 Open access peer-reviewed chapter
Open access peer-reviewed chapter
Abstract
Glioma is a malignant brain tumor associated with a poor outcome. Attempts at surgical removal of the tumor are the first approach. Additional necessary treatment strategies including cranial irradiation and systemic or local chemotherapy each have serious side effects and provide relatively minimal survival benefits. Antigenic differences between normal and malignant cells of the cancer patient form the rationale for clinical immunotherapeutic strategies. Cytokines such as IL-15 or IL-2 that stimulate an antitumor immune response have been shown to have a particularly high potential for use in immunotherapy against various tumors. In this chapter studies with either a poxvirus genetically engineered to secrete IL-15 or allogeneic fibroblasts engineered to secrete IL-2 are shown to be an effective treatment strategy in prolonging survival in mice with malignant intracerebral tumors upon injection of the treatment cells into the brain. Future studies with these treatment strategies in patients with intracerebral tumors are urgently needed.
Keywords
- immunotherapy
- IL-2
- IL-15
- brain tumors
- prolonged survival
1. Introduction
1.1 Limitation of current brain tumor treatments
Some increase in the ability to diagnose and surgically treat primary brain tumors has been achieved, although the mortality and overall survival of patients with these tumors has not improved over many years [1]. The present standard treatment modalities following surgery to remove the tumors followed by radiation therapy and chemotherapy each have significant side effects. The long-term survivors are few and often left with cognitive deficits and other disabilities [2, 3]. Gliomas are resistant to adjuvant treatments including radiation and cytotoxic chemotherapies making it difficult to treat these tumors [4, 5]. Novel therapies are urgently needed.
1.2 Principles of brain tumor immunology
Antigenic differences between normal and malignant cells of the cancer patient form the rationale for clinical immunotherapeutic strategies. Several different strategies have been attempted to enhance the anti-tumor immune responses in mice and patients with intracerebral neoplasms. Immunization with dendritic cells provided with derivatives of tumor cells or transfected with tumor-RNA can result in the development of immune responses against antigens expressed by the tumor cells [6, 7]. Immunization in patients with dendritic cells transfected with mRNA from malignant glioma has been found to elicit tumor-specific CD8+ cytotoxic T-lymphocyte (CTL) responses against the patient’s tumor [8]. Novel and more specific targets such as glioma stem cells have been shown to improve the success of dendritic cell immunotherapy [9]. Although results of dendritic cell immunotherapy have demonstrated promise in animal models, clinical trials have documented only short benefits in a limited number of patients [10].
Another strategy involves a vaccine prepared by transfer of a cDNA expression library derived from tumor cells into an allogeneic mouse fibroblast cell line expressing a cytokine such as IL-2, which appears to have great potential in the development of an antitumor immune response in the treatment of an intracerebral tumor in mouse models [11]. Upon transfer of the cDNA-expression library from the tumor cells into a highly immunogenic fibroblast cell line, genes specifying an array of tumor antigens are expressed. The transferred DNA integrates into the genome of the recipient cells and replicates as the cells divide. The transfected fibroblasts can be expanded to obtain quantities for repeated immunizations of the patient. This strategy should be capable of inducing immunity to a broad array of tumor antigens that characterize the patient’s tumor. Enough DNA to prepare the vaccine can be obtained from small amounts of tumor tissue (4 mm), enabling treatment at an early stage of the disease.
In many aggressive tumors, such as gliomas, progression is enhanced by local immunosuppression associated with the accumulation of regulatory T-cells (Treg) and myeloid-derived suppressor cells (MDSC) [12]. The lack of response to treatment in glioma patients may be attributed to the immunosuppressive T-cells that normally prevent autoimmunity when the human immune response is evoked [13]. Various cytokines including interleukin-10 and transforming growth factor-β have been implicated in the stimulation of Tregs. The targeting of immune checkpoints that regulate the immune system is emerging as a potent and viable cancer therapy [14]. Immunosuppressive mediators such as IL-10, TGF-β and prostaglandin can inhibit the function of the immune system and promote the growth of tumors [15]. Reversing the immunosuppressive tumor microenvironment is one of the keys to the success of tumor treatment.
There are several immunomodulatory cytokines including IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21 which belong to the family of four α-helix bundle cytokines [16]. The development of IL-2 has been a sentinel force in the development of immunotherapy in cancer [17]. However, the use of IL-2 is limited by toxicity and the expansion of regulatory T-cells. These limitations can be overcome with the use of other T-cell stimulatory agents such as IL-15 which also has been in clinical development. IL-15 has been shown to have a particularly high potential for use in immunotherapy against various tumors [18]. Furthermore, IL-15 unlike IL-2 does not contribute to the maintenance of regulatory T-cells [19].
A variety of tumor vaccination strategies have been attempted including modification of neoplastic cells to stimulate anti-tumor immune responses. Immunization with tumor cells modified to secrete cytokines such as IL-2, IFN-γ and GM-CSF has resulted in the development of MHC-restricted anti-tumor immune responses in animal models [20, 21, 22, 23, 24, 25, 26, 27, 28]. Tumor regression has been documented in experimental animals receiving immunotherapy alone, which suggests that this treatment strategy may be effective.
1.3 Potential applications of oncolytic viruses in brain tumor therapy
An oncolytic virus is a type of virus, either engineered or in nature, which may infect and lyse tumor cells but not affect normal cells [29]. There are many oncolytic viruses which include herpes simplex virus (HSV), adenovirus, reovirus, poliovirus (PV), vaccinia virus (VV), myxoma virus (vMyx), measles virus, vesicular stomatitis virus (VSV) and newcastle disease virus [30]. There are multiple potential mechanisms contributing to the selectivity of oncolytic viruses for tumor cells over normal cells. First, viruses can enter tumor cells by binding with certain receptors which are overexpressed on the tumor cells’ surface. For instance, HSV binds herpes virus entry mediator (HVEM) or nectin-1, VV binds glycosaminoglycans (GAGs) and VSV binds low-density lipoprotein receptor (LDLR) to enter host cells.
Second, some of the hyper-activated signaling pathways in tumor cells over normal cells may facilitate virus infection. Hyper-activation of AKT (serine/threonine kinase) is commonly found in most cancer cells which is a requirement for vMyx infection [31, 32]. EGFR activation, common in cancer cells, contributes to a productive infection by the attenuated vaccinia virus JX-594 [33]. Third, deficiency of tumor cells to Type I interferon responses minimize the anti-viral immune responses allowing oncolytic viruses replication [34, 35]. Fourth, dysfunction of tumor suppressor genes, such as p53, ataxia telangiectasia (ATM) and retinoblastoma protein (Rb) can potentially compromise cellular antiviral activity by accumulating genomic instability and blocking the apoptotic response [36] which contributes to the permissiveness of cancer cells.
Once oncolytic viruses infect tumor cells, they may contribute to the anti-tumor response by a direct cytotoxic effect on tumor cells and consequent release of tumor antigens which could stimulate anti-tumor immune responses [37]. When the virus is engineered to express an immunostimulatory cytokine [38], it becomes a vector for local expression of potent immune-activating agents, attracting immune cells into the tumor microenvironment (TME) while limiting inflammation that systemic delivery of the cytokine might produce.
Many oncolytic viruses have already been tested in several preclinical and clinical trials. T-VEC (also known as Talimogene laherparepvec or OncoVEXGM−CSF) is the first oncolytic virus approved for the treatment of advanced melanoma by the U.S. Food and Drug Administration (FDA) in 2015 [39]. T-VEC is an engineered oncolytic herpes simplex virus type 1 (HSV-1), whose neurovirulence factor ICP34.5 is replaced by the gene of human granulocyte-macrophage colony-stimulating factor (hGM-CSF) and the viral ICP47 gene is deleted [40], to prevent neuronal involvement [41] and enhance anti-tumor efficacy. An OPTiM phase III clinical trial showed the efficacy of T-VEC to target patients with early metastatic melanoma (stage IIIB/C-IVM1a) [42]. It also showed enhanced antitumor activity when T-VEC was combined with pembrolizumab (anti-programmed death-ligand 1 antibody; PD-1 blockade) in a phase II clinical trial [43]. In addition, G47∆, a triple-mutated, third-generation oncolytic HSV-1 [44] was demonstrated with a high safety profile and high anti-tumor efficacy (1-year survival rate 92.3 versus 15%) when targeting human glioblastoma in a phase II clinical trial [45].
Poliovirus is another potential candidate for oncolytic virotherapy. The recombinant nonpathogenic polio–rhinovirus chimera (PVSRIPO) is a neuro-attenuated recombinant poliovirus (Sabin vaccine strains), whose internal ribosomal entry site (IRES) was replaced with human rhinovirus type 2 (HRV2) [46]. The result from a phase I clinical trial, where 61 patients with recurrent World Health Organization (WHO) grade IV malignant glioma were intratumorally infused with PVSRIPO, confirmed the safety of PVSRIPO used in the brain and showed significantly higher survival rate at 24 and 36 months after virus infusion [47]. Studies also showed that PVSRIPO has the potential to show therapeutic effects on breast cancer, prostate cancer [48] and neuroblastoma [49].
Poxvirus, a group of large, enveloped DNA viruses associated with diseases that generate poxes in the skin, can also be a good choice for oncolytic virotherapy since the entire replication of poxvirus happens in viral factories within the cytoplasm of infected cells with no integration of viral DNA into host genome which is safe for host cells [50]. Poxviruses can take multiple large foreign genes into their genomes [51] which supports the feasibility of further arm poxviruses (e.g., adding genes of tumor antigen or immune-enhancing cytokines to poxviruses). vvDD vaccinia virus is a new strain of poxvirus which was attenuated by double deletion of thymidine kinase and vaccinia growth factor. A preclinical study showed great anti-tumor efficacy when mice bearing MC38 colon cancer or ID8 ovarian cancer were treated with IL15 armed vvDD vaccinia virus. In addition, when combined with PD-1 blockade, IL15 armed vvDD vaccinia virus leads to dramatic tumor regression [52]. It has been reported that IL15 armed myxoma virus (another poxvirus) cured 83% of mice bearing orthotopic glioma when combined with adoptive T-cell therapy, rapamycin and celecoxib [53].
Despite the promising results, some concerns still need attention when using oncolytic viruses. One major concern for oncolytic virotherapy is the safety issue of the oncolytic virus. For example, vvDD vaccinia virus which has undergone two phase I clinical trials and has been found safe in humans [54, 55] can still be fatal for hosts if it accidentally enters the cerebral lateral ventricle [56]. Therefore, it is essential to study the safety profile of the oncolytic virus thoroughly before moving to clinical trials. Another concern is the development of the anti-viral immune responses mediated by neutralizing antibodies [57] and immune cells such as macrophages [58] and natural killer [59] (NK) cells which can diminish the ability of the virus to enhance anti-tumor immunity.
2. Pre-clinical experimental findings
2.1 Survival of mice with intracerebral glioma upon treatment with fibroblasts engineered to secrete cytokines
Gl261 cells are a glioma cell line of C57Bl/6 mouse origin (H-2b). LM fibroblasts are derived from C3H/He mice and express H-2k determinants. The potential development of an antitumor immune response was explored using fibroblasts engineered to secrete either IL-2 or IL-2 and interferon- injected into the brain in mice with an intracerebral (i.c.) glioma [60]. Glioma cells were mixed with cells secreting one or two of the cytokines of interest and subsequently were injected i.c. into the right frontal lobe of C57BL/6 mice which are syngeneic with G1261 cells. The results (Figure 1) demonstrate that IL-2-secreting fibroblasts were capable of prolonging survival in mice with a right frontal glioma upon i.c. injection of the treatment cells (P < 0.025). The results were more dramatic upon i.c. injection of mice with glioma treated with LM-IL-2/interferon- double cytokine-secreting cells (P < 0.005). The i.c. injection of mice with equivalent numbers of LM-IL-2 cells without tumors lived many months and did not demonstrate ill effects or neurologic deficit.

Figure 1.
Treatment of intracerebral glioma in C57Bl/6 mice by immunization with allogeneic cytokine-secreting fibroblasts. The C57Bl/6 mice (8 per group) were injected i.c. with a mixture of 106 cells of one of the cell types and 105 Gl261 glioma cells. Gl261 is a malignant glial tumor syngeneic in C57Bl/6 mice. The median lengths of survival were as follows (in days): Mice with nonimmunized glioma cells, 16.9 ± 1.9; glioma plus LM cells, 20.0 ± 4.5; glioma plus LM-IL-2 cells, 23.4 ± 6.8; glioma plus LM-IFN-(cells, 18.0 ± 1.8; glioma plus LM-IL-2/IFN-γ cells, 28.1 ± 5.8. Probability values were: Nonimmunized vs. LM-IL-2, p < 0.025; nonimmunized or LM vs. LM-IL-2/IFN-γ, p < 0.005; LM-IL-2 vs. LM-IL-2/IFN-γ, p < 0.05.
2.2 Immunocytotoxic studies from spleen cells with mice treated with allogeneic cytokine-secreting fibroblasts
A chromium release assay was used to determine the reactivity of spleen cells from the immunized mice to chromium-labeled Gl261 glioma cells. The results [60] demonstrate a significantly elevated chromium release when spleen cells from the mice with i.c. Gl261 cells treated with cytokine-secreting fibroblasts were co-incubated with chromium-labeled Gl261 cells. This data documents the development of a systemic anti-tumor immune response in the mice injected with the cytokine-secreting cells. Antibody depletion studies reveal that the antitumor immunity was mediated both by CD8+ and NK/LAK cells.
2.3 Treatment of glioma in mice treated with IL-15 secreting cells
Two oncolytic poxviruses, vvDD vaccina virus and myxoma virus, were engineered to express the fusion protein IL15Ra-IL15 (53). Viral gene expression was confirmed in the murine glioma by staining for M-T7 (a myxoma-encoded protein) and IL-15 (Figure 2A). Mice with glioma were treated with either of these two viruses supplemented by rapamycin, celecoxib and adoptive T-cell therapy (tumor-specific CD8+ T-cells). Rapamycin was used to enhance the spread and replication of oncolytic viruses [61, 62, 63], while celecoxib should reduce the immunosuppressive tumor microenvironment by inhibiting the production of prostaglandins (mainly PGE2) [15, 64]. Direct injection of vvDD-IL15-Rα into the lateral cerebral ventricles was uniformly fatal, whereas mice that received intracerebral vMyx-IL15Rα-tdTr injection recovered from the virus infection [56]. This suggests that vvDD vaccinia virus may not be a safe choice to treat tumors inside of the brain, whereas myxoma virus could be a potential alternative. Mice that received intracerebral vMyx-IL15Rα-tdTr injection recovered from the virus infection [56].
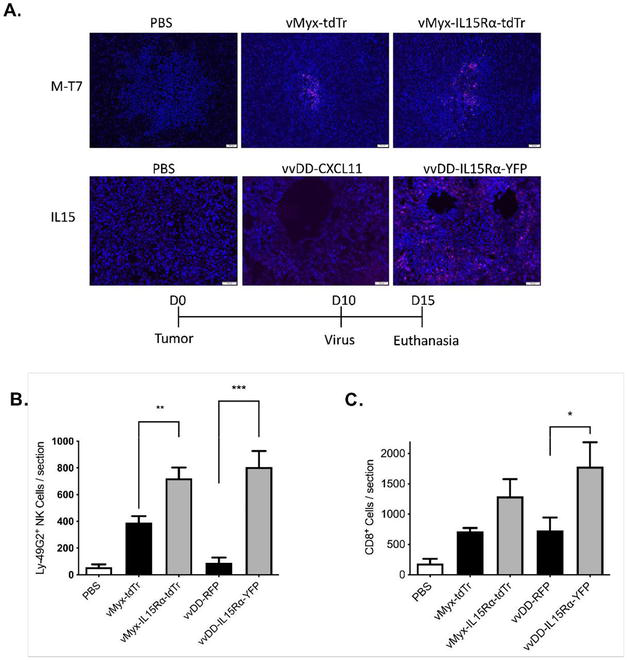
Figure 2.
To explore the anti-tumor efficacy of myxoma virus expressing IL15, the combination treatment (vMyx-IL15Rα-tdTr, rapamycin, celecoxib and adoptive T-cell transfer therapy) was applied to tumor-bearing mice. An increased number of infiltrating NK and CD8+ T-cells was detected in the tumor specimens (Figure 2B,C) indicating that the IL15Rα-IL15 fusion protein is biologically functional and could attract NK and CD8+ T-cells into the tumor site. The prolongation of survival (P < 0.01 compared to untreated animals or animals that did not receive Celecoxib) (Figure 3) indicated that myxoma virus (vMyx-IL15Rα-tdTr) supplemented by rapamycin and the prostaglandin inhibitor celecoxib could provide a safe and effective anti-tumor treatment in mice with intracerebral glioma upon injection of the treatment cells into the brain or lateral ventricles. The efficacy of this novel combination treatment strategy in glioma-bearing mice following tumor resection is being explored [65].

Figure 3.
The therapeutic effect of the full combination treatment using vMyx-IL15Rα-tdTr. Survival of tumor-bearing mice treated with intratumoral vMyx-IL15Rα-tdTr virus injection, rapamycin and celecoxib, adoptive transfer of CD8+ T-cells and TriVax booster. Mice received tumor on day 0, 1 μL vMyx-IL15Rα-tdTr virus (2 × 106 pfu) or PBS injection, rapamycin with or without celecoxib on day 7 (medication treatment continued until day 73), adoptive transfer of CD8+ T-cells on day 9, TriVax booster on day 10. Mice that received the full combination treatment survived longer compared to other groups. p < 0.05 for each comparison.
2.4 Immunization with allogeneic cytokine-secreting fibroblasts transfected with DNA from breast cancer in treatment of C3H mice with intracerebral breast cancer
A tumor vaccine was constructed by transfer of DNA from a breast cancer cell line (SB-5b) that arose in C3H mice (H-2K) into cytokine-secreting mouse fibroblasts (H-2K). The application of these cells as a potential treatment of an intracerebral breast neoplasm arising in C3H mice was investigated. The cells were modified to express H-2Kb determinants, and this should also ensure rejection. Studies with these cells revealed that there was a prolongation of survival (Figure 4) in C3H mice with intracerebral breast cancer upon treatment by immunization with fibroblasts secreting either IL-2 or GM-CSF transfected with DNA from the same spontaneous breast neoplasm (P < 0.05) [66].

Figure 4.
Treatment of C3H/He mice with intracerebral SB-5b breast carcinoma with mice immunized with cytokine-secreting allogeneic fibroblasts transfected with DNA from a spontaneous breast neoplasm (SB-5b). C3H/He mice (nine animals/group) were injected with a mixture of 1.0 × 104 SB-5b cells and 1.0 × 106 cytokine-secreting fibroblasts transfected with tumor DNA or with an equivalent number of non-secreting cells transfected with tumor DNA (LMKb/SB5b). Mean survival time (MST) in days: Media control, 23.0 ± 1.9; LMKb/SB5b, 27.3 ± 6.3; LMKbGMCSF/SB5b, 30.0 ± 9.5; LMKbIL-2/SB5b, 36.6 ± 7.0; LMKbIL-18/SB5b, 28.4 ± 4.8. Probability values were as follows: LMKbIL-2/SB5b vs. LMKb/SB5b or media control, P < 0.005; LMKbIL-2/SB5b vs. LMKbIL-18/SB5b, P < 0.025; LMKbIL-2/SB5b vs. LMKbGMCSF/SB5b, P < 0.05; LMKbGMCSF/SB5b vs. media control, P < 0.05.
2.5 The proportion of splenic T-cells reactive with SB-5b tumor cells in mice immunized with transfected cytokine-secreting fibroblasts
An ELISPOT-IFN-γ assay was used to estimate the development of splenic T-cells reactive with SB-5b cells in mice immunized with transfected fibroblasts modified to secrete IL-2 or GM-CSF. The assay was performed 6 weeks after the i.c. injection of the mixture of SB-5b cells and the transfected fibroblasts. The findings in these studies revealed that the highest proportion of T-cells reactive with SB-5b cells was in surviving mice injected with fibroblasts modified to secrete IL-2 (Figure 5A). Lesser numbers of spots were found in T-cells from mice injected with SB-5b cells and non-secreting transfected fibroblasts or SB-5b cells and transfected fibroblasts modified to secrete GM-CSF.

Figure 5.
The proportion of T-cells responsive to tumor cells in mice bearing an i.c. tumor immunized i.c. with DNA-transfected cells modified to secrete IL-2, GM-CSF or IL-18. ELISPOT assay detecting INF- secretion by spleen cells reactive with SB-5b cells in mice immunized with transfected fibroblasts modified to secrete IL-2 or GM-CSF. These animals survived for six weeks following the initial intracerebral injection of SB-5b tumor. The results indicated that the highest proportion of T-cells from the spleens reactive with SB-5b cells were in surviving mice injected with fibroblasts modified to secrete IL-2. Lesser numbers of spots were found in T-cells from mice injected with SB-5b cells and non-secreting transfected fibroblasts or SB-5b cells and transfected fibroblasts modified to secrete GM-CSF. The effect of mAbs against T-cell subsets or NK/LAK cells on the anti-tumor cytotoxic activities of spleen cells from C3H/He mice injected i.c. with a mixture of SB-5b tumor cells and the cytokine-secreting cells transfected with tumor DNA. The animals were injected with a mixture of 1.0 × 104 SB-5b tumor cells and 1.0 × 106 cytokine-secreting fibroblasts transfected with tumor DNA. Two weeks after the injection, mononuclear cells from the spleens of the immunized mice were used for the ELISPOT assay detecting INF- producing cells. The overall P-value between unstimulated vs. tumor cell stimulated group is P < 0.001. The error bars represent one standard deviation.
In additional experiments, animals with i.c. breast cancer were treated i.c. with LMKbIL-2/SB5b cells. An ELISPOT assay was done after 2 weeks using the spleen cells to detect IFN-γ secretion in the presence or absence of SB-5b tumor cells and antibodies against various T-cell subsets. These studies revealed that CD4+, CD8+ and NK/LAK cells were responsible for the antitumor immune response (Figure 5B). The overall P-value between the unstimulated vs. the tumor cell stimulated group is P < 0.001.
2.6 Development of an enrichment strategy for a more potent vaccine
A strategy was developed to enrich the vaccine for efficacy by identifying cell populations that were the most highly immunogenic [11]. Populations with higher numbers of immunogenic cytokine-secreting cells transfected with tumor DNA were identified by their stronger antitumor immune response against SB5b cells in C3H/He mice. Two sub-pools that stimulated immunity to the greatest (immunohigh pool) and least (immunolow pool) after three rounds of enrichment were identified and used in further studies.
2.7 Importance of various treatment cells in the development of the antitumor immune response
To analyze the development of systemic anti-tumor immunity in tumor-free mice injected i.c. with cells from the various treatment groups, ELISPOT IFN-γ assays for measurement of responding T-cells were done using cells from the cervical lymph nodes and spleens from the injected mice. A micro cannula was placed into the right frontal lobe of naive C3H/He mice. Following the cannula insertion, the animals were injected into the brain through the cannula with 1.0 × 106 cells from the immunohigh pool on 2 days separated by 1 week. As controls, the same procedure was followed except that the cells from the non-enriched master pool or cells from the immunolow pool were substituted for cells from the immunohigh pool. As additional controls, the tumor-bearing mice were injected into the brain with equivalent numbers of non-DNA-transfected LMKb cells or the mice were injected with SB5b tumor cells alone. Mice injected with SB5b tumor cells received only one injection. The data revealed that the highest number of responding T-cells were in the cervical lymph nodes (6A) or spleens (6B) of mice injected i.c. with cells from the immunohigh pool (P < 0.05) (Figure 6).

Figure 6.
Increased numbers of responding T-cells were detected in the spleens and cervical lymph nodes of naïve mice which were injected i.c. with cells from the various treatment groups. To determine if systemic anti-tumor immunity was generated in tumor-free mice injected i.c. with cells from the immunohigh pool, cervical lymph node and spleen cells from the injected mice were analyzed by ELISPOT IFN-γ assays for responding T-cells. Naïve C3H/He mice received 2 i.c. injections at weekly intervals of 1.0 × 106 cells from the immunohigh pool. One week after the second injection, mononuclear cells from the spleens and cervical lymph nodes of the immunized mice were analyzed for the presence of T-cells responsive to the breast cancer cells. As controls, an equivalent number of cells from the non-selected master pool or cells from the immunolow pool were substituted for cells from the immunohigh pool. As additional controls, the same protocol was followed except that the mice were injected i.c. with equivalent numbers of SB5b cells, with LMKb cells or with media. Mice injected with SB5b tumor cells received only one injection. The results illustrated in this figure indicate that the highest number of responding cervical lymph nodes (A) or spleen cells (B) were in mice injected i.c. with cells from the immunohigh pool (p < 0.05 versus the number of responding spleen cells in mice injected with cells from the master pool and
2.8 Stimulation of T-cell subsets in the spleens of mice with i.c. breast cancer following treatment of the tumor with cells from the immunohigh pool
The number of responding T-cells in the spleens of mice with i.c. breast cancer treated with Cytokine-secreting cells from the various treatment groups was determined using ELISPOT IFN-γ assays. A micro cannula was placed into the right frontal lobe of C3H/He mice, and SB5b cells (1.0 × 105 in 30 μl) were introduced into the brain through the cannula. On days 2 and 9 following the introduction of tumor, the animals were injected through the cannula into the tumor bed with cells from the immunohigh pool. The results (Figure 7) reveal that the strongest anti-tumor immune response developed in the spleens of mice with i.c. breast cancer treated i.c. with cells from the immunohigh pool (p < 0.05) versus the number of responding spleen cells treated with the master pool or any of the other groups.

Figure 7.
The effect of antibodies against various T-cell subsets on the cytotoxic response in mice with i.c. breast cancer treated with cells of the various treatment groups. A cannula was placed into the right frontal lobe of C3H/He mice. One day later, the animals received an i.c. injection through the cannula of 1.0 × 104 SB5b breast carcinoma cells. On days 2 and 9 following the introduction of tumor, the animals were injected through the cannula into the tumor bed with 1.0 × 106 cells from the immunohigh pool of transfected cells. As controls, the same procedure was followed except for equivalent numbers of cells from the immunolow pool, the non-selected master pool or non-transfected LMKb cells were substituted for cells from the immunohigh pool. Two weeks after the injection of tumor cells, mononuclear cells from the spleens of the mice were co-incubated with (stimulated) or without (un-stimulated) SB5b cells. The ratio of spleen cells: SB5b cells = 10:1, and the number of INF- spots/106 spleen cells is measured. Reduced numbers of spots were detected in spleen cells from mice with i.c. breast cancer injected with cells from the immunohigh pool (
The effect of antibodies against various T-cell subsets on the responding T-cell response was used to determine the types of cells activated for antitumor immunity in the spleens of mice treated with cells from the immunohigh pool using the protocol described above. ELISPOT IFN-γ assays were used for this analysis. The antitumor immune response was inhibited to the greatest extent by antibodies against CD4+ cells (Figure 7). The results were less dramatic if the spleen cells were incubated in the media containing CD8+ or NK/LAK antibodies.
2.9 Decreased number of T-reg cells in the spleens of mice with i.c. breast cancer treated with cells from the immunohigh pool
Potent inhibition of antitumor immunity is regulated by T-reg cells [67]. The success of immunotherapeutic protocols depends in part upon the relative numbers of T-reg cells and cytotoxic T lymphocytes in tumor-bearing animals and patients. Quantitative RT-PCR for Foxp3, a transcription factor characteristic of T-reg cells, was used to estimate the relative proportions of T-reg cells in the spleens and brains of mice with i.c. breast cancer injected into the tumor bed with cells from the immunohigh pool of transfected cells. Naïve C3H/He mice were injected i.c. with 5.0 × 104 SB5b cells along with 1.0 × 106 cells from the immunohigh pool of transfected cells. The animals received a second i.c. injection of cells from the immunohigh pool through the same burr hole. As controls, the same procedure was followed except that the mice were injected with equivalent numbers of SB5b cells and cells from the non-enriched master pool or the immunolow pool. The results (Figure 8A,B) indicate that Foxp3+ T-reg cells were relatively deficient in the spleens but not in the brains of animals injected with cells from the immunohigh pool. An analysis by FACS of spleen cells from mice injected i.c. with cells from the immunohigh pool also revealed a relative deficiency of CD4+/CD25+/Foxp3+ T-cells and a corresponding increase in the relative numbers of CD8+ cells in the spleen (Figure 8C) from these animals.

Figure 8.
T-reg cells in the spleens of mice injected with i.c. breast cancer and allogeneic fibroblasts from the various treatment groups. RT-PCR for Foxp3 in the brains and spleens of mice injected i.c. with a mixture of breast cancer cells and cells from the immunohigh pool of transfected cells. C3H/He mice received an injection of 5.0 × 104 SB5b cells and 1.0 × 106 cells from the immunohigh pool of transfected cells through a small burr hole. One week later the animals received a second injection of cells from the immunohigh pool through the same small burr hole. As controls, the same protocol was followed except that the mice were injected with equivalent numbers of SB5b cells along with cells from the immunolow pool, the non-selected master pool, non-transfected LMKb cells or the mice were injected with SB5b cells alone. The animals that received SB5b tumor cells were given only one injection. One week after the last injection, mononuclear cells from the tumor bed and the spleen were analyzed by RT-PCR for the expression of Foxp3. Expression of Foxp3 by cells from the tumor bed. The intensity of the amplified 400 bp Foxp3 band, relative to that of the internal control, GAPDH. An analysis by FACS of the spleens of the injected animals shown below revealed a relative deficiency of CD4+/CD25+/Foxp3+ T-reg T-cells and a corresponding increase in the relative numbers of CD8+ cells in the spleens of mice injected i.c. with cells from the immunohigh pool. Two weeks following the first injection of tumor and treatment cells, mononuclear cells from the spleens of three immunized mice were harvested and pooled for analysis by FACS. Each of the following steps was performed on ice. Tissues were minced and finely homogenized using a razor blade in 5 ml of serum-free DMEM. The homogenate was spun down and re-suspended in fresh, ice-cold serum-free DMEM. Single-cell suspension was obtained by filtration through 70-μm nylon mesh into sterile 50-ml conical tubes. The cells were centrifuged to form a pellet and the supernatant was poured off. The supernatant was subsequently removed and the cell pellet was re-suspended in ACK lysis buffer for 5 minutes, and spun down. The pellet was washed in 5 ml of PSS and spun down. All pellets were re-suspended in 5 ml of FACS buffer (5% bovine serum albumin and 5% heat-inactivated horse serum), incubated (4°C) for 5 min, spun down to a pellet and resuspended in 5 ml of PBS. Data is expressed as a percentage of gated cells from the entire population of spleen cells.
3. Prolonged survival of mice with i.c. breast cancer upon treatment by injection into the tumor bed with cells from the immunohigh pool
To explore the potential prolongation of survival in mice with i.c. breast cancer treated by cells from the immunohigh pool, C3H/He mice were injected i.c. with 5.0 × 104 SB5b cells and 1.0 × 106 cells from the immunohigh pool. The findings from these studies (Figure 9) revealed that mice with i.c. breast cancer treated with cells from the immunohigh pool survived significantly longer than untreated mice (

Figure 9.
Survival of mice with i.c. breast cancer treated by immunization with cells from the immunohigh pool of transfected cells. C3H/He mice (eight animals/group) were injected with 5.0 × 104 SB5b cells and 1.0 × 106 cells from the immunohigh pool through a small burr hole. At the same time animals with i.c. tumor were injected s.c. (subcutaneously) with an equivalent number of cells from the immunohigh pool alone or both i.c. and s.c. with cells from the immunohigh pool. Mean survival time (MST) in days: Injected with SB5b alone, 12.7 ± 1.0; injected with SB5b and cells from immunohigh pool s.c., 15.6 ± 3.9; injected with SB5b cells and cells from immunohigh pool i.c., 15.4 ± 3.3; injected with SB5b cells and cells from the immunohigh pool i.c. and s.c., 17.4 ± 5.9. Probability values were as follows:
4. Conclusions
Patients with primary or metastatic tumors in the brain continue to experience a limited survival which has not improved over many years. There is an urgent need for new and more effective forms of treatment for these patients. Immunotherapy, which is designed to stimulate an antitumor immune response to various tumors is beginning to show potential for several different types of cancer. Cytokine expression in tumors is a strategy to stimulate potent activation of the immune system. The use of cytokines has become more common in the treatment of patients with high-grade gliomas, particularly with IL-2 and IL-15 [68, 69]. A potential advantage of IL-15 is that there is less activation of immune inhibitory Tregs associated with this cytokine [19]. Preliminary results, however, have not demonstrated a robust efficacy for prolonging survival in patients with brain tumors using various cytokines including IL-2 or IL-15 [70].
In the studies presented here, a significant prolongation of survival was observed with two different oncolytic poxviruses expressing the IL15Rα-IL15 fusion protein in combination with a prostaglandin synthesis inhibitor to block immunosuppression. Oncolytic viruses may contribute to the anti-tumor response by a direct cytotoxic effect on cancer cells associated with the release of tumor antigens that may stimulate an immune response. Previous studies have shown that the oncolytic poxvirus, myxoma virus, is safe to use in mice even when directly injected into the cerebral ventricles.
When the virus is engineered to express a cytokine such as IL-15, it can stimulate local expression of potent immune-activating agents, while avoiding systemic inflammation that parenteral delivery of the cytokine would produce. In this study, IL15 was chosen because it activates and maintains the function of NK and CD8+ T-cells [71] with less vascular leakage potential [72] and less activation of Tregs.
In addition, there is less activation-induced cell death for CD8+ effector T-cells with this cytokine. In summary, the data suggests that a poxvirus genetically engineered to secrete IL-15 along with an anti-immuno-suppressant is an effective treatment strategy in prolonging survival in mice with a malignant glioma.
In other studies, it was shown that the enhanced immunotherapeutic properties of a vaccine prepared by transfer of a cDNA expression library derived from tumor cells into a mouse fibroblast cell line engineered to secrete IL-2 also appears to have significant potential in the treatment of a brain tumor. The cDNA integrates spontaneously into the genome of the recipient cells followed by replication and expression. The vaccine can be prepared from small amounts of tumor tissue, enabling treatment at an early stage of the disease, when tumor tissue is available in only limited amounts and the tumor is most susceptible to immune-based therapy. A unique enrichment strategy has also been developed such that the proportion of active immunotherapeutic cells in the vaccine is increased.
The use of cells from the enriched vaccine was associated with the development of a strong antitumor immune response. Cells from the (immunohigh) pool were injected into the tumor bed of mice with intracerebral breast cancer, and this resulted in a prolongation of survival in the treated mice. ELISPOT IFN-γ assays revealed significantly elevated anti-tumor immune responses in spleen cell populations derived from tumor-bearing mice injected i.c. with cells from the immunohigh pools. The injection of cells from various control groups including cells from the immunolow pools did not reveal significant anti-tumor immune responses. The predominant cell type activated in mice immunized with cells from the the immunohigh pool were CD4+ T-cells. Injection of cells from the immunohigh pool into the tumor bed also resulted in a reduction of the relative numbers of T-reg cells with a potential prevention of the impaired anti-tumor immune responses frequently found in patients with malignant brain tumors.
The goal of tumor treatment would be the removal of every tumor cell. It is unlikely that a single therapy can achieve this goal in the case of brain tumors. However, immunotherapy in combination with surgical removal of the tumor, radiation therapy and chemotherapy will likely be involved in the treatment of patients with brain tumors. The development of DNA-based tumor vaccines in combination with cytokine secretion is a novel strategy that does not require antigen identification or protein purification and yet can elicit a potent and long-lasting activation of the immune response, which will lead to the rejection of the tumor. From a practical point of view, these vaccines are easy to prepare, and they are relatively inexpensive. Only small quantities of tumor-derived DNA are needed, which can be obtained from small surgical specimens. The enrichment strategy represents a unique approach to isolating highly immunogenic pools of transfected cells which leads to the development of enhanced anti-tumor immunity. Thus DNA-based cytokine-secreting vaccines offer several unique advantages, which support their further development for cancer immunotherapy in general and specifically for the treatment of patients with malignant intracerebral tumors. The use of viruses engineered to secrete cytokines remains another treatment option for further study regarding the management of these tumors.
References
- 1.
Ries LAG, Kosary CL, Hankey BF, Miller BA, Edwards BK. SEER Cancer Statistics Review, 1973-1995. Bethesda, MD: National Cancer Institute; 1988 - 2.
Imperato JP, Paleologos NA, Vick NA. Effects of treatment on long-term survivors with malignant atrocytomas. Annals of Neurology. 1990; 28 :818-822 - 3.
Heimans JJ, Taphoorn MJ. Impact of brain tumor treatment on quality of life. Journal of Neurology. 2002; 249 :955-960 - 4.
Belanich M, Randall T, Pastor MA, Kibitel JT, Alas LG, Dolan ME, et al. Intracellular localization and intercellular heterogeneity of the human DNA repair protein O(6)-methylguanine-DNA methyltransferase. Cancer Chemotherapy and Pharmacology. 1996; 37 :547-555 - 5.
Hotta T, Saito Y, Fujita H, Mikami T, Kurisu K, Kiya K, et al. O6-alkylguanine-DNA alkyltransferase activity of human malignant glioma and its clinical implications. Journal of Neuro-Oncology. 1994; 21 :135-140 - 6.
Insug O, Ku G, Ertl HCJ, Blaszczyk-Thurin M. A dendritic cell vaccine induces protective immunity to intracranial growth of glioma. Anticancer Research. 2002; 22 :613-622 - 7.
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: Moving beyond current vaccines. Nature Medicine. 2004; 10 :909-915 - 8.
Kobayashi T, Yamanaka R, Homma J, Tsuchiya N, Yajima N, Yoshida S, et al. Tumor mRNA-loaded dendritic cells elicit tumor-specific CD8+ cytotoxic T cells in patients with malignant glioma. Cancer Immunology, Immunotherapy. 2003; 52 :632-637 - 9.
Finocchiaro G, Pellegatta S. Immunotherapy with dendritic cells loaded with glioblastoma stem cells: From preclinical to clinical studies. Cancer Immunology and Immunotherapy. 2016; 65 :101-109 - 10.
Reardon DA, Gilbert MR, Wick W, Liau L. Immunotherapy for neuro-oncology: The critical rationale for combinatorial therapy. Neuro Oncology. 2015; 17 (Suppl. 7):vii32-vii40 - 11.
Lichtor T, Glick RP, Feldman LA, Osawa G, Hardman J, O-Sullivan I, Cohen EP. Enhanced immunity to intracerebral breast cancer in mice immunized with a cDNA-based vaccine enriched for immunotherapeutic cells. Journal of Immunotherapy. 2008; 31 :18-27 - 12.
Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Research. 2016; 76 :5671-5682 - 13.
Ooi YC, Tran P, Ung N, Thill K, Trang A, Fong BM, et al. The role of regulatory T-cells in glioma immunology. Clinical Neurology and Neurosurgery. 2014; 119 :125-132 - 14.
Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-Li and TGF-β. Science Translational Medicine. 2018; 10 (424):eaan5488. DOI: 10-1126/scitranlmed.aan5488 - 15.
Mostofa AGM, Punganuru SR, Madala HR, Al-Obaide M, Srivenugopal KS. The process and regulatory components of inflammation in brain oncogenesis. Biomolecules. 2017; 7 (2):34. DOI: 10.3390/biom7020034 - 16.
Steel JC, Waldmann A, Morris JC. Interleukin-15 biology and its therapeutic implications in cancer. Trends in Permacology Sciences. 2012; 33 :35-41 - 17.
Wrangle JM, Patterson A, Johnson CB, Neitzke DJ, Mehrotra S, Denlinger CE, et al. IL-2 and beyond in cancer immunotherapy. Journal of Interferon & Cytokine Research. 2018; 38 :45-68 - 18.
Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunological Reviews. 2008; 222 :357-368 - 19.
Berger C, Berger M, Hackman RC, Gough M, Elliott C, Jensen MC, et al. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood. 2009; 114 :2417-2426 - 20.
Gansbacher B, Bannerji R, Daniels B, Zier K, Cronin K, Gilboa E. Retroviral vector-mediated gamma-interferon gene transfer into tumor cells generates potent and long-lasting antitumor immunity. Cancer Research. 1990; 50 :7820-7825 - 21.
Colombo MP, Ferrari G, Stoppacciaro A, Parenza M, Rodolfo M, Mavilio F, et al. Granulocyte colony-stimulating factor gene transfer suppressed tumorigenicity of a murine adenocarcinoma in vivo . The Journal of Experimental Medicine. 1991;173 :889-897 - 22.
Golumbek PT, Lazenby AJ, Levitsky HI, Jaffee LM, Karasuyama H, Baker M, et al. Treatment of established renal cancer by tumor cells engineered to secrete interleukin-4. Science. 1991; 254 :713-716 - 23.
Mullen CA, Coale MM, Levy AT, Stetler-Stevenson WG, Liotta LA, Brandt S, et al. Fibrosarcoma cells transduced with the IL-6 gene exhibit reduced tumorigenicity, increased immunogenicity, and decreased metastatic potential. Cancer Research. 1992; 52 :6020-6024 - 24.
Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific and long-lasting anti-tumor immunity. Proceedings of the National Academy of Sciences of the United States of America. 1993; 90 :3539-3543 - 25.
Connor J, Bannerji R, Saito S, Heston W, Fair W, Gilboa E. Regression of bladder tumors in mice treated with interleukin 2 gene-modified tumor cells. The Journal of Experimental Medicine. 1993; 177 :1127-1111 - 26.
Cavallo F, Pierro FD, Giovarelli M, Gulino A, Vacca A, Stoppacciaro A, et al. Protective and curative potential of vaccination with interleukin-2-gene-transfected cells from a spontaneous mouse mammary adenocarcinoma. Cancer Research. 1993; 53 :5067-5070 - 27.
Tahara H, Zeh HJ, Storkus WJ, Pappo I, Watkins SC, Gubler U, et al. Fibroblasts genetically engineered to secrete interleukin 12 can suppress tumor growth and induce antitumor immunity to a murine melanoma in vivo . Cancer Research. 1994;54 :182-189 - 28.
Yu JS, Wei MX, Chiocca EA, Martuza RL, Tepper RI. Treatment of glioma by engineered interleukin 4-secreting cells. Cancer Research. 1993; 53 :3125-3128 - 29.
Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nature Reviews. Immunology. 2018; 18 :498-513 - 30.
Jayawardena N, Poirier JT, Burga LN, Bostina M. Virus-receptor interactions and virus neutralization: Insights for oncolytic virus development. Oncolytic Virotherapy. 2020; 9 :1-15 - 31.
Wang G, Barrett JW, Stanford M, Werden SJ, Johnston JB, Gao X, et al. Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proceedings of the National Academy of Sciences. 2006; 103 :4640-4645 - 32.
Werden SJ, McFadden G. The role of cell signaling in poxvirus tropism: The case of the M-T5 host range protein of myxoma virus. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 2008; 1784 :228-237 - 33.
Parato KA, Breitbach CJ, Le Boeuf F, Wang J, Storbeck C, Ilkow C, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Molecular Therapy. 2012; 20 :749-758 - 34.
Bartee E, Mohamed MR, Lopez MC, Baker HV, McFadden G. The addition of tumor necrosis factor plus Beta interferon induces a novel synergistic antiviral state against poxviruses in primary human fibroblasts. Journal of Virology. 2009; 83 :498-511 - 35.
Bartee E, McFadden G. Human cancer cells have specifically lost the ability to induce the synergistic state caused by tumor necrosis factor plus interferon-beta. Cytokine. 2009; 47 :199-205 - 36.
Kim M, Williamson CT, Prudhomme J, Bebb DG, Riabowol K, Lee PWK, et al. The viral tropism of two distinct oncolytic viruses, reovirus and myxoma virus, is modulated by cellular tumor suppressor gene status. Oncogene. 2010; 29 :3990-3996 - 37.
Rahman MM, McFadden G. Oncolytic virotherapy with myxoma virus. Journal of Clinical Medicine. 2020; 9 :171. DOI: 10.3390/jcm9010171 - 38.
Guo ZS, Lu B, Guo Z, Giehl E, Feist M, Dai E, et al. Vaccinia virus-mediated cancer immunotherapy: Cancer vaccines and oncolytics. Journal for Immunotherapy of Cancer. 2019; 7 (1):6. DOI: 10.1186/s40425-018-0495-7 - 39.
Johnson DB, Puzanov I, Kelley MC. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy. 2015; 7 :611-619 - 40.
Bommareddy PK, Patel A, Hossain S, Kaufman HL. Talimogene laherparepvec (T-VEC) and other oncolytic viruses for the treatment of melanoma. American Journal of Clinical Dermatology. 2017; 18 :1-15 - 41.
Liu BL, Robinson M, Han ZQ , Branston RH, English C, Reay P, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Therapy. 2003; 10 :292-303 - 42.
Franke V, Berger DMS, Klop WMC, van der Hiel B, van de Wiel BA, Ter Meulen S, et al. High response rates for T-VEC in early metastatic melanoma (stage IIIB/C-IVM1a). International Journal of Cancer. 2019; 145 :974-978 - 43.
Kelly CM, Antonescu CR, Bowler T, Munhoz R, Chi P, Dickson MA, et al. Objective response rate among patients with locally advanced or metastatic sarcoma treated with talimogene laherparepvec in combination with pembrolizumab: A phase 2 clinical trial. JAMA Oncology. 2020; 6 :402-408 - 44.
Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proceedings of the National Academy of Sciences. 2001; 98 :6396-6401 - 45.
Todo T, ATIM-14. Results of phase II clinical trial of oncolytic herpes virus G47Δ in patients with glioblastoma. Neuro-Oncology . 2019;21 (Issue Supplement):vi4 - 46.
Gromeier M, Alexander L, Wimmer E. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proceedings of the National Academy of Sciences. 1996; 93 :2370-2375 - 47.
Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. The New England Journal of Medicine. 2018; 379 :150-161 - 48.
Holl EK, Brown MC, Boczkowski D, McNamara MA, George DJ, Bigner DD, et al. Recombinant oncolytic poliovirus, PVSRIPO, has potent cytotoxic and innate inflammatory effects, mediating therapy in human breast and prostate cancer xenograft models. Oncotarget. 2016; 7 :79828-79841 - 49.
Toyoda H, Yin J, Mueller S, Wimmer E, Cello J. Oncolytic treatment and cure of neuroblastoma by a novel attenuated poliovirus in a novel poliovirus-susceptible animal model. Cancer Research. 2007; 67 :2857-2864 - 50.
Fields BN, Knipe DM, Howley PM. Fields Virology. Philadelphia, PA, USA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2013 - 51.
Smith GL, Moss B. Infectious poxvirus vectors have capacity for at least 25 000 base pairs of foreign DNA. Gene. 1983; 25 :21-28 - 52.
Kowalsky SJ, Liu Z, Feist M, Berkey SE, Ma C, Ravindranathan R, et al. Superagonist IL-15-armed oncolytic virus elicits potent antitumor immunity and therapy that are enhanced by PD-1 blockade. Molecular Therapy. 2018; 26 :2476-2486 - 53.
Tang B, Guo ZS, Bartlett DL, Yan DZ, Schane CP, Thomas DL, et al. Synergistic combination of oncolytic virotherapy and immunotherapy for glioma. Clinical Cancer Research. 2020; 26 :2216-2230 - 54.
Zeh HJ, Downs-Canner S, McCart JA, Guo ZS, Rao UN, Ramalingam L, et al. First-in-man study of Western reserve strain oncolytic vaccinia virus: Safety, systemic spread, and antitumor activity. Molecular Therapy. 2015; 23 :202-214 - 55.
Downs-Canner S, Guo ZS, Ravindranathan R, Breitbach CJ, O'Malley ME, Jones HL, et al. Phase 1 study of intravenous oncolytic poxvirus (vvDD) in patients with advanced solid cancers. Molecular Therapy. 2016; 24 :1492-1501 - 56.
Tang B, Guo ZS, Bartlett DL, Liu J, McFadden G, Shisler JL, et al. A cautionary note on the selectivity of oncolytic poxviruses. Oncolytic Virotherapy. 2019; 8 :3-8 - 57.
Niemann J, Woller N, Brooks J, Fleischmann-Mundt B, Martin NT, Kloos A, et al. Molecular retargeting of antibodies converts immune defense against oncolytic viruses into cancer immunotherapy. Nature Communications. 2019; 10 :3236 - 58.
Fulci G, Dmitrieva N, Gianni D, Fontana EJ, Pan X, Lu Y, et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Research. 2007; 67 :9398-9406 - 59.
Alvarez-Breckenridge CA, Yu J, Price R, Wojton J, Pradarelli J, Mao H, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nature Medicine. 2012; 18 :1827-1834 - 60.
Lichtor T, Glick RP, Kim TS, Hand R, Cohen EP. Prolonged survival of mice with glioma injected intracerebrally with double cytokine-secreting cells. Journal of Neurosurgery. 1995; 83 :1038-1044 - 61.
Lun XQ , Jang J-H, Tang N, Deng H, Head R, Bell JC, et al. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clinical Cancer Research. 2009; 15 :2777-2788 - 62.
Stanford MM, Barrett JW, Nazarian SH, Werden S, McFadden G. Oncolytic virotherapy synergism with signaling inhibitors: Rapamycin increases myxoma virus tropism for human tumor cells. Journal of Virology. 2007; 81 :1251-1260 - 63.
Thomas DL, Doty R, Tosic V, Liu J, Kranz DM, McFadden G, et al. Myxoma virus combined with rapamycin treatment enhances adoptive T cell therapy for murine melanoma brain tumors. Cancer Immunology, Immunotherapy. 2011; 60 :1461-1472 - 64.
Seibert K, Masferrer JL. Role of inducible cyclooxygenase (COX-2) in inflammation. Receptor. 1994; 4 :17-23 - 65.
Tang B, Foss K, Lichtor T, Phillips H, Roy E. Resection of orthotopic murine brain glioma. Neuroimmunology and Neuroinflammation. 2021; 8 :64-69. DOI: 10.20517/2347-8659.2020.28 - 66.
Lichtor T, Glick RP, Lin H, Sullivan OI, Cohen EP. Intratumoral injection of IL-secreting syngeneic/allogeneic fibroblasts transfected with DNA from breast cancer cells prolongs the survival of mice with intracerebral breast cancer. Cancer Gene Therapy. 2005; 12 :708-714 - 67.
Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased regulatory T-cell function amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Research. 2006; 66 :3294-3202 - 68.
Zhenjiang L, Rao M, Luo X, Valentini D, Von Landenberg A, Meng Q , et al. Cytokine networks and surviving peptide-specific cellular immune responses predict improved survival in patients with gliobastoma multiforme. eBioMedicine. 2018; 33 :49-56 - 69.
Zhan Q , Zhang HJ, Wang Om Zhu G, Jin G, Fusheng L. Glioma-associated mesenchymal stem cells-mediated PD-L1 expression is attenuated by AD5-Ki67-15 in GBM treatment. Stem Cell Research & Therapy. 2022; 13 :284 - 70.
Medikonda R, Pant A, Lim M. Immunotherapy as a new therapeutic approach for brain and spinal cord tumors. Advances in Experimental Medicine and Biology. 2023; 1394 :73-84 - 71.
Jakobisiak M, Golab J, Lasek W. Interleukin 15 as a promising candidate for tumor immunotherapy. Cytokine & Growth Factor Reviews. 2011; 22 :99-108 - 72.
Munger W, DeJoy SQ , Jeyaseelan R, Torley LW, Grabstein KH, Eisenmann J, et al. Studies evaluating the antitumor activity and toxicity of interleukin-15, a new T cell growth factor: Comparison with Interleukin-2. Cellular Immunology. 1995; 165 :289-293