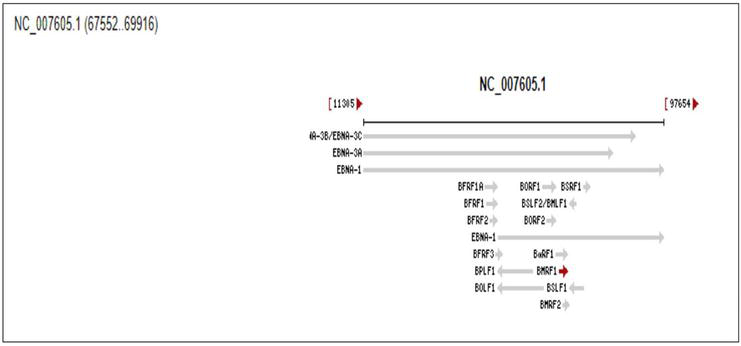
High titers of anti-EBV antibodies against viral capsid antigen (VCA) expressed on the surface of viral particles, and early diffuse (EAd/BMRF1) in patients with undifferentiated NPC seen in the endemic region with high incidence, suggest a tight association of EBV infection with occurrence of the cancer arising in the coating epithelium of nasopharynx [8, 9], and also, the frequent entering to lytic cycle of EBV harbored in the lesion of NPC, replicating in the tumor cells. The detection of the antiserum against the two EBV antigens whose coding gene cluster scheme is indicated in Figure 1 is diagnostic of NPC in combination of other factors like genetic predisposition.
Figure 1.
Genetic mapping of EAd coding gene BMRF1. The gene ID is 3,783,718, with sequence spanning the nucleotides 67,552–69,916 on genome DNA of EBV (NC_007605.1) with full length of 171 kb. Retrieved from https://www.ncbi.nlm.nih.gov/gene/3783718 on 4th July, 2023.
EBV is not identified in other forms of head and neck cancers, with exception of salivary gland tumors [10]. The replication site in the lesion is constituted with stratified squamous epithelium with differentiating properties; the microenvironment is speculated to be favorable for a program of lytic replication adopted by invading EBV. It is believed that the intracellular niche of less differentiated is suitable for EBV to establish infection; EBV genome is identified in poorly or undifferentiated but not well-differentiated cases of cancer of nasopharynx.
2. The switch from latency to lytic cycle as a characteristic process in the life cycle of herpesviruses
The life cycle of EBV, the first human tumorigenic virus ever being identified, has been defined as latency and lytic or productive infection by the early seminal work by George and Eva Klein. Up to now, at least 80 proteins encoded by the viral genome have been discovered, and a number of them still remain to be definitively identified [11]. During latent infection of EBV, the integrated viral genome synthesizes six nuclear antigens, termed EBV determined nuclear antigens (EBNAs), and they are alternatively called EBNA1-6 or EBNA 1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA-leader protein, together with three membrane integral proteins, namely latent membrane protein 1, 2A, and 2B (LMP1, 2A, 2B) [11]. Some EBNAs are expressed in lymphocytes with similar phenotype of lymphoblastoid cell lines (LCL) and of antigen activated in immunocompromised individuals, manifested as lymphoproliferative disorders; their transforming potential may play a role of genesis of malignancies. A latent EBV antigen, EBNA3C, alternatively called EBNA6 is mainly expressed in immunocompromised hosts due to its high immunogenicity. It has been described to be required for in vitro transformation of B cells. EBNA3C interacts with various cellular and viral factors to act as a transcriptional coregulatory. It has been revealed that EBNA-3C primarily targets two important cellular pathways—cell cycle and apoptosis. During EBV latency, EBNA-3C promotes B-cell lymphomagenesis by seizing cellular pathways [12].
One of the integral proteins encoded by EBV, LMP1, behaves as a constitutively active surface receptor to stimulate proliferation through the engagement of intracellular signaling pathways, notably of transcription factor NF-κB [13, 14], and JNK [15], its C-terminal activation regions (CTARs) of LMP1 selectively activate STAT family proteins, such as STAT3, STAT5, and STAT1 [16, 17]. LMP1 is therefore classified as a viral oncoprotein. It has been reported that LMP1 is detected in considerable amount of NPC cases, which are clinically immunocompetent [18, 19].
The latent infection of EBV is switched to lytic phase, which is activated by some chemicals in vitro, like sodium butyrate (NaB). The switch of latent infection to lytic cycle in EBV and other human herpesviruses has been studied in cultured B cell lines; it has been shown that the activation of lytic cycle in EBV is controlled by two EBV-encoded transcription factors, ZEBRA and Rta (reviewed in Ref. [20]). Distinctive genomic products are expressed in two phases of the EBV life cycle, latent and lytic cycle, contributing to the initiation and maintenance of the cycle [20]. The main products of EBV-encoded proteins and non-coding RNAs are listed in Table 1.
Time phase/gene exp. profile
Latent
Lytic cycle
IE
E
L
Proteins
EBNA 1, EBNA 2, EBNA 3s, EBNA LP LMP1, 2A, 2B
BZLF1 BRLF1
BMRF1 BALF2 BMLF2/3
VCA etc.
Non-coding RNAs
EBER1, 2 BARTs
Table 1.
The life cycle of EBV and expression profile of its genomic product.
The life cycle is divided into latent and lytic cycle; the latter is comprised of three phases, immediate early (IE), early (E), and late (L). During the three time courses IE, E, and L, the genomic products expressed are categorized as transactivators, replication factors, and structural proteins respectively.
The mapping of the genomic products expressed in EBV lytic cycle, contributing to its initiation and maintenance [20] on the linear structure of EBV genome is indicated in Figure 1. The two EBV-derived transcriptional activators are essential for the viral replication; strains with deletion of BZLF1 or BRLF1 in the genome are not competent for replication of viral DNA or production of mature virions [21]. At the initial step of lytic cycle, early genes-encoded proteins are upregulated.
Replication of EBV during lytic cycle is regulated by two immediate-early genes, BZLF1 and BRLF1 (Figure 2). They code for viral transcriptional activators Z and R, termed Zta and Rta, respectively [22]. EBV-encoded BZLF1 gene, a switch from latent infection to lytic infection, is expressed as early as 1.5 h after EBV infection in Burkitt’s lymphoma-derived, EBV-negative Akata and Daudi cells and primary B lymphocytes. Since BZLF1 mRNA is expressed even when the cells are infected with EBV in the presence of anisomycin, an inhibitor of protein synthesis, its expression does not require prerequisite protein synthesis, indicating that BZLF1 is expressed as an immediate-early gene following primary EBV infection of B lymphocytes [22].
Figure 2.
The genetic cluster of the immediate early lytic genes BRLF1 and BZLF1. The gene ID for the two genes are 3,783,727 and 3,783,744, spanning nucleotides 89,838–93,925(complement) and 89,838–90,943(complement). The number on the scale above the figure indicates the position of nucleotides within the genomic sequence. They code for lytic proteins Rta and Zta, respectively. Retrieved from https://www.ncbi.nlm.nih.gov/gene/3783727 and https://www.ncbi.nlm.nih.gov/gene/3783744 on 4th July 2023.
Late genes coding for structural proteins is expressed after replication of EBV DNA. The lytic genes of EBV respond to Zta and Rta at varying extent [23]. They are activated by either Rta or Zta primarily [24].
The expression of either Zta or Rta induces other lytic proteins and disrupts viral latency [25, 26]; activation of Rta and Zta has been found in vitro that their promoters are responsive to cross-linking of B-cell receptor (BCR), chemicals such as phorbol esters, and inophores, in addition to NaB.
Rta in EBV is homologous to open-reading frame 50 (ORF 50) within the genome of Kaposi Sarcoma-associated herpesvirus (KSHV), known as human herpesvirus 8 (HHV8). It activates lytic cycle through two distinct phases: The upstream events control the expression of lytic cycle activator genes encoded by the viral genome, and the downstream events drive the viral lytic cycle to express and hence initiate the replication of viral DNA.
The lytic cycle of EBV, and also of KSHV, is activated by a panel of chemicals like NaB. It is a classic inhibitor of histone deacetylase, which prevents removing acetyl group from modified amino acid. It has been shown that NaB disrupts latency of EBV, to allow the virus to enter lytic cycle. In case of EBV, protein synthesis is required when lytic cycle is activated by EBV [27].
An EBV immediate-early gene BZLF1 codes for a protein ZEBRA, which drives the lytic cycle, and this is the major downstream mediator of EBV lytic cycle activation. It acts both as a transcription activator and an essential replication protein. It has been shown that these two functions are separated by phosphorylation of ZEBRA at its casein kinase 2 (CK2) site. The activation of lytic replication by ZEBRA but not through induction of expression of early lytic cycle genes requires phosphorylation by CK (Figure 2) [28].
Zp, the promoter of the EBV immediate-early gene BZLF1, contains several phorbol ester response elements. Study on the role of PKC pathway in lytic cycle suggests that in lytic cycle induction by histone deacetylase (HDAC) inhibitors involves protein kinase C (PKC) [28], which is a group of enzymes that regulates the activation of activator protein-1, NF-кB and cAMP responsive element binding protein to stimulate the expression of a panel of genes. When activated by the phorbol ester TPA, B95–8 cells were activated into the lytic cycle a PKC-dependent mechanism but not by HDAC inhibitors such as n-butyrate and trichostatin A (TSA). In cells in which the immediate-early promoters Zp and Rp were simultaneously activated by the HDAC inhibitors, TPA by itself failed to activate lytic gene expression. Inhibition of PKC activity by bisindolylmaleimide I did not block lytic cycle activation in the EBV-positive Burkitt lymphoma cells by n-butyrate or TSA.
In an extensive exploration of the mechanism underlying the different responses, the variable role of the PKC pathway found in different cell lines could not be accounted for by significant polymorphisms in the promoters of the immediate-early genes, or by differences in the nucleosomal organization of EBV DNA in the region of Zp or Rp [29].
3. The regulation of latency-to-lytic phase by viral and cellular machinery: the implication of transcription factors
The transcriptional activators Z (Zta) and R (Rta) are encoded by two immediate-early genes, BZLF1 and BRLF1 of EBV, to regulate EBV replication. Zta is a basic zipper (bZIP) protein with DNA binding capacity; it binds and activates promoters containing AP1 sites (TGASTCA) and related sequences called Zta-responsive elements [30]. The EBV-encoded transcriptional activator, Rta has a length of 605-amino-acid (aa) residues; it is an acidic transactivator protein; its homolog has not been found in any DNA-binding proteins in the cell. Its homologs, however, are present in all gamma-herpesviruses, and the greatest homology exists in their N-terminal DNA binding domains (DBDs) on the amino(N)-terminus with up to 40% similarity between EBV and KSHV [30].
Another EBV-encoded transcriptional activator, Rta binds to a particular sequence, Rta response elements (RREs), conforming to the consensus GNCCN9GGNG, to activate various promoters through a direct mechanism [31, 32, 33, 34]. It also activates other promoters lacking the sequence of RREs through an indirect mechanism(s). Such target sequences include their own promoter (Rp). Rta activates the sequences through Sp1/Sp3 binding sites [35].
The Zta promoter (Zp) activates many promoters via the ZII cyclic AMP(cAMP) response element [36]; and the BALF5 DNA polymerase gene, a lytic EBV gene, through USF and E2F binding sites [37, 38]. For the promoter Rp, it has been proposed that Rta is targeted indirectly to Sp1/Sp3 sites through an interaction with MCAF1, an Sp1-associated factor [39]. This mechanism closely mirrors that of KSHV Rta, which activates promoters indirectly via an interaction with the DNA-binding protein RBP-Jκ [40, 41, 42].
Rta can also activate promoters indirectly through the activation of mitogen-activated protein kinase and phosphatidylinositol-3 kinase, resulting in phosphorylation of ATF2 bound to the ZII element and activation of Zta expression [43, 44].
The induction of viral transcriptional activators Zta and Rta contributes to the switch between the latent and the lytic cycle of EBV infection. In EBV-infected B cells, their expression is likely regulated by CD4+ T cell-derived cytokines. It has been shown that during the first 3 days of culture interleukin-21 decreases constitutive expression of Rta and its target EA-D in EBV-infected B cell lines. In some cell lines, this is followed by a strong increase in the expression of Zta, Rta, and EA-D during the prolonged culture. Additionally, there is evidence suggesting that IL-21-mediated JAK/STAT signals regulate increased expression of Zta [45].
It has been observed that high level of STATs notably remains the state of latent infection of EBV [46]. Protein inhibitor of activated STAT(PIAS) proteins has been identified as negative regulators of STAT signaling. The PIAS proteins exert the inhibitory effect through modification activity of SUMO E3 ligase on ubiquitination of protein substrate. Up to more than 60 proteins, most of them being transcription factors, either positively or negatively regulated by members of the PIAS family through multiple mechanisms, have been identified in biochemical studies [47, 48].
A member of this family, PIAS1 is a restriction factor of EBV, acting to inhibit viral and cellular transcription factors. The interactions of PIAS1 with interferon regulatory factor 8 (IRF8) have been demonstrated and PIAS1 has been shown to inhibit lytic gene activation mediated by IRF8 [45, 49]. In the population refractory to lytic cycle induction, Stat3 and Fos transcripts were preferentially upregulated. Expression of both factors was increased folds compared to untreated cells.
In EBV harboring Burkitt lymphoma cells, the regulation of lytic cycle entry is investigated. When latently infected with EBV, the cells express high levels of STAT3 protein, predominantly in unphosphorylated form. When exposed to NaB, a lytic cycle-inducing agent and a prototype inhibitor of HDAC, entry of lytic cycle entry is induced. The cells were treated with IFN-γ determining whether STAT3 is phosphorylated at the tyrosine residue Y705 in this cell line or if this pathway was defective. Since the increase of Stat3 transcript occurs primarily in refractory cells, the levels in STAT3 protein were examined present in this population. Increased level of STAT3 protein in the refractory population is relative to the untreated cells in a manner of time course dependency after treatment with NaB is identified. STAT3 protein, however, is not significantly upregulated in the subpopulation of lytic cells [20, 21].
The exact molecular mechanisms, such as underlying posttranscriptional processes, to control latency of EBV, the reactivation and progression of the lytic cycle remain to be fully elucidated [50, 51]. Master transcriptional regulators of plasma cell differentiation, including Blimp-1/PRDM1, also activate the promoters of EBV genes BZLF1 (Zp) and BRLF1 (Rp) [51, 52]. Transcription factors such as ATFs, Sp1/3, MEF2D, XBPs, family members of cAMP-responsive element-binding protein (CREB), AP1 heterodimers of c-Fos and phosphorylated c-Jun, and HIF1α interact with Zp in response to challenge of antigen or oxidative stress; Zp further contains cis-regulatory elements to exert autoregulation [50, 51, 52, 53, 54, 55]. Repressors of Zp are repressed by molecules like the zinc-finger E-box-binding proteins encoded by ZEB1 and ZEB2 and the polycomb protein Yin Yang 1 (YY1) [50, 51, 56, 57, 58]. Notably, microRNAs (miRNAs) from the miR-200 family (miR-200b and miR-429 expressed in epithelial cells) which post-transcriptionally silence ZEB1/2 expression are capable of enhancing EBV reactivation through increased Zp activity [59, 60].
It has been shown that high levels of host STAT3 curtail the susceptibility of latently infected cells to lytic cycle activation signals. Cellular PCBP2 [poly(C)-binding protein 2], an RNA-binding protein, has been identified as a transcriptional target of STAT3 in refractory cells. It has been demonstrated that single cells expressing high levels of PCBP2 are refractory to lytic activation of EBV, both spontaneous and induced, lytic susceptibility is regulated by STAT3 via cellular PCBP2, and suppression of PCBP2 levels is sufficient to increase EBV lytic cells in number [61]. The findings would guide efforts to improve oncolytic therapy for EBV-associated cancers.
Host miRNAs in the EBV lytic cycle are profiled. Among small RNAs in reactivated Burkitt lymphoma cells, several miRNAs, such as miR-141, are identified. They are induced upon BCR cross-linking. Notably, EBV encodes a viral miRNA, miR-BART9, with sequence homology to miR-141. Their molecular targets and experimentally validated multiple candidates are commonly regulated by both miRNAs. Targets included B cell transcription factors and known regulators of EBV immediate-early genes, leading us to hypothesize that these miRNAs modulate kinetics of the lytic cascade in B cells. Through functional assays, we identified roles for miR-141 and EBV miR-BART9 and one specific target, FOXO3, in progression of the lytic cycle [62, 63].
4. Lytic cycle activation contributes to viral tumorigenicity
Cellular factors like E2F6, E2F1, tumor suppressor gene (TSG) coding product Rb, and enzymes catalyzing removal of acetyl group from histones and non-histone proteins, histone deacetylase 1 (HDAC1), and HDAC2 are associated with EBV-encoded nuclear antigens, to regulate such events like gene expression, based on the next-generation sequencing (NGS) analysis. A complex of E2F-Rb-HDAC exhibits similar distributions located in genomic regions of EBV-positive cells and its long-range regulatory regions are associated with oncogenic super-enhancers [64].
The transforming EBV latent antigens cooperatively hijack this complex, to bind KLFs gene loci on host genomic sequence, to facilitate the expression of KLF14 gene in LCLs. These results demonstrate that EBV latent antigens have been demonstrated to function as master regulators of this multi-subunit repressor complex (E2F-Rb-HDAC) with tumor suppressive potential, to reverse its activities antagonistic and facilitate downstream gene expression to contribute to the viral transforming protein-mediated lymphomagenesis [64].
To date, several cytogenetic lesions have been reported to be of crucial alterations in the occurrence of NPC, an EBV-associated tumor endemic of certain regions in the world, including southern China, Southeast Asia, and Greenland inhabited by Inuit and North Africa [65]. In Asian NPC, loss of homozygosity (LOH) is frequently detected at several chromosomal regions, particularly the locations 3p, 9p, 11q, 13q, and 14q [66, 67, 68, 69, 70, 71]. It has been reported that the anomaly in 3p is among key changes during occurrence of NPC [66]. A high frequency of LOH in 3p was also found in normal nasopharyngeal epithelial cells and precancerous lesions in individuals from endemic areas, suggesting that the inactivation of TSGs in this chromosome might be an early event in the genesis of NPC [66]. Coding genes for p16/CDKN2A and RASSF1A on 9p and 3p respectively have been recognized as main TSGs in NPC [66, 67, 68, 69, 70, 71]. Mutability of RASSF1A has been reported in NPC [67]. CCND1 coding for cyclin D1(cycD1) has been observed to be amplified in NPC epithelium; the role played by cycD1 in EBV-mediated transformation has been documented [72].
Another 3p21-mapped TSG, BLU codes for a zinc finger motif containing protein ZMYND10 with tumor suppressive potential have been identified. Its inactivation due to epigenetic approach has been observed in clinical specimens, as well as passaged cell lines of NPC. Its promoter hypermethylation was detected in the primary tumors of NPC, but not in normal nasopharyngeal epithelium and cells of immortalized normal epithelial cells [73, 74]. The expression of BLU is correlated with a favorable prognosis, prolonged survival of NPC patients [75].
The cytogenetic aberrations suggested that genome instability is closely associated with the development of NPC. Given that both chemicals and the virus have been shown to be co-carcinogens in the development of cancer [76], it would be interesting to examine the interplay between EBV and chemical carcinogens and their effects on the genome instability of NPC.
With regard to the dietary association with the genesis of NPC, traditionally consuming food from high-risk areas of NPC is found to contain EBV inducers and mutagens, as well as N-nitrosamines [77]. Moreover, it has also been shown that various chemicals aforementioned as inducers of EBV lytic cycle, like phorbol esters and n-butyrate present in several herbal medicines and food sources, can induce the EBV lytic cycle and may be involved in the tumorigenesis of NPC [78, 79, 80].
The regions of China with a high annual incidence of NPC were colocalized with those where herbal drugs containing phorbol esters are commonly used [81]. These results suggest that chemical carcinogens may contribute to the carcinogenesis of NPC. However, the underlying mechanism has not been extensively studied yet [65]. The synergy of chemicals including EBV lytic cycle inducers 12-O-tetradecanoylphorbol-13-acetate (TPA) and NaB in enhancing EBV reactivation and genome instability for implication of NPC genesis has been investigated with an EBV-negative nasopharyngeal carcinoma cell line and an EBV-positive NPC cell derived from the same line. The results of expression profile analysis indicate that many carcinogenesis-related genes were altered after recurrent EBV reactivation, and several aberrations are observed in correspondence to alterations in NPC. Cooperation between chemical carcinogens enhances the reactivation of EBV leading to alteration of cancer hallmark gene expression with resultant enhancement of tumorigenesis in NPC [82].
5. Perspectives: application in therapeutic strategy for treating virally associated tumors
EBV infection is associated with a number of human tumors of lymphoid and epithelial origin; the tightest association has been seen with NPC, in virtually all cases, and the viral DNA is isolated from the tumor cells. EBV is mainly present in NPC cells with a pattern of latent infection. Inhibition of virus replication, however, is not efficacious in treating EBV-associated malignancies. Instead, activation of EBV replication is potentially therapeutic, because virus replication can directly kill EBV-infected tumor cells, sensitize them to nucleoside analogues, and stimulate immune-mediated killing via increased virus antigen expression in tumor cells [83, 84]. When EBV is triggered to enter the reproductive lytic phase, immunogenic proteins, in addition to only a few viral proteins and non-coding small RNAs expressed at latent stage, are expressed. The expression of lytic antigens provokes a stronger and more effective immune response [85]. Importantly, viral kinases expressed as lytic cycle proteins render tumor cells to sensitive for certain antiviral treatment, like (val)ganciclovir [86].
The intentional induction of the lytic form of EBV infection combined with ganciclovir (GCV) treatment has been proposed as a novel regimen for anti-EBV positive tumor therapy [87]. EBV-encoded BGLF4 with thymidine kinase activity is expressed only during the lytic form of infection. Its catalytic activity converts a nucleoside analogue, GCV into its active, cytotoxic form. Gemcitabine and doxorubicin induce lytic EBV infection in EBV-transformed B cells in vitro and in vivo. The combination of gemcitabine or doxorubicin and GCV has significantly more cytostatic effective for EBV-driven lymphoproliferative disease in SCID mice than chemotherapy alone. The results suggest that the addition of GCV, due to the presence of active enzyme, may enhance the therapeutic efficacy of gemcitabine- or doxorubicin-containing chemotherapy regimens, for treating EBV-driven lymphoproliferative disease in patients.
In addition to EBV-associated BL, specifically target EBV-positive cells for destruction have been tested as a therapeutic approach for tumors of epithelial origin. The efficacy of adenovirus vectors expressing the BZLF1 or BRLF1 proteins for treatment of EBV-positive such tumors have been examined. The BZLF1 and BRLF1 vectors induced preferential killing of EBV-positive gastric carcinoma cells in vitro and NPC tumors. The antitumor effect of the BZLF1 and BRLF1 adenovirus vectors was not significantly affected by adding ganciclovir. These results suggest lytic cycle of EBV induction of a potential cancer therapy against EBV-related tumors [88].
Virus-targeted lytic induction treatment in EBV-associated malignancies aims at evoking more potent immune responses and induce susceptibility to achieve a goal in the virally targeting therapy. Clinically, this strategy was initially attempted in EBV-associated lymphoma [89] and has now been studied in subsequent years [90, 91]. Different agents, such as HDAC inhibitors, chemotherapeutics, radiation, phorbol esters, and butyrates, have been tested as inducers of the lytic phase of EBV in associated tumor cell lines [86, 87, 88]. Until now, only a few clinical proof-of-principle studies on viral lytic induction have been performed [88, 92].
The infection of several viruses, including two human herpesviruses, EBV and KSHV/HHV-8, has been found to be associated with carcinogenesis. The tightest association has been seen with EBV in case of BL and NPC. EBV adopts a distinctive pattern of life cycle, with latency and lytic cycle. During lytic cycle, the viral replication leads to host cells rupture and release of mature viral particles, to allow establishment of new infection of latency. The viral-encoded products play a role in achievement of malignancy, and understanding of regulation and drug targeting of viral lytic cycle will contribute to improve anticancer therapy.
The authors would like to dedicate the manuscript to the late Professor George Klein (1925-2016), a pioneering tumor biologist who made tremendous contribution in the fields of chemical and viral carcinogenesis together with discoveries in cancer cytogenetics and oncogenes; his life time efforts have enabled tumor biology to evolve as an independent science from a branch of cell biology. We are proud of being on his student chain.
References
1.Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from burkitt's lymphoma. Lancet. 1964;1(7335):702-703. DOI: 10.1016/s0140-6736(64)91524-7
2.Zur Hausen H, Schulte-Holthausen H, Klein G, Henle W, Henle G, Clifford P, et al. EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature. 1970;228:1056-1058
3.Pattle SB, Farrell PJ. The role of Epstein–Barr virus in cancer. Expert Opinion on Biological Therapy. 2006;6(11):1193-1205. DOI: 10.1517/14712598.6.11.1193
5.Brady G, MacArthur GJ, Farrell PJ. Epstein-Barr virus and Burkitt lymphoma. Journal of Clinical Pathology. 2007;60(12):1397-1402. DOI: 10.1136/jcp.2007.047977
6.Pathmanathan R, Prasad U, Sadler R, Flynn K, Raab-Traub N. Clonal proliferations of cells infected with Epstein–Barr virus in preinvasive lesions related to nasopharyngeal carcinoma. The New England Journal of Medicine. 1995;333:693-698
7.Osato T, Imai S. Epstein-Barr virus and gastric carcinoma. Seminars in Cancer Biology. 1996;7(4):175-182. DOI: 10.1006/scbi.1996.0024
8.Paramita DK, Fachiroh J, Artama WT, van Benthem E, Haryana SM, Middeldorp JM. Native early antigen of Epstein-Barr virus, a promising antigen for diagnosis of nasopharyngeal carcinoma. Journal of Medical Virology. 2007;79(11):1710-1721. DOI: 10.1002/jmv.20987
9.Liu MY, Huang YT, Sheen TS, Chen JY, Tsai CH. Immune responses to Epstein-Barr virus lytic proteins in patients with nasopharyngeal carcinoma. Journal of Medical Virology. 2004;73(4):574-582. DOI: 10.1002/jmv.20128
10.Palser AL, Grayson NE, White RE, Corton C, Correia S, Ba Abdullah MM, et al. Genome diversity of Epstein-Barr virus from multiple tumor types and normal infection. Journal of Virology. 2015;89:5222-5237. DOI: 10.1128/jvi.03614-14
11.Houldcroft CJ, Kellam P. Host genetics of Epstein-Barr virus infection, latency and disease. Reviews in Medical Virology. 2015;25:71-84. DOI: 10.1002/rmv.1816
12.Saha A, Robertson ES. Impact of EBV essential nuclear protein EBNA-3C on B-cell proliferation and apoptosis. Future Microbiology. 2013;8(3):323-352. DOI: 10.2217/fmb.12.147
13.Miller WE, Mosialos G, Kieff E, Raab-Traub N. Epstein-Barr virus LMP1 induction of the epidermal growth factor receptor is mediated through a TRAF signaling pathway distinct from NF-kappaB activation. Journal of Virology. 1997;71(1):586-594. DOI: 10.1128/JVI.71.1.586-594.1997
14.Eliopoulos AG, Stack M, Dawson CW, Kaye KM, Hodgkin L, Sihota S, et al. Epstein-Barr virus-encoded LMP1 and CD40 mediate IL-6 production in epithelial cells via an NF-kappaB pathway involving TNF receptor-associated factors. Oncogene. 1997;14(24):2899-2916. DOI: 10.1038/sj.onc.1201258
15.Eliopoulos AG, Blake SM, Floettmann JE, Rowe M, Young LS. Epstein-Barr virus-encoded latent membrane protein 1 activates the JNK pathway through its extreme C terminus via a mechanism involving TRADD and TRAF2. Journal of Virology. 1999;73(2):1023-1035. DOI: 10.1128/JVI.73.2.1023-1035.1999.6
16.Eliopoulos AG, Young LS. LMP1 structure and signal transduction. Seminars in Cancer Biology. 2001;11(6):435-444
17.Tsao SW, Tramoutanis G, Dawson CW, Lo AK, Huang DP. The significance of LMP1 expression in nasopharyngeal carcinoma. Seminars in Cancer Biology. 2002;12(6):473-487
18.Young LS, Dawson CW, Clark D, Rupani H, Busson P, Tursz T, et al. Epstein-Barr virus gene expression in nasopharyngeal carcinoma. The Journal of General Virology. 1988;69(Pt 5):1051-1065. DOI: 10.1099/0022-1317-69-5-1051
19.Tsurumi T, Fujita M, Kudoh A. Latent and lytic Epstein-Barr virus replication strategies. Reviews in Medical Virology. 2005;15(1):3-15. DOI: 10.1002/rmv.441
20.Fåhraeus R, Fu HL, Ernberg I, Finke J, Rowe M, Klein G, et al. Expression of Epstein-Barr virus-encoded proteins in nasopharyngeal carcinoma. International Journal of Cancer. 1988;42(3):329-338. DOI: 10.1002/ijc.2910420305
21.Miller G, El-Guindy A, Countryman J, Ye J, Gradoville L. Lytic cycle switches of oncogenic human gammaherpesviruses. Advances in Cancer Research. 2007;97:81-109. DOI: 10.1016/S0065-230X(06)97004-3
22.Miller IG Jr, El-Guindy A. Regulation of Epstein-Barr virus lytic cycle activation in malignant and nonmalignant disease. Journal of the National Cancer Institute. 2002;94:1733-1735
23.Wen W, Iwakiri D, Yamamoto K, Maruo S, Kanda T, Takada K. Epstein-Barr virus BZLF1 gene, a switch from latency to lytic infection, is expressed as an immediate-early gene after primary infection of B lymphocytes. Journal of Virology. 2007;81(2):1037-1042. DOI: 10.1128/JVI.01416-06
24.Feederle R, Kost M, Baumann M, Janz A, Drouet E, Hammerschmidt W, et al. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. The EMBO Journal. 2000;19:3080-3089
25.Lu CC, Jeng YY, Tsai CH, Liu MY, Yeh SW, Hsu TY, et al. Genome-wide transcription program and expression of the Rta responsive gene of Epstein-Barr virus. Virology. 2006;345:358-372
26.Ragoczy T, Miller G. Role of the Epstein-Barr virus RTA protein in activation of distinct classes of viral lytic cycle genes. Journal of Virology. 1999;73:9858-9866
27.Ragoczy T, Heston L, Miller G. The Epstein-Barr virus Rta protein activates lytic cycle genes and can disrupt latency in B lymphocytes. Journal of Virology. 1998;72:7978-7984
28.Zalani S, Holley-Guthrie E, Kenney S. Epstein-Barr viral latency is disrupted by the immediate-early BRLF1 protein through a cell-specific mechanism. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9194-9199
29.Gorres KL, Daigle D, Mohanram S, Miller G. Activation and repression of Epstein-Barr virus and Kaposi's sarcoma-associated herpesvirus lytic cycles by short- and medium-chain fatty acids. Journal of Virology. 2014;88(14):8028-8044. DOI: 10.1128/JVI.00722-14
30.Gradoville L, Kwa D, El-Guindy A, Miller G. Protein kinase C-independent activation of the Epstein-Barr virus lytic cycle. Journal of Virology. 2002;76(11):5612-5626. DOI: 10.1128/jvi.76.11.5612-5626.2002
31.Chen Y, Fachko DN, Ivanov NS, Skalsky RL. B cell receptor-responsive miR-141 enhances Epstein-Barr virus lytic cycle via FOXO3 inhibition. mSphere. 2021;6:e00093-e00021. DOI: 10.1128/mSphere.00093-21
32.Calderwood MA, Holthaus AM, Johannsen E. The Epstein-Barr virus LF2 protein inhibits viral replication. Journal of Virology. 2008;82(17):8509-8519
33.Chen LW, Chang PJ, Delecluse HJ, Miller G. Marked variation in response of consensus binding elements for the Rta protein of Epstein-Barr virus. Journal of Virology. 2005, 2005;79:9635-9650
34.Gruffat H, Duran N, Buisson M, Wild F, Buckland R, Sergeant A. Characterization of an R-binding site mediating the R-induced activation of the Epstein-Barr virus BMLF1 promoter. Journal of Virology. 1992;66:46-52
35.Manet E, Rigolet A, Sergeant A. The enhancer factor R of Epstein-Barr virus (EBV) is a sequence-specific DNA binding protein. Nucleic Acids Research. 1990;18:6835-6843
36.Gruffat H, Sergeant A. Characterization of the DNA-binding site repertoire for the Epstein-Barr virus transcription factor R. Nucleic Acids Research. 1994;22(7):1172-1178
37.Ragoczy T, Miller G. Autostimulation of the Epstein-Barr virus BRLF1 promoter is mediated through consensus Sp1 and Sp3 binding sites. Journal of Virology. 2001;75:5240-5251
38.Speck SH, Chatila T, Flemington E. Reactivation of Epstein-Barr virus: Regulation and function of the BZLF1 gene. Trends in Microbiology. 1997;5:399-405
39.Furnari FB, Adams MD, Pagano J, JS. Regulation of the Epstein-Barr virus DNA polymerase gene. Journal of Virology. 1992;66:2837-2845
40.Liu C, Sista ND, Pagano JS. Activation of the Epstein-Barr virus DNA polymerase promoter by the BRLF1 immediate-early protein is mediated through USF and E2F. Journal of Virology. 1996;70:2545-2555
41.Chang LK, Chung JY, Hong YR, Ichimura T, Nakao M, Liu ST. Activation of Sp1-mediated transcription by Rta of Epstein-Barr virus via an interaction with MCAF1. Nucleic Acids Research. 2005;33:6528-6539
42.Carroll KD, Bu W, Palmeri D, Spadavecchia S, Lynch SJ, Marras SA, et al. Kaposi’s sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP-Jk/CSL to activate the notch pathway. Journal of Virology. 2006;80:9697-9709
43.Liang Y, Chang J, Lynch SJ, Lukac DM, Ganem D. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-J (CSL), the target of the notch signaling pathway. Genes & Development. 2002;16:1977-1989
44.Liang Y, Ganem D. Lytic but not latent infection by Kaposi’s sarcoma-associated herpesvirus requires host CSL protein, the mediator of notch signaling. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8490-8495
45.Adamson AL, Darr D, Holley-Guthrie E, Johnson RA, Mauser A, Swenson J, et al. Epstein-Barr virus immediate-early proteins BZLF1 and BRLF1 activate the ATF2 transcription factor by increasing the levels of phosphorylated p38 and c-Jun N-terminal kinases. Journal of Virology. 2000;74:1224-1233
46.Kenney SC, Mertz JE. Regulation of the latent-lytic switch in Epstein-Barr virus. Seminars in Cancer Biology. 2014;26:60-68
47.Shuai K, Liu B. Regulation of JAK–STAT signaling in the immune system. Nature Reviews Immunology. 2003;3:900-911
48.Shuai K. Modulation of STAT signaling by STAT-interacting proteins. Oncogene. 2000;19:2638-2644
49.Zhang K, Lv D-W, Li RB. Cell receptor activation and chemical induction trigger caspase-mediated cleavage of PIAS1 to facilitate Epstein-Barr virus reactivation. Cell Reports. 2017;21:3445-3457
50.Darr CD, Mauser A, Kenney S. Epstein-Barr virus immediate-early protein BRLF1 induces the lytic form of viral replication through a mechanism involving phosphatidylinositol-3 kinase activation. Journal of Virology. 2001;75:6135-6142
51.Konforte D, Paige CJ. Interleukin-21 regulates expression of the immediate-early lytic cycle genes and proteins in Epstein-Barr virus infected B cells. Virus Research. 2009;144:339-343
52.McKenzie J, El-Guindy A. Epstein-Barr virus lytic cycle reactivation. Current Topics in Microbiology and Immunology. 2015;391:237-261. DOI: 10.1007/978-3-319-22834-1_8
53.Reusch JA, Nawandar DM, Wright KL, Kenney SC, Mertz JE. Cellular differentiation regulator BLIMP1 induces Epstein-Barr virus lytic reactivation in epithelial and B cells by activating transcription from both the R and Z promoters. Journal of Virology. 2015;89:1731-1743. DOI: 10.1128/JVI.02781-14
54.Liu S, Liu P, Borras A, Chatila T, Speck SH. Cyclosporin A-sensitive induction of the Epstein-Barr virus lytic switch is mediated via a novel pathway involving a MEF2 family member. The EMBO Journal. 1997;16:143-153. DOI: 10.1093/emboj/16.1.143
55.Liu S, Borras AM, Liu P, Suske G, Speck SH. Binding of the ubiquitous cellular transcription factors Sp1 and Sp3 to the ZI domains in the Epstein-Barr virus lytic switch BZLF1 gene promoter. Virology. 1997;228:11-18. DOI: 10.1006/viro.1996.8371
56.Bhende PM, Dickerson SJ, Sun X, Feng WH, Kenney SC. X-box-binding protein 1 activates lytic Epstein-Barr virus gene expression in combination with protein kinase D. Journal of Virology. 2007;81:7363-7370. DOI: 10.1128/JVI.00154-07
57.Murata T, Sato Y, Nakayama S, Kudoh A, Iwahori S, Isomura H, et al. TORC2, a coactivator of cAMP-response element-binding protein, promotes Epstein-Barr virus reactivation from latency through interaction with viral BZLF1 protein. The Journal of Biological Chemistry. 2009;284:8033-8041. DOI: 10.1074/jbc.M808466200
58.Murata T, Noda C, Saito S, Kawashima D, Sugimoto A, Isomura H, et al. Involvement of Jun dimerization protein 2 (JDP2) in the maintenance of Epstein-Barr virus latency. The Journal of Biological Chemistry. 2011;286:22007-22016. DOI: 10.1074/jbc.M110.199836
59.Yu X, Wang Z, Mertz JE. ZEB1 regulates the latent-lytic switch in infection by Epstein-Barr virus. PLoS Pathogens. 2007;3:e194. DOI: 10.1371/journal.ppat.0030194
60.Montalvo EA, Cottam M, Hill S, Wang YJ. YY1 binds to and regulates cis-acting negative elements in the Epstein-Barr virus BZLF1 promoter. Journal of Virology. 1995;69:4158-4165. DOI: 10.1128/JVI.69.7.4158-4165.1995
61.Koganti S, Clark C, Zhi J, Li X, Chen EI, Chakrabortty S, et al. Cellular STAT3 functions via PCBP2 to restrain Epstein-Barr virus lytic activation in B lymphocytes. Journal of Virology. 2015;89(9):5002-5011. DOI: 10.1128/JVI.00121-15
62.Ellis-Connell AL, Iempridee T, Xu I, Mertz JE. Cellular microRNAs 200b and 429 regulate the Epstein-Barr virus switch between latency and lytic replication. Journal of Virology. 2010;84:10329-10343. DOI: 10.1128/JVI.00923-10
63.Lin Z, Wang X, Fewell C, Cameron J, Yin Q , Flemington EK. Differential expression of the miR-200 family microRNAs in epithelial and B cells and regulation of Epstein-Barr virus reactivation by the miR-200 family member miR-429. Journal of Virology. 2010;84:7892-7897. DOI: 10.1128/JVI.00379-10
64.Pei Y, Wong JH-Y, Jha HC, Tian T, Wei Z, Robertson ES. Epstein-Barr virus facilitates expression of KLF14 by regulating the cooperative binding of the E2F-Rb-HDAC complex in latent infection. Journal of Virology. 2020;94:e01209-e01220. DOI: 10.1128/JVI.01209-20
65.Lo KW, To KF, Huang DP. Focus on nasopharyngeal carcinoma. Cancer Cell. 2004;5:423-428
66.Lo KW, Teo PM, Hui AB, To KF, Tsang YS, Chan SY, et al. High resolution allelotype of microdissected primary nasopharyngeal carcinoma. Cancer Research. 2000;60:3348-3353
67.Huang DP, Lo KW, van Hasselt CA, Woo JK, Choi PH, Leung SF, et al. A region of homozygous deletion on chromosome 9p21-22 in primary nasopharyngeal carcinoma. Cancer Research. 1994;54:4003-4006
68.Lo KW, Tsao SW, Leung SF, Choi PHK, Lee JCK, Huang DP. Detailed deletion mapping on the short arm of chromosome 3 in nasopharyngeal carcinomas. International Journal of Oncology. 1994;4:1359-1364
69.Chen J, Fu L, Zhang LY, Kwong DL, Yan L, Guan XY. Tumor suppressor genes on frequently deleted chromosome 3p in nasopharyngeal carcinoma. Chinese Journal of Cancer. 2012;31(5):215-222
70.Chan AS, To KF, Lo KW, Ding M, Li X, Johnson P, et al. High frequency of chromosome 3p deletion in histologically normal nasopharyngeal epithelia from southern Chinese. Cancer Research. 2000;60:5365-5370
71.Kashuba VI, Pavlova TV, Grigorieva EV, Kutsenko A, Yenamandra SP, Li J, et al. High mutability of the tumor suppressor genes RASSF1 and RBSP3 (CTDSPL) in cancer. PLoS One. 2009;4(5):e5231. DOI: 10.1371/journal. pone. 00052 31
72.Tsang CM, Yip YL, Lo KW, Deng W, To KF, Hau PM, et al. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(50):E3473-E3482. DOI: 10.1073/pnas.12026 37109
73.Liu XQ , Chen HK, Zhang XS, Pan ZG, Li A, Feng QS, et al. Alterations of BLU, a candidate tumor suppressor gene on chromosome 3p21.3, in human nasopharyngeal carcinoma. International Journal of Cancer. 2003;106:60-65
74.Qiu GH, Tan LK, Loh KS, Lim CY, Srivastava G, Tsai ST, et al. The candidate tumor suppressor gene BLU, located at the commonly deleted region 3p21.3, is an E2F-regulated, stress-responsive gene and inactivated by both epigenetic and genetic mechanisms in nasopharyngeal carcinoma. Oncogene. 2004;23:4793-4806
75.Fang L, Shi L, Wang W, Chen Q , Rao X. Identifying key genes and small molecule compounds for nasopharyngeal carcinoma by various bioinformatics analysis. Medicine. 2021;100(37):e27257
76.Haverkos HW. Viruses, chemicals and co-carcinogenesis. Oncogene. 2004;23:6492-6499
77.Bouvier G, Poirier S, Shao YM, Malaveille C, Ohshima H, et al. Epstein-Barr virus activators, mutagens and volatile nitrosamines in preserved food samples from high-risk areas for nasopharyngeal carcinoma. IARC Scientific Publications. 1991;105:204-209
78.Zur Hausen H, O’Neill FJ, Freese UK, Hecker E. Persisting oncogenic herpesvirus induced by the tumour promotor TPA. Nature. 1978;272:373-375
79.Luka J, Kallin B, Klein G. Induction of the Epstein-Barr virus (EBV) cycle in latently infected cells by n-butyrate. Virology. 1979;94:228-231
80.Rickinson AB, Kieff E. Epstein-Barr virus. In: Knipe DM, Howley PM, editors. Fields’ Virology. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 2575-2627
81.Hirayama T, Ito Y. A new view of the etiology of nasopharyngeal carcinoma. Preventive Medicine. 1981;10:614-622
82.Fang C-Y, Huang S-Y, Wu C-C, Hsu H-Y, Chou S-P, et al. The synergistic effect of chemical carcinogens enhances Epstein-Barr virus reactivation and tumor progression of nasopharyngeal carcinoma cells. PLoS One. 2012;7(9):e44810. DOI: 10.1371/journal.pone.0044810
83.Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: Lessons from Epstein–Barr virus. Annual Review of Immunology. 2007;25:587-617
84.Moore SM, Cannon JS, Tanhehco YC, Hamzeh FM, Ambinder RF. Induction of Epstein–Barr virus kinases to sensitize tumor cells to nucleoside analogues. Antimicrobial Agents and Chemotherapy. 2001;45(7):2082-2091
85.Feng WH, Hong G, Delecluse HJ, Kenney SC. Lytic induction therapy for Epstein-Barr virus-positive B-cell lymphomas. Journal of Virology. 2004;78:1893-1902
86.Westphal E, Mauser A, Raab-Traub N, Gulley ML, Busson P, Kenney SC. Use of adenovirus vectors expressing Epstein-Barr virus (EBV) immediate-early protein BZLF1 or BRLF1 to treat EBV-positive tumors. Journal of Virology. 2002;76(21):10951-10959. DOI: 10.1128/jvi.76.21.10951-10959.2002
87.Faller DV, Mentzer SJ, Perrine SP. Induction of the Epstein–Barr virus thymidine kinase gene with concomitant nucleoside antivirals as a therapeutic strategy for Epstein–Barr virus-associated malignancies. Current Opinion in Oncology. 2011;13(5):360-367
88.Perrine SP, Hermine O, Small T, Suarez F, O’Reilly R, Boulad F, et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein–Barr virus-associated lymphoid malignancies. Blood. 2007;109(6):2571-2578
89.Ghosh SK, Perrine SP, Faller DV. Advances in virus-directed therapeutics against Epstein-Barr virus-associated malignancies. Advances in Virology. 2012;2012:509296
90.Lima RT, Seca H, Bras S, Nascimento MS, Vasconcelos MH. Treatment of Akata EBV-positive cells with doxorubicin causes more EBV reactivation than treatment with etoposide. Chemotherapy. 2011;57(3):195-203
91.Li JH, Huang D, Sun BF, Zhang X, Middeldorp J, Klamut H, et al. Efficacy of ionizing radiation combined with adenoviral p53 therapy in EBV-positive nasopharyngeal carcinoma. International Journal of Cancer. 2000;87(4):606-610
92.Stevens SJ, Zwaan CM, Verkuijlen SA, Middeldorp JM. Epstein–Barr virus (EBV) serology for predicting distant metastases in a white juvenile patient with nasopharyngeal carcinoma and no clinical response to EBV lytic induction therapy. Head & Neck. 2006;28(11):1040-1045
Written By
Xiangning Zhang, Zhe Zhang and Pankaj Trivedi
Submitted: 04 July 2023Reviewed: 11 August 2023Published: 21 November 2023
 Open access peer-reviewed chapter
Open access peer-reviewed chapter