Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
The major histocompatibility complex in humans, known as the human leukocyte antigen (HLA) and located on chromosome 6, is the most polymorphic genetic system in humans. The biological role of the HLA class I and class II molecules is to present processed peptides to CD8+ and CD4+ T lymphocytes, respectively. These cells can also respond to foreign (allogeneic) HLA molecules (direct allo-recognition) or to foreign HLA-derived peptides (indirect allo-recognition), respectively. Thus, the HLA system controls the acceptance or rejection of transplanted foreign tissues and organs (allografts). High-resolution HLA typing is routinely performed to provide HLA matching in hematopoietic stem cell transplantation to prevent allograft rejection and graft-versus-host disease. In contrast, low-resolution HLA typing is routinely performed in solid organ transplantation to provide HLA matching but, most importantly, to allow for the detection of donor-specific antibodies to prevent antibody-mediated allograft rejection. The capability to amplify DNA by polymerase chain reaction has facilitated the clinical application of molecular techniques and, currently, several molecular HLA typing methods are now available in the histocompatibility laboratory. Herein, we describe the different molecular HLA typing techniques and the different levels of HLA typing resolution used for clinical purposes.
Department of Laboratory Medicine and Pathology, Mayo Clinic, Phoenix, Arizona, USA
Katrin Hacke
Department of Laboratory Medicine and Pathology, Mayo Clinic, Phoenix, Arizona, USA
*Address all correspondence to: jaramillo.andres@mayo.edu
1. Introduction
The major histocompatibility complex (MHC) in humans, known as the human leukocyte antigen (HLA) system, controls the acceptance or the rejection of transplanted foreign tissues and organs (allografts). Immunocompetent CD8+ and CD4+ T lymphocytes respond to foreign (allogeneic) HLA molecules (direct recognition) or to foreign HLA-derived peptides (indirect recognition), respectively [1, 2]. Of note, allorecognition of HLA molecules may activate up to 10% of the total T lymphocyte pool. The extent of T lymphocyte activation varies according to the level of HLA disparity between donor and recipient: the greater the level of HLA disparity, the stronger the extent of the response.
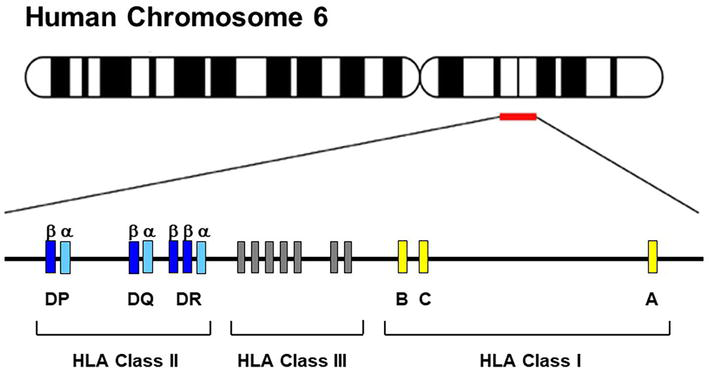
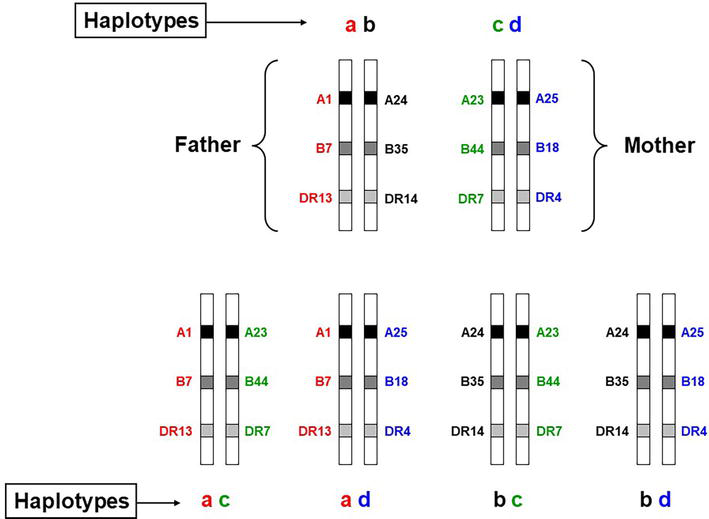
HLA class I molecules (A, B, C) are expressed on all nucleated cells and are recognized by CD8+ T lymphocytes. Alternatively, HLA class II molecules (DR, DQ, DP) are only expressed on the surface of antigen-presenting cells and are recognized by CD4+ T lymphocytes (Table 1) [1, 2]. The HLA class II molecules are also expressed in a variety of other cell types, such as endothelial cells, upon stimulation with interferon-γ. Every individual inherits nine clinically relevant HLA alleles (three HLA class I and six HLA class II) from each parent. Such antigens are co-dominantly expressed on the cell surface. The entire set of clinically relevant HLA-A, B, C, DRB1, DRB3/B4/B5, DQA1, DQB1, DPA1, and DPB1 genes encoded on chromosome 6 is called a “haplotype” (Figure 1). According to Mendelian laws, the HLA “haplotypes” are inherited en block from each parent (Figure 2). This concept is of high importance in clinical hematopoietic stem cell transplantation (HSCT) where the first donor of choice is an HLA identical sibling. Based on the Mendelian laws of inheritance, everyone has only a 25% probability of being HLA identical to their siblings (Table 2).
Region
Gene products
Tissue location
Function
Class I
HLA-A, B, C
Nucleated cells
Recognition of tumor and virus-infected cells by CD8+ T lymphocytes
Class II
HLA-DR, DQ, DP
Antigen-presenting cells: B lymphocytes, macrophages, dendritic cells, endothelial cells
Recognition of foreign antigens by CD4+ T lymphocytes
Table 1.
Expression of HLA genes.
Figure 1.
The human leukocyte antigen complex on chromosome 6. The HLA class I region is 3-6 kb long and the HLA class II region is 4-11 kb long. The HLA class III is not part of the polymorphic HLA system.
Figure 2.
Inheritance of HLA haplotypes. HLA genes are inherited en block from each parent according to Mendelian laws.
HLA
A
B
C
DRB1
DQB1
Degree of matching
Patient
01:01
07:02
07:02
13:02
06:04
25:01
18:01
12:03
04:04
03:02
Father
01:01
07:02
07:02
13:02
06:04
Haplo-identical
24:02
35:03
12:03
14:01
05:03
Mother
23:01
44:03
04:01
07:01
02:02
Haplo-identical
25:01
18:01
12:03
04:04
03:02
Sibling 1
01:01
07:02
07:02
13:02
06:04
HLA-identical
25:01
18:01
12:03
04:04
03:02
Sibling 2
01:01
07:02
07:02
13:02
06:04
Haplo-identical
23:01
44:03
04:01
07:01
02:04
Sibling 3
24:02
35:03
12:03
14:01
05:03
Haplo-identical
25:01
18:01
12:03
04:04
03:02
Sibling 4
24:02
35:03
12:03
14:01
05:03
Two haplotype mismatch
23:01
44:03
04:01
07:01
02:02
Table 2.
Family segregation analysis for HLA genes.
Several organs and tissues such as kidney, heart, lung, liver, pancreas, skin, bone marrow, hematopoietic stem cells, cornea, and vascularized composite tissue (allografts composed of multiple different tissues such as a hand allograft, which consist of muscle, skin, bone, blood vessels, nerves, and connective tissue) can be transplanted. As mentioned above, all transplanted organs and tissues are called allografts indicating genetic differences between donor and recipient. HLA matching (compatibility) between donor and recipient increases the chance for successful long-term allograft survival [3, 4, 5]. For example, if the donor and recipient are not HLA-matched, the recipient’s T and B lymphocytes will recognize the foreign donor cells as non-self and will develop an immune response against the allograft resulting in immune-mediated rejection and possible loss of function.
Since HLA mismatches induce the activation of an alloreactive immune response, transplant programs try to match as many as possible donor and recipient HLA molecules. As shown in Table 3, better-matched kidney allografts have better survival rates [3, 4, 5, 6]. Thus, it is recommended to determine the HLA typing of prospective donor-recipient pairs before transplantation to have a better immunological risk assessment. Optimal HLA matching between donor and recipient prevents allograft rejection in solid organ transplantation (SOT) and prevents both allograft rejection and graft versus host disease (GVHD) in hematopoietic stem cell transplantation (HSCT). Current clinical histocompatibility testing consists of molecular HLA typing and testing for the presence of circulating donor-specific anti-HLA antibodies. The required level of HLA typing resolution varies depending on the transplant type. Potential SOT donors and recipients are typed by means of low-resolution molecular HLA typing techniques. In contrast, potential HSCT donors and recipients require high-resolution HLA typing. Currently, histocompatibility laboratories have replaced serological typing methods with molecular typing methods. These new molecular techniques have significantly improved the histocompatibility laboratory’s accuracy and efficiency [7, 8, 9].
Influence of HLA matching on kidney graft survival.
Data obtained from the Organ Procurement and Transplantation Network (OPTN): Kidney allografts transplanted between 2008 and 2015 [6].
CI, confidence intervals.
Several studies have shown that a small but important percentage of kidney allografts are lost during the first-year post-transplantation due to cellular and/or antibody-mediated rejection despite optimal HLA matching (Table 3) [3, 4, 5]. This observation suggests the presence of mismatches in other non-HLA minor histocompatibility antigenic systems that also play an important role in allograft rejection [10]. In addition, these data also suggest that low-resolution HLA typing may no longer be considered adequate for SOT patients [11, 12]. Thus, it has been established that better HLA matching results in better allograft survival as well as lower GVHD rates and less need for immunosuppression after transplantation. Hence, HLA typing plays an important role in donor selection and risk assessment in both SOT and HSCT.
The contribution of the MHC to allograft rejection was first proposed by Bover [13] who observed that skin allografts between identical twins were not rejected like those from genetically different individuals. Then, the MHC genes involved in the allograft rejection process were first described in mice by Gorer [14] Subsequently, Snell [15] used mouse cell lines to further define a locus, which was called H for histocompatibility. Gorer [14] referred to the gene products of locus H as antigens II and the combined term H-2 was consequently used for the mouse MHC. The HLA system was subsequently discovered in the 1950s. Several investigators independently observed that sera from previously transfused individuals and from multiparous women contained antibodies that agglutinated leukocytes [16]. This observation led to the development of serological typing methods that identified a single locus that was subsequently split into two loci: HLA-A and HLA-B. At first, several techniques were used for serological typing but the microlymphocytotoxicity assay became the most widely employed [17, 18, 19, 20]. Subsequently, it was observed that, when cultured together in a “mixed lymphocyte culture” (MLC), lymphocytes from unrelated individuals matched at HLA-A and HLA-B loci, showed a robust proliferative response [21, 22]. This observation led to the discovery of an additional locus initially called HLA-D [23, 24, 25]; it was subsequently shown that mismatches at HLA-DR and HLA-DQ contributed to the lymphocyte activation observed in the “mixed lymphocyte culture”. Soon after, extensive serological studies led to the discovery of HLA-C [26]. Later, HLA-DP, originally called the “secondary B cell” (SB) antigen, was discovered by means of a secondary stimulation assay, called the “primed lymphocyte test” (PLT), that showed the recognition of another HLA molecule different from those recognized in the primary “mixed lymphocyte culture” [27, 28].
The HLA genetic system is composed of three regions containing genes of different classes (I, II, and III) located on chromosome 6 (Figure 1) [29]. The class I region contain genes encoding for HLA-A, B, and C. The class II regions contain genes encoding for HLA-DR, DQ, and DP. Moreover, the class III region is located between the class I and class II and contains genes encoding for molecules involved in immune function that are not targets for allorecognition (Figure 1) [29]. Several cytokine genes such as tumor necrosis factor (TNF) are found in the class III region. In addition to the main HLA system, the extended MHC system covers 8 Mb and includes the hemochromatosis (HFE) gene, the farthest telomeric locus in the system, and the tapasin (TAPBP) gene, the farthest centromeric locus of the system. Additionally, several genes that encode for proteins involved in antigen processing and presentation (HLA-DO, TAP-1, and subunits of the immune proteasome) are located in the class II region [29].
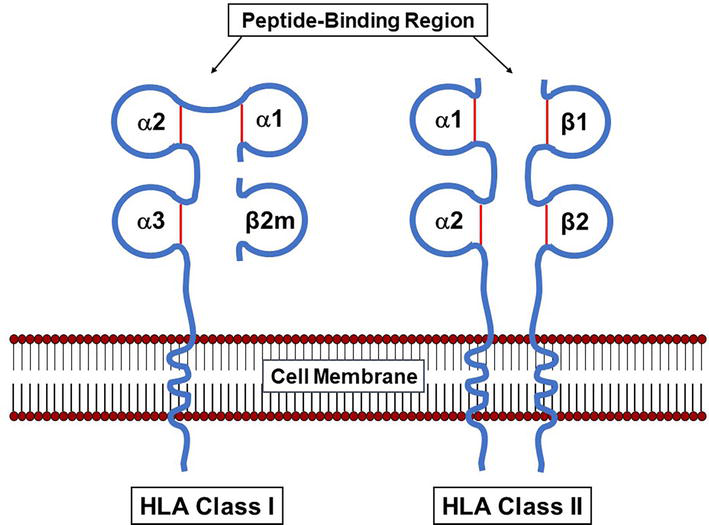
As mentioned above, HLA class I and class II molecules are expressed differently in different tissues (Table 1) [30, 31, 32, 33]. Despite the discovery that HLA molecules are the main mediators of transplant rejection, allorecognition is not their main function. HLA molecules are expressed on the surface of cells of the immune system, allowing for cell-cell interactions during the development of an immune response [1, 2]. As shown in Figure 3, HLA class I molecules is composed of a long protein of 45 kDa (heavy chain) associated with a smaller protein of 12 kDa called the β2-microglobulin (β2m) encoded by a gene located on chromosome 15. The two chains are associated on the cell surface by non-covalent bonds. The heavy chain has a transmembrane polypeptide, anchoring the complex on the cell surface. HLA class II molecules are composed of two transmembrane proteins, the α chain of 33–35 kDa and the β chain of 26–28 kDa. The two chains associate forming a groove that bind peptide fragments (approximately 15 amino acids long) derived from extracellular proteins that have been processed by the cell (extracellular antigens). In contrast, HLA class I molecules bind peptide fragments (approximately 9 amino acids long) derived from proteins synthesized within the cell (intracellular antigens). HLA class I and class II molecules present the bound peptides to CD8+ and CD4+ T lymphocytes, respectively. The different HLA molecules vary in their efficiency to bind a particular peptide fragment, resulting in a range of immune responses to a given peptide. The different affinity of HLA molecules for a given peptide can affect symptoms of disease, for example, the susceptibility of individuals carrying a particular HLA genotype to develop acquired immunodeficiency syndrome (AIDS) after human immunodeficiency virus infection (HIV) [34].
Figure 3.
Structure of the HLA class I and class II molecules. The HLA class I molecules consist of one polymorphic heavy chain (α) associated with a light chain called the β2-microglobulin (β2m). The HLA class II molecules consist of two polymorphic chains (α and β).
The HLA system is the most polymorphic genetic system of the human genome. As mentioned above, HLA polymorphisms were first defined phenotypically by acceptance or rejection of tissue and/or by reaction with defined alloantibodies (serological typing methods). Subsequently, molecular typing methods showed HLA polymorphisms that range from a single nucleotide change to a loss or gain of an entire genetic region. The identification of HLA polymorphisms was originally performed by serological and cell proliferation assays [18, 19, 20, 25]. These assays were successfully used to initially characterize the HLA system; however, despite their extensive application, these have significant limitations in terms of accuracy and reproducibility [35]. Also, alloreactive antisera are usually in limited supply and both serological and cellular HLA typing assays require live cells. Overall, the main limitation of serological HLA typing is its inability to recognize minor polymorphic differences capable to activate CD4+ or CD8+ T lymphocytes.
A general characteristic of the HLA genes is that the distal membrane domains are highly polymorphic, while the proximal membrane domains, the transmembrane, and cytoplasmic domains have very low or no polymorphisms. The heavy chain of the HLA class I molecule is composed of 3 extra-cellular domains and both the α and β chains of the HLA class II molecule contain 2 extracellular domains (Figure 3). Of note, the HLA genes, like all eukaryotic genes, contain both coding (exons) and non-coding (introns) regions. The HLA class I genes contain 8 exons while the HLA class II genes contain 6 or 7 exons [1, 2]. The wide application of molecular typing techniques has allowed for the characterization of thousands of HLA alleles [36]. The current number of HLA alleles are shown in Table 4 and can also be found on the IPD-IMGT/HLA Database [37, 38, 39]. As mentioned above, analysis of the nucleotide sequences of the HLA genes indicates that most of the polymorphisms are found in exons 2 and 3 of the HLA class I genes and in exon 2 of the HLA class II genes. These exons encode for the distal membrane domains called the “peptide-binding region” [1, 2, 37, 40, 41]. It has been observed that most nucleotide polymorphisms within the “peptide-binding regions” involve changes that induce a change in the corresponding amino acid sequence (non-synonymous substitutions) and has a high level of correlation with phenotype differences detected by serological and cellular methods [35]. However, serological equivalents are not available for all described alleles [37, 38, 39]. Additionally, it is difficult to predict the serological specificities of selected alleles with polymorphisms corresponding to more than one antigenic group [42, 43].
HLA specificities identified by serological and molecular methods (assigned as of September 2022).
Data obtained from the IPD-IMTG/HLA Database [37, 38, 39].
It has been observed that most of the polymorphisms are restricted to some segments of the gene called variable regions. Alleles pairs associated with the same serotype (e.g., A*02:01, A*02:02) differ only by a few nucleotides while distinguishing sequences are observed in alleles of other serotypes, indicating the patchwork nature of HLA polymorphisms. The significant HLA polymorphism probably evolved from the existence of a few allelic lineages followed by short segmental exchanges to increase the number of alleles of a given locus. It appears that most of the HLA polymorphisms was generated by this mechanism. Then, selected natural selection events must have been necessary for new alleles to reach a significant population frequency. Nevertheless, it should be noted that many alleles arose from single-point mutations.
With the understanding of the significant level of HLA polymorphism and the improvement of molecular techniques, several molecular HLA typing methods have been developed [44, 45, 46]. These methods have focused on the detection of polymorphisms in exons 2 and 3 of HLA class I genes and in exon 2 of the HLA class II genes. The application of these molecular techniques has resulted in accurate and reproducible HLA typing methods suitable for clinical application [8, 44, 47, 48]. The wide application of these methods has led to the identification of many new alleles that were previously undetectable with the serological and cellular methods [37, 38, 39]. The molecular HLA typing methods are widely used and take advantage of the effortlessness of DNA amplification by polymerase chain reaction (PCR). The more widely used molecular HLA typing methods currently used in histocompatibility laboratories are: (1) amplification with sequence-specific primers (SSP), (2) hybridization with sequence-specific oligonucleotide probe hybridization (SSOPH), and (3) direct analysis the DNA sequence (sequence-based typing, SBT) by means of Sanger sequencing or next-generation sequencing (NGS).
A standard nomenclature for serologically defined HLA specificities has been established by the World Health Organization (WHO) nomenclature committee [37, 38, 39]. A list of the serologically defined HLA molecules accepted by the WHO is shown in Table 5 and can also be found on the IPD-IMGT/HLA Database [37, 38, 39]. The WHO official nomenclature refers to serologically defined antigens by a number following the gene name, for example, HLA-A2 indicates the HLA-A antigen 2. Subtypes from a broad specificity are followed by the number of the parental antigen in parentheses. For example, HLA-A24(9) indicates the HLA-A antigen 24 derived from the parental HLA-A antigen 9. In this regard, the derived antigens are called “split” specificities. As mentioned above, the current numbers of HLA specificities identified by serological and molecular methods are shown in Table 4 and can also be found on the IPD-IMGT/HLA Database [37, 38, 39].
Broad antigen specificities are listed in parentheses.
Associated antigens (e.g., A2, A203, A210) are listed together.
Data obtained from the IPD-IMGT/HLA Database [37, 38, 39].
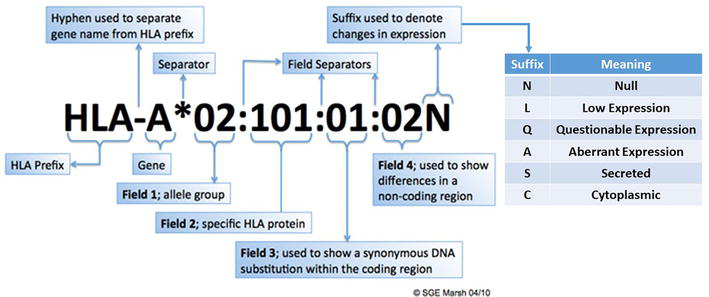
With the introduction of molecular HLA typing methods in the 1980s, HLA typing at the DNA level required nomenclature for specific DNA sequences [37, 38, 39]. A revised nomenclature is used for designating alleles (Figure 4) [49, 50]. Each HLA allele has a unique number corresponding to up to four sets of digits separated by colons. All alleles receive at least a four-digit name, which corresponds to the first two sets of digits; longer names are assigned only when necessary. The gene name, such as HLA-A, is followed by an asterisk and the allele family number (which often corresponds to the serological typing); for example, A*02 (Figure 4). The second set of digits is used to list the subtypes of the allele family, the numbers have been assigned in the order in which DNA sequences have been found. Alleles whose numbers differ in the two sets of digits must differ in one or more nucleotide(s) that change the amino acid sequence of the encoded protein; for example, A*02:01 and A*02:02 (Figure 4). Alleles that differ by a synonymous nucleotide substitution in the exons (changes in the DNA sequence that do not change the amino acid sequence), also called silent mutations or non-coding substitutions, are distinguished using the third set of digits; for example, A*02:01:01 and A*02:01:02 (Figure 4). Alleles that differ by a nucleotide substitution in the introns, or in the 5′ or 3′ untranslated regions that flank the exons and introns, are distinguished using the fourth set of digits; for example, A*02:01:01:01 and A*02:01:01:03 (Figure 4).
Figure 4.
HLA nomenclature. The gene is indicated after the HLA prefix and hyphen. Then an asterisk is used as a separator before the first field, which is represented by two digits. A colon is used to separate each of the fields, with up to four fields listed. The second field indicates the specific HLA protein, while the third field is used to describe a nucleotide change within the coding region that does not change the encoded amino acid. The fourth field describes changes in non-coding regions. A suffix may be utilized to indicate changes in expression, and the potential suffixes are listed in the table. The figure is courtesy of Professor Steven G.E. Marsh, Anthony Nolan Research Institute, London, United Kingdom [38].
In addition to the unique allele number, there are additional suffixes that may be added to an allele to indicate its expression status (Figure 4) [37, 38, 39]. Alleles that have been shown not to be expressed have been given the suffix “N” for “Null”; for example, HLA-B*13:07 N is not expressed due to a 15-base pair deletion in exon 2. “Null” alleles can also be the result of premature stop codon mutations, nonsense, frameshift, and splice site. In addition, the suffix “L” indicates a “Low” cell surface expression as compared to normal expression levels. The suffix “Q” indicates a “Questionable” cell surface expression, given that the allele carries a mutation that has been shown to affect normal expression levels. The suffix “A” indicates an “Aberrant” cell surface expression, specifically where there is doubt as to whether the allele product has any cell surface expression. The suffix “S” indicates an allele product that is a “Secreted” molecule with no cell surface expression. The suffix “C” indicates an allele product that is present in the “Cytoplasm” with no cell surface expression. A list of the “Null” and alternatively expressed alleles can be found on the IPD-IMGT/HLA Database [37, 38, 39].
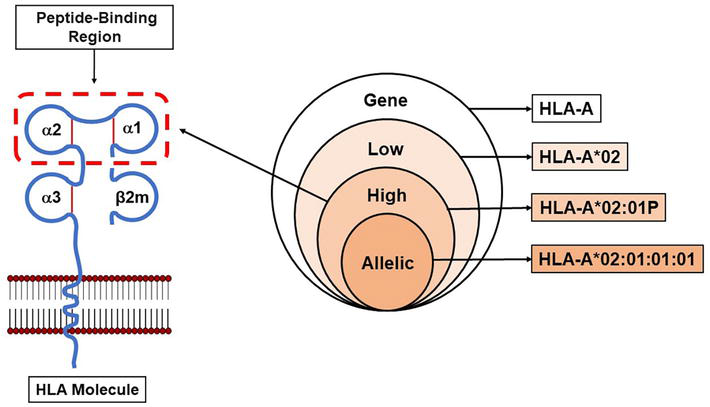
HLA specificities can be identified to varying degrees of resolution depending on the methodology used [8]. In this regard, the definitions for low, high, and allelic resolution typing were compiled by the “Harmonization of Histocompatibility Typing Terms Working Group”, to define a language for histocompatibility laboratories to report HLA typing results [40, 41]. An allelic-resolution HLA typing result is consistent with a single allele as defined by the WHO HLA Nomenclature Report [37, 38, 39]. An allele is defined as a unique nucleotide sequence for a gene defined using all the digits in a current allele name; for example, A*02:01:01:01 (Figure 5). A high-resolution HLA typing result is defined as a group of alleles that encode the same protein sequence for the region of the HLA molecule called the “peptide-binding region” (Figure 5). The “peptide-binding region” includes domains 1 and 2 of the HLA class I α chain (encoded by exons 2 and 3) and domain 1 of the HLA class II α and β chains (encoded by exon 2) [1, 2]. This group of alleles is designated by an upper case “P” which follows the 2-field allele designation of the lowest numbered allele in the group; for example, A*02:01P (Figure 5). A low-resolution HLA typing result is defined as the digits composing the first field in the DNA-based nomenclature; for example, A*02 (Figure 5). If the resolution corresponds to a serologic equivalent, this typing result should also be called low-resolution; for example, A2 [40, 41].
Figure 5.
HLA typing resolution. The Venn diagram illustrates increasing levels of HLA typing resolution. The figure on the left shows the “peptide-binding region” of an HLA class I molecule. High-resolution HLA typing defines the specific DNA sequence of the ‘peptide-binding region”. Allelic resolution defines a single allele as defined by a unique DNA sequence for the HLA gene. Adapted from Nunes et al. [40, 41].
As mentioned above, histocompatibility testing was traditionally performed using serological methods utilizing (1) sera with known HLA specificity to identify HLA antigens and (2) using cells with known HLA antigens to identify anti-HLA antibodies in patient sera [17, 18, 19, 20]. Although serological methods yield only low-resolution HLA typing results, there are some advantages to these methods; serological methods are relatively rapid and will reveal immunologically relevant epitopes. Also, serological methods can be used to confirm “Null” alleles detected by molecular methods.
6.2 Molecular typing
Several molecular HLA typing methods are now widely available to the histocompatibility laboratory [8, 44, 46, 51]. These methods can be broadly classified into two categories: (1) methods in which the polymorphisms are identified directly by the PCR process, without further steps, for example SSP; and (2) methods that generate PCR products containing polymorphisms that can be identified by a secondary technique, for example SSOPH, Sanger sequencing, and NGS.
The use of specific techniques depends on the histocompatibility laboratory’s requirements for HLA typing resolution and volumes. The different methods have different requirements in terms of skills of testing personnel, instruments, and turn-around time. Currently, most histocompatibility laboratories use a combination of HLA typing techniques to obtain the desired level of resolution. Currently, low-resolution HLA typing is sufficient for SOT while HSCT requires allelic- or high-resolution HLA typing depending on the internal policies of the transplant program (Figure 6) [52]. In recent years, it has been shown that alloantibodies may also recognize allelic differences; therefore, in some cases, high-resolution HLA typing may also be required for SOT [7, 11].
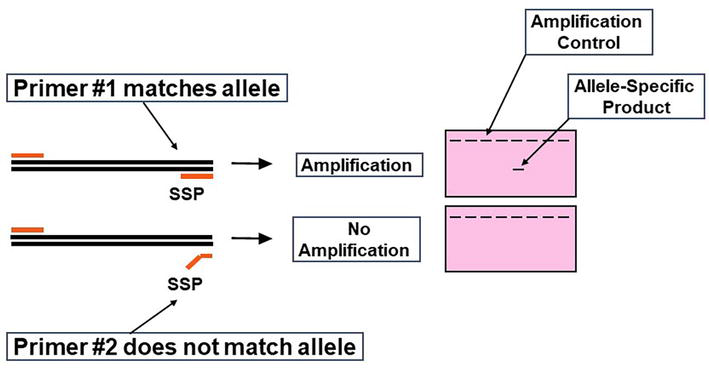
Figure 6.
Principle of sequence-specific primers. HLA alleles are amplified by PCR using SSP. The PCR products are then detected by agarose gel electrophoresis or real-time PCR amplification plots. An amplification control is included with each reaction to detect false negative results due to amplification failure.
6.2.1 Steps for molecular typing
Molecular HLA typing techniques involve three general steps: (1) extraction of genomic DNA, (2) amplification of segments of the gene of interest and (3) detection of the sequence polymorphisms that define the alleles.
6.2.1.1 DNA extraction
Genomic DNA is usually extracted in the histocompatibility laboratory from peripheral blood leukocytes. Only a few micrograms of genomic DNA are sufficient to perform any of the molecular HLA typing methods described below. The most widely used DNA extraction method in the histocompatibility laboratory is the “salting-out” method. This is a simple and non-toxic DNA extraction method developed by Miller et al. [53] During this method, proteinase K and RNase are added to the sample after cell lysis. Proteinase K is used to digest proteins and remove contamination from the sample. In this regard, addition of proteinase K rapidly inactivates nucleases that might otherwise degrade the DNA during purification. Then, saturated NaCl is added to precipitate the proteins. Then, the sample is centrifuged, and the DNA, present in the supernatant, is transferred to a second tube to be centrifuged again, and subsequently washed with 70% ethanol. High yields of high-quality DNA are obtained with this method in a relatively fast and inexpensive manner.
6.2.1.2 DNA Amplification
Once it is established that there is enough quantity of high-quality DNA. The PCR mixture, containing genomic DNA, primers, Taq DNA polymerase, and four types of deoxynucleotide triphosphate (dNTP) (adenine, dATP; cytosine, dCTP; guanine, dGTP; and thymidine, dTTP), is prepared. Then, the PCR mixture is subjected to repeated cycles of heating to 94–96°C for double-stranded DNA denaturation; cooling down to the corresponding temperature for primer annealing and lastly, warming up to 72°C for an optimal Taq DNA polymerase activity to integrate the complementary dNTP to the single-stranded DNA. After the first amplification cycle, the DNA copies serve as templates to allow exponential growth of the PCR product. Because both DNA strands need to be amplified, the primers for those strands should be designed to include intervening segments between the two strands. To ensure the optimal efficiency of the PCR reaction, the number of cycles and the duration of incubation periods at each temperature should be based on the length of the DNA fragments and GC content from both the DNA fragments and the primers.
6.2.1.3 Detection of sequence polymorphisms
6.2.1.3.1 Sequence-specific primers
SSP is a rapid method that uses sets of primer pairs to amplify genomic DNA. The efficiency of the amplification reaction is controlled by the primers that amplify conserved sequences of a selected gene. The 3′ end of the primer must match the template for recognition by the Taq DNA polymerase. By designing primers with polymorphic sequences at the 3′ end, successful amplification can be used to type specific HLA alleles (allelic-resolution) or a group of alleles (low- or high-resolution). The HLA typing results are interpreted by analyzing the amplification pattern, detected on an agarose gel electrophoresis (Figure 6).
SSP reactions can be set up in a 96-well plate format with different allele-specific primer sets in each well. Each PCR mixture contains sequence-specific primers and a set of amplification control primers. The amplification control primers should yield a PCR product for every specimen (except the negative control). The sequence-specific primers should only yield a product if the specimen has the HLA specificity matching the primers’ sequences. The amplification control primers are designed to yield a PCR product of different size from the product of the HLA-specific primers. The two PCR products are then resolved by agarose gel electrophoresis or real-time PCR. Specimens will yield two PCR products (amplification control and HLA-specific product) only from those PCR reactions containing primers matching the sample’s HLA specificity (Figure 6). PCR reactions containing primers that do not match the sample’s HLA specificity should only show the amplification control. This method can easily be implemented, HLA typing can be performed in 4–5 h, and it is adequate for low-volume histocompatibility laboratories.
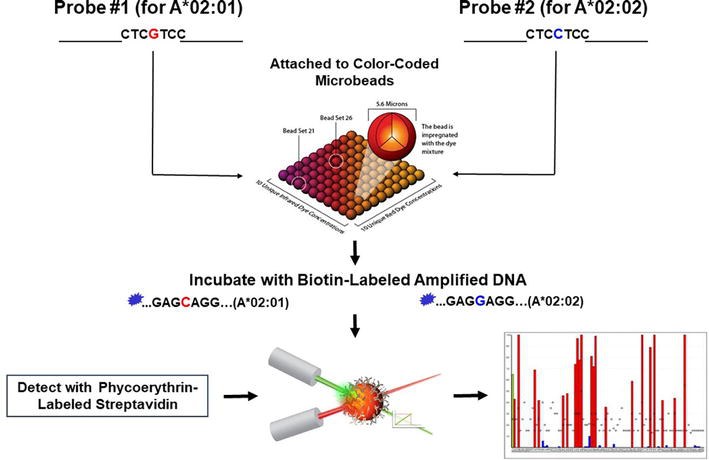
Specific PCR amplification of an HLA locus and the subsequent probing of the PCR product with probes immobilized on an array of Luminex® microbeads is the method known as SSOPH. The Luminex® assay uses color-coded microbeads of defined spectral properties conjugated to sequence-specific probes and incubated with the samples in a 96-well microplate (Figure 7). The Luminex® technology is a bead-based assay that allows for multiplex detection of up to 100 analytes simultaneously. For this method, the HLA locus is first amplified using primers flanking the polymorphic sequences and labeled with biotin at the 5′ end. The probes are short (approximately 20 nucleotides long) single-stranded DNA fragments designed to hybridize to a specific HLA specificity (Figure 7). The probe sequences, based on sequences found on the IPD-IMGT/HLA Database, are aligned so that the polymorphic sequences are in the middle of the probe’s sequence [37, 38, 39]. The amplified biotin-labeled DNA binds to the probes over during a pre-selected period of incubation (Figure 7). After the first incubation, the microbeads are washed, and a second incubation follows in the presence of a biotin-specific molecule, streptavidin, labeled with a fluorescent dye, phycoerythrin (Figure 7). After the second incubation, the microbeads are washed and ready to analyze. The microbead array is then classified as phycoerythrin-positive or negative in a Luminex® flow cytometry instrument. Different patterns of phycoerythrin-positive microbeads define the different HLA alleles (Figure 7). The number of probes used in the assay depends on the desired HLA typing resolution. Of note, because of multiple recombination events generating polymorphism in the HLA system, many of the sequences in the polymorphic regions are not allele-specific, i.e., several sequences are shared by many HLA alleles. Therefore, since selected probes detect multiple HLA specificities, hybridization profiles are usually highly complex, and a software is needed for accurate interpretation of the results (Figure 7).
Figure 7.
Principle of sequence-specific oligonucleotide probe hybridization. HLA genetic regions are amplified by PCR using generic primers covalently bound to biotin at the 5′ end. The amplified and labeled DNA bound to the probes immobilized on Luminex® microbeads is then detected with a biotin-specific molecule, streptavidin, conjugated with phycoerythrin. The microbead array is then classified as phycoerythrin-positive or negative by a Luminex® flow cytometry instrument. Different patterns of phycoerythrin-positive and negative microbeads define specific HLA alleles.
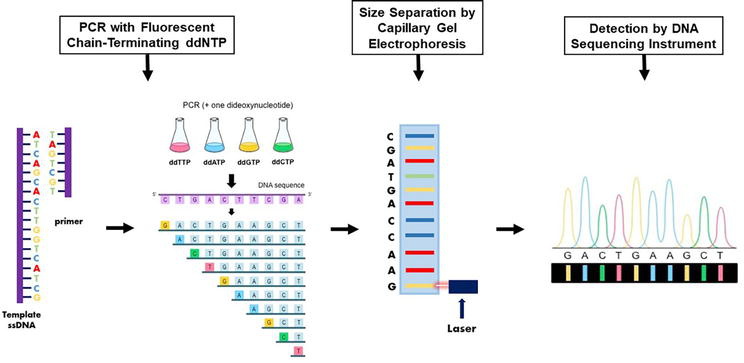
6.2.1.3.3 Sequence-based typing by sanger sequencing
The most accurate method for HLA typing is the direct identification of the DNA sequence. Until recently, the most widely used sequence-based HLA typing method was the “chain termination method” that is performed by the random incorporation of four types of dideoxynucleotide triphosphate (ddNTP) (adenine, ddATP; cytosine, ddCTP; guanine, ddGTP; and thymidine, ddTTP) labeled with four different fluorescent dyes (Figure 8) [54]. This method is named after the inventor of this ground-breaking technology, Dr. Frederick Sanger, who developed this method in the 1970s [54]. In the second step, the chain-terminated DNA fragments are separated by size via gel electrophoresis; the DNA samples are loaded into one end of a gel matrix, and an electric current is applied; DNA is negatively charged, so the DNA fragments are pulled toward the positive electrode on the opposite side of the gel. Because all DNA fragments have the same charge per unit of mass, the speed at which the DNA fragments move will be determined only by size. The smaller a DNA fragment is, the less friction it will experience as it moves through the gel, and the faster it will move. As a result, the labeled dideoxynucleotides attached to the DNA fragments will be arranged from smallest to largest, and they will be detected first reading the gel from bottom to top (Figure 8). The last step involves reading the gel to determine the DNA sequence. Since DNA polymerase only synthesizes DNA in the 5′–3′ direction starting at a provided primer, each terminal ddNTP will correspond to a specific nucleotide in the original DNA sequence (e.g., the shortest DNA fragment must terminate at the first nucleotide from the 5′ end, the second-shortest DNA fragment must terminate at the second nucleotide from the 5′ end, etc.). Therefore, by reading the gel bands from the smallest to largest DNA fragment, the 5′–3′ sequence of the original DNA strand can be determined. A computer reads each band of the gel, in order, using fluorescence to call the identity of each terminal ddNTP. Briefly, a laser excites the fluorescent terminal ddNTP in each band, and a computer detects the resulting light emitted. Because each of the four ddNTP is labeled with a different fluorescent dye, the light emitted can be directly tied to the identity of the terminal ddNTP. The output is called a chromatogram, which shows the fluorescent peak of each nucleotide along the length of the template DNA (Figure 8).
Figure 8.
Principle of sequence-based typing by Sanger sequencing. HLA genetic regions are amplified by PCR using locus-specific primers. The PCR products are then purified from unused PCR reaction components, sequencing reactions are performed using forward and reverse sequencing primers and these reactions are loaded onto the automated DNA sequencer to detect the nucleotide sequence of the targeted genes.
When a single PCR reaction is performed in a particular sample, simultaneous amplification and sequencing of both alleles are obtained and heterozygous nucleotide assignments are observed at positions where both alleles have different nucleotides. Some heterozygous genotypes displaying the same sequencing pattern result in ambiguous (alternative) sequencing results. Performing additional tests targeting only one of the possible alleles, either by additional sequencing primers, SSP, and/or SSOPH usually resolves these ambiguous results, enabling the separation of the two alleles in heterozygous samples.
6.2.1.3.4 Sequence-based typing by next-generation sequencing
HLA typing by Sanger sequencing is restricted by its low throughput and high cost. In addition, HLA typing ambiguities, resulting from the inability to phase heterozygous nucleotide positions, requires time-consuming follow-up testing to resolve the ambiguous HLA typing results. Short-read NGS, also called second-generation sequencing has begun to alleviate these disadvantages for HLA typing. The common element of short-read NGS technologies is massive sequencing of short (250–800 basepairs long), clonally amplified DNA fragments sequenced in parallel [55]. Several NGS-based HLA typing kits are commercially available, all of which have achieved similar accuracy and straightforward multiplexing workflows covering the clinically relevant HLA class I and class II genes.
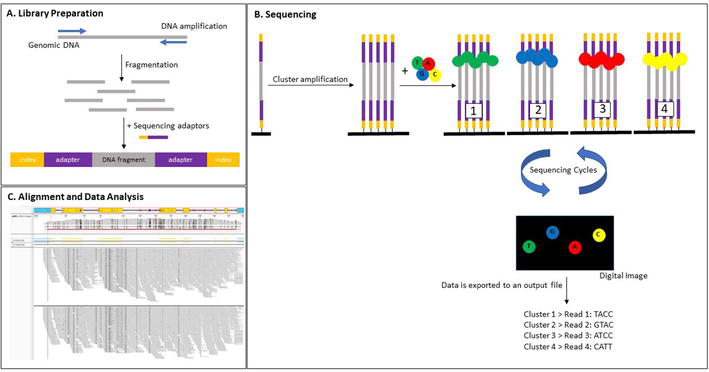
The current NGS workflow includes the following steps: DNA isolation, library preparation, and sequencing (Figure 9). DNA can be extracted from anticoagulated whole blood or buccal swabs utilizing commercial kits. High quality DNA, free of biological and chemical contaminants, is crucial for sequencing success. Due to limitations in the sensitivity of standard spectrophotometry, fluorometers are advantageous for analyzing DNA concentration, but cannot provide absorbance ratios. A necessity for NGS is a good quality library. During library preparation DNA templates are obtained which are compatible with the sequencing instrument utilized for NGS. Library enrichment protocols in general depend on one of the following methods: short-range PCR, long range PCR, or hybrid capture. For PCR-based library enrichment protocols, multiplex primers are used to amplify the HLA loci of interest. Short-range PCR-based enrichment for HLA typing focuses on the amplification of exons encoding the “peptide-binding region” and has the advantage of faster sequencing and higher depth of coverage for the enriched genomic regions. However, the possible loss of phasing over longer segments of DNA has been observed with this methodology resulting in typing ambiguities seen with Sanger sequencing [56]. Long-range PCR substitutes a longer library preparation time to accomplish amplification of whole HLA genes including introns, upstream and downstream flanking sequences, and in some cases, untranslated genomic regions. Thus, long-range PCR improved issues of HLA typing ambiguities observed with short amplicon sequencing. It should be noted, however, that long-range PCR-based protocols have the potential to show allele imbalance or dropout because of where the primers anneal and the sequence composition in these genomic regions. Consequently, it has been recommended that homozygous HLA alleles or typing results suspected to be affected by allele dropout should be typed by another molecular methodology to ensure correct results. The most recent library enrichment methodology utilizes HLA locus complementary oligonucleotide probes which hybridize to the locus of interest. These probes are bound to magnetic beads, which allow for the selection and enrichment of HLA genes during the library preparation step [57]. The benefit of hybrid capture-based library enrichment is that there is no need for long-range PCR, therefore saving PCR time and avoiding PCR amplification errors. However, this methodology has an intrinsic bias in capturing AT/GC-rich DNA regions, and highly repetitive sequences may be underrepresented. Since HLA class II genes carry large intronic areas consisting of repeat elements, it is challenging to design oligonucleotide probes with sufficient specificity to capture these sequences. Consequently, probe-based assays only cover the full exons of HLA class II genes. The lack of coverage in introns can result in typing ambiguities for HLA class II genes, but mainly in the 4th field of typing.
Figure 9.
Principle of sequence-based typing by next-generation sequencing. (A) The sequencing library is prepared by fragmenting the amplified genomic DNA and ligating specialized adapters with indices to both fragment ends. (B) The sequencing library is loaded into a flow cell and the fragments are hybridized to the flow cell surface. Each bound fragment is amplified into a clonal cluster. Sequencing reagents, including fluorescent-dye labeled nucleotides, are added and the first nucleotide is incorporated. The flow cell is imaged and the emitted light from each fragment cluster is recorded. The emission wavelength and intensity are used to identify the incorporated nucleotide. This cycle is repeated several times. (C) Reads are aligned to a reference sequence from the IPD-IMGT/HLA Database [37, 38, 39]. After alignment, differences between the reference genome and the newly sequenced reads can be identified.
Library preparation includes the additional steps of fragmentation, end-repair, adaptor ligation, and size selection. Fragmentation shears DNA into the ideal NGS platform-specific size range. DNA fragmentation can be achieved by sonication, transposase “tagmentation”, or heat treatment with divalent metal cations. Importantly, shorter DNA fragments offer high-quality sequencing data, whereas longer fragments provide distal phasing information. Subsequent end-repair of the DNA fragments prepares the sequencing library for adaptor ligation. Adaptors comprise NGS platform-specific sequences for DNA fragment recognition by the instrument and as well as a unique sequence, called an index or barcode, for labeling of an individual sample thereby allowing several patient samples to be pooled and sequenced concurrently in a single run (multiplex sequencing). Following adaptor ligation, a size selection step enriches DNA fragments within a defined size range and removes contaminations to improve sequencing quality and efficiency. Size selection can be accomplished by utilizing bead-based or electrophoretic-based methodologies. Bead-based approaches allow for simultaneously concentrating the sequencing library, while electrophoretic-based methods enhance precision. Alternatively, DNA fragments can be generated by utilizing an on-bead “tagmentation” protocol, which combines the library preparation steps of DNA normalization, fragmentation, and size selection. Following “tagmentation”, a PCR step is performed to incorporate the sequencing adapters and barcodes for patient sample identification. This workflow is straightforward and fast, allowing sequencing libraries to be generated in less than 90 min, with less than 15 min of hands-on time [51].
Since NGS produces substantial amounts of data, efficient bioinformatics data analysis, and data management are critical for successful implementation of NGS in the HLA laboratory. Primary sequencing data analysis is performed by the instrument and involves base-calling for each clonally amplified DNA fragment. Quality control procedures including read filtering and trimming, also take place during this step. Sequencing data is saved together with the quality matrix as a FASTQ file. For HLA genotyping, specific software programs are commercially available for the final analysis steps. As a result of the highly polymorphic nature of HLA genes, alignment to the human reference genome is inefficient for precisely determining the HLA alleles present within a patient sample, and instead depend on alignment to the IPD-IMGT/HLA Database, which contains the sequences of all currently characterized HLA alleles [37, 38, 39]. Another quality indicator at this step is coverage, which includes depth of coverage (number of times a base is sequenced) and breadth of coverage (percentage of a reference genome covered). Importantly, coverage may not be consistent throughout the amplicon, and lack of coverage in key regions such as exons may affect the accuracy of the HLA typing result. Commercially available HLA software analysis programs designed for HLA-typing, typically have built-in filters to define the minimum coverage needed for accurate HLA genotyping, although some situations may allow for a lower threshold, such as when the polymorphisms of two alleles of a locus are phased, or when the region with low coverage, such as introns and untranslated genomic regions, does not affect the HLA typing. An important concern for HLA typing data analysis is the evaluation of adequate allele balance to detect issues related to allele dropout, caused either by preferential amplification due to technical issues or the patient’s disease state whereby one of the two alleles has been eradicated (loss of heterozygosity).
More recently, novel long-read methods, also known as third-generation sequencing technologies have been established, which generate sequences >10 kb directly from genomic DNA. While early iterations of these methods were challenged with inaccuracies, latest improvements have permitted much higher accuracy and offer the advantage to sequence large DNA fragments in comparison to NGS. Long-read sequencing is particularly advantageous for HLA typing because it would facilitate complete phasing of alleles and mitigate further the challenge of HLA typing ambiguities [58].
It has been established that HLA incompatibility is an important risk factor for early graft loss after SOT and for development of GVHD and lack of graft engraftment after HSCT. As a result of improvement in immunosuppressive protocols, surgical techniques, and the management of peri- and post-operative clinical complications, short-term allograft survival has greatly increased. However, long-term allograft survival is still hampered by the development of chronic rejection [59, 60]. The current hypothesis is that chronic allograft rejection is the result of a common lesion in which different inflammatory triggers such as rejection episodes, ischemia-reperfusion injury, and infection will lead to a similar histological and clinical outcome. Evidently, the process of chronic rejection is mainly the combined effect of an indolent immune response as well as non-immunologic factors [10, 59, 61, 62, 63, 64, 65, 66]. In this regard, a growing body of evidence has demonstrated that chronic rejection is mainly due to the immune response developed against mismatched HLA molecules of the allograft. This is supported by the observation that the main risk factors for the development of chronic rejection after SOT are HLA mismatches between donor and recipient and the severity and frequency of humoral and/or cellular acute rejection episodes. Moreover, the main risk factor for GVHD after HSCT is the presence of HLA mismatches between donor and recipient.
Several studies have shown that the development of de novo donor-specific anti-HLA antibodies is associated with the development of chronic rejection and allograft disfunction after SOT [62, 67, 68, 69, 70]. Related studies have also shown that pre-existing donor-specific anti-HLA antibodies are associated with early allograft dysfunction after SOT [62, 68, 69]. It was previously suggested that the development of de novo donor-specific anti-HLA antibodies was as an epiphenomenon to the activation of the cellular immune response during the development of chronic rejection. However, there is now compelling evidence that donor-specific anti-HLA antibodies play an important role in the chronic rejection process. In this regard, several studies have demonstrated that anti-HLA antibodies induced intracellular signal transduction in both endothelial and epithelial cells resulting in cellular activation, proliferation, and apoptosis as well as the production of cytokines, chemokines, and fibrogenic growth factors [65, 67, 71].
Chronic rejection after SOT and GVHD after HSCT are the result of a failure of current immunosuppressive protocols to control the alloreactive immune response primarily to mismatched HLA molecules. In this regard, it has been suggested that CD4+ T lymphocytes activated through the indirect allorecognition pathway are less responsive to conventional immunosuppression as compared to those activated through the direct allorecognition pathway [72]. To better prevent chronic allograft rejection and GVHD, immunosuppressive protocols that better block the indirect allorecognition pathway are needed. As organ donation rates plateau, extending the life of transplanted organs is of paramount importance. This objective can only be accomplished through better understanding of the immunological processes that occur during the chronic allograft rejection and GVHD.
References
1.Klein J, Sato A. The HLA system. First of two parts. The New England Journal of Medicine. 2000;343(10):702-709
2.Klein J, Sato A. The HLA system. Second of two parts. The New England Journal of Medicine. 2000;343(11):782-786
3.Kim JJ, Fuggle SV, Marks SD. Does HLA matching matter in the modern era of renal transplantation? Pediatric Nephrology. 2021;36(1):31-40
4.Yacoub R, Nadkarni GN, Cravedi P, He JC, Delaney VB, Kent R, et al. Analysis of OPTN/UNOS registry suggests the number of HLA matches and not mismatches is a stronger independent predictor of kidney transplant survival. Kidney International. 2018;93(2):482-490
5.Opelz G, Wujciak T, Dohler B, Scherer S, Mytilineos J. HLA compatibility and organ transplant survival. Collaborative Transplant Study. Reviews in Immunogenetics. 1999;1(3):334-342
6.Organ Procurement and Transplantation Network (OPTN) Database. Available from: https://optn.transplant.hrsa.gov/
7.Huang Y, Dinh A, Heron S, Gasiewski A, Kneib C, Mehler H, et al. Assessing the utilization of high-resolution 2-field HLA typing in solid organ transplantation. American Journal of Transplantation. 2019;19(7):1955-1963
8.Smith AG, Pereira S, Jaramillo A, Stoll ST, Khan FM, Berka N, et al. Comparison of sequence-specific oligonucleotide probe vs next generation sequencing for HLA-A, B, C, DRB1, DRB3/B4/B5, DQA1, DQB1, DPA1, and DPB1 typing: Toward single-pass high-resolution HLA typing in support of solid organ and hematopoietic cell transplant programs. Hla. 2019;94(3):296-306
9.Marino SG, Jaramillo A, Fernández-Viña MA. The human major histocompatibility complex and DNA-based typing of human leukocyte antigens for transplantation. In: O'Gorman MRG, Donnenberg AD, editors. Handbook of Human Immunology. 2nd ed. New York, NY, USA: CRC Press - Taylor & Francis Group; 2008. pp. 541-564
10.Lefaucheur C, Louis K, Philippe A, Loupy A, Coates PT. The emerging field of non-human leukocyte antigen antibodies in transplant medicine and beyond. Kidney International. 2021;100(4):787-798
11.Senev A, Emonds MP, Van Sandt V, Lerut E, Coemans M, Sprangers B, et al. Clinical importance of extended second field high-resolution HLA genotyping for kidney transplantation. American Journal of Transplantation. 2020;20(12):3367-3378
12.Zavyalova D, Abraha J, Rao P, Morris GP. Incidence and impact of allele-specific anti-HLA antibodies and high-resolution HLA genotyping on assessing immunologic compatibility. Human Immunology. 2021;82(3):147-154
13.Bover KH. Homoisotransplantation von Epidermis bei eineiigen Zwillingen. Beiträge zur Klinischen Chirurgie. 1927;141:442-447
14.Gorer PA. The genetic and antigenic basis for tumor transplantation. The Journal of Pathology and Bacteriology. 1937;44:691-697
15.Snell GD. Methods for the study of histocompatibility genes. Journal of Genetics. 1948;49(2):87-108
16.Payne R. The development and persistence of leukoagglutinins in parous women. Blood. 1962;19:411-424
17.Terasaki PI, Mandell M, Vandewater J, Edgington TS. Human blood lymphocyte cytotoxicity reactions with allogenic antisera. Annals of the New York Academy of Sciences. 1964;120:322-334
18.Terasaki PI, McClelland JD. Microdroplet assay of human serum cytotoxins. Nature. 1964;204:998-1000
19.Terasaki PI, Mickey MR. Histocompatibility-transplant correlation, reproducibility, and new matching methods. Transplantation Proceedings. 1971;3(2):157-171
20.Terasaki PI, Rich NE. Quantitative determination of antibody and complement directed against lymphocytes. Journal of Immunology. 1964;92:128-138
21.Bach FH, Amos DB. Hu-1: Major histocompatibility locus in man. Science. 1967;156(3781):1506-1508
22.Bach FH, Kisken WA. Predictive value of results of mixed leukocyte cultures for skin allograft survival in man. Transplantation. 1967;5(4, Suppl):1046-1052
23.Williams RC Jr, Emmons JD, Yunis EJ. Studies of human sera with cytotoxic activity. The Journal of Clinical Investigation. 1971;50(7):1514-1524
24.Yunis EJ, Amos DB. Three closely linked genetic systems relevant to transplantation. Proceedings of the National Academy of Sciences of the United States of America. 1971;68(12):3031-3035
26.Thorsby E, Sandberg L, Lindholm A, Kissmeyer-Nielsen F. The HL-A system: Evidence of a third sub-locus. Scandinavian Journal of Haematology. 1970;7(3):195-200
27.Shaw S, Duquesnoy RJ, Smith PL. Population studies of the HLA-linked SB antigens. Immunogenetics. 1981;14(1–2):153-162
28.Shaw S, Johnson AH, Shearer GM. Evidence for a new segregant series of B cell antigens that are encoded in the HLA-D region and that stimulate secondary allogenic proliferative and cytotoxic responses. The Journal of Experimental Medicine. 1980;152(3):565-580
29.Shiina T, Hosomichi K, Inoko H, Kulski JK. The HLA genomic loci map: Expression, interaction, diversity and disease. Journal of Human Genetics. 2009;54(1):15-39
30.Boegel S, Lower M, Bukur T, Sorn P, Castle JC, Sahin U. HLA and proteasome expression body map. BMC Medical Genomics. 2018;11(1):36
31.Pellegrino MA, Belvedere M, Pellegrino AG, Ferrone S. B peripheral lymphocytes express more HLA antigens than T peripheral lymphocytes. Transplantation. 1978;25(2):93-95
32.Yamamoto F, Suzuki S, Mizutani A, Shigenari A, Ito S, Kametani Y, et al. Capturing differential allele-level expression and genotypes of all classical HLA loci and haplotypes by a new capture RNA-Seq method. Frontiers in Immunology. 2020;11:941
33.Yarzabek B, Zaitouna AJ, Olson E, Silva GN, Geng J, Geretz A, et al. Variations in HLA-B cell surface expression, half-life and extracellular antigen receptivity. eLife. 2018;7:e34961
34.MacDonald KS, Fowke KR, Kimani J, Dunand VA, Nagelkerke NJ, Ball TB, et al. Influence of HLA supertypes on susceptibility and resistance to human immunodeficiency virus type 1 infection. The Journal of Infectious Diseases. 2000;181(5):1581-1589
35.Noreen HJ, Yu N, Setterholm M, Ohashi M, Baisch J, Endres R, et al. Validation of DNA-based HLA-A and HLA-B testing of volunteers for a bone marrow registry through parallel testing with serology. Tissue Antigens. 2001;57(3):221-229
36.Leen G, Stein JE, Robinson J, Maldonado Torres H, Marsh SGE. The HLA diversity of the Anthony Nolan register. Hla. 2021;97(1):15-29
38.HLA Nomenclature Database. Available from: https://hla.alleles.org/
39.IPD-IMGT/HLA Database. Available from: https://www.ebi.ac.uk/ipd/imgt/hla/
40.Nunes E, Heslop H, Fernandez-Vina M, Taves C, Wagenknecht DR, Eisenbrey AB, et al. Definitions of histocompatibility typing terms. Blood. 2011;118(23):e180-e183
41.Nunes E, Heslop H, Fernandez-Vina M, Taves C, Wagenknecht DR, Eisenbrey AB, et al. Definitions of histocompatibility typing terms: Harmonization of Histocompatibility Typing Terms Working Group. Human Immunology. 2011;72(12):1214-1216
42.Muller CA, Engler-Blum G, Gekeler V, Steiert I, Weiss E, Schmidt H. Genetic and serological heterogeneity of the supertypic HLA-B locus specificities Bw4 and Bw6. Immunogenetics. 1989;30(3):200-207
43.Voorter CE, van der Vlies S, Kik M, van den Berg-Loonen EM. Unexpected Bw4 and Bw6 reactivity patterns in new alleles. Tissue Antigens. 2000;56(4):363-370
44.Bravo-Egana V, Sanders H, Chitnis N. New challenges, new opportunities: Next generation sequencing and its place in the advancement of HLA typing. Human Immunology. 2021;82(7):478-487
45.Dunn PP. Human leucocyte antigen typing: Techniques and technology, a critical appraisal. International Journal of Immunogenetics. 2011;38(6):463-473
46.Moyer AM, Gandhi MJ. Human leukocyte antigen (HLA) testing in pharmacogenomics. Methods in Molecular Biology. 2022;2547:21-45
47.Allen ES, Yang B, Garrett J, Ball ED, Maiers M, Morris GP. Improved accuracy of clinical HLA genotyping by next-generation DNA sequencing affects unrelated donor search results for hematopoietic stem cell transplantation. Human Immunology. 2018;79(12):848-854
48.Lind C, Ferriola D, Mackiewicz K, Heron S, Rogers M, Slavich L, et al. Next-generation sequencing: The solution for high-resolution, unambiguous human leukocyte antigen typing. Human Immunology. 2010;71(10):1033-1042
49.Marsh SGE. Nomenclature for factors of the HLA system, update July, August and September 2022. International Journal of Immunogenetics. 2022;49(6):385-416
50.Marsh SGE, WHO Nomenclature Committee for Factors of the HLA System. Nomenclature for factors of the HLA system, update July, August and September 2022. Hla. 2022;100(6):673-710
51.Hu T, Chitnis N, Monos D, Dinh A. Next-generation sequencing technologies: An overview. Human Immunology. 2021;82(11):801-811
52.National Marrow Donor Program (NMDP) Policies. Available from: https://bioinformatics.bethematchclinical.org/policies/
53.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research. 1988;16(3):1215
54.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 1977;74(12):5463-5467
55.Tucker T, Marra M, Friedman JM. Massively parallel sequencing: The next big thing in genetic medicine. American Journal of Human Genetics. 2009;85(2):142-154
56.Cornaby C, Schmitz JL, Weimer ET. Next-generation sequencing and clinical histocompatibility testing. Human Immunology. 2021;82(11):829-837
57.Brown NK, Merkens H, Rozemuller EH, Bell D, Bui TM, Kearns J. Reduced PCR-generated errors from a hybrid capture-based NGS assay for HLA typing. Human Immunology. 2021;82(4):296-301
58.Logsdon GA, Vollger MR, Eichler EE. Long-read human genome sequencing and its applications. Nature Reviews. Genetics. 2020;21(10):597-614
59.Loupy A, Lefaucheur C. Antibody-mediated rejection of solid-organ allografts. New England Journal of Medicine. 2018;379(12):1150-1160
60.Tinckam KJ, Chandraker A. Mechanisms and role of HLA and non-HLA alloantibodies. Clinical Journal of the American Society of Nephrology. 2006;1(3):404-414
61.Haas M, Loupy A, Lefaucheur C, Roufosse C, Glotz D, Seron D, et al. The Banff 2017 Kidney Meeting Report: Revised diagnostic criteria for chronic active T cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. American Journal of Transplantation. 2018;18(2):293-307
62.Jaramillo A, Fernandez FG, Kuo EY, Trulock EP, Patterson GA, Mohanakumar T. Immune mechanisms in the pathogenesis of bronchiolitis obliterans syndrome after lung transplantation. Pediatric Transplantation. 2005;9(1):84-93
63.Lefaucheur C, Viglietti D, Mangiola M, Loupy A, Zeevi A. From humoral theory to performant risk stratification in kidney transplantation. Journal of Immunology Research. 2017;2017:5201098
64.Loupy A, Hill GS, Jordan SC. The impact of donor-specific anti-HLA antibodies on late kidney allograft failure. Nature Reviews. Nephrology. 2012;8(6):348-357
65.Maruyama T, Jaramillo A, Narayanan K, Higuchi T, Mohanakumar T. Induction of obliterative airway disease by anti-HLA class I antibodies. American Journal of Transplantation. 2005;5(9):2126-2134
66.Robson KJ, Ooi JD, Holdsworth SR, Rossjohn J, Kitching AR. HLA and kidney disease: From associations to mechanisms. Nature Reviews. Nephrology. 2018;14(10):636-655
67.Jin YP, Jindra PT, Gong KW, Lepin EJ, Reed EF. Anti-HLA class I antibodies activate endothelial cells and promote chronic rejection. Transplantation. 2005;79(3 Suppl):S19-S21
68.Reinsmoen NL, Nelson K, Zeevi A. Anti-HLA antibody analysis and crossmatching in heart and lung transplantation. Transplant Immunology. 2004;13(1):63-71
69.Terasaki PI, Ozawa M. Predicting kidney graft failure by HLA antibodies: A prospective trial. American Journal of Transplantation. 2004;4(3):438-443
70.Valenzuela NM, Reed EF. Antibody-mediated rejection across solid organ transplants: Manifestations, mechanisms, and therapies. The Journal of Clinical Investigation. 2017;127(7):2492-2504
71.Jaramillo A, Smith CR, Maruyama T, Zhang L, Patterson GA, Mohanakumar T. Anti-HLA class I antibody binding to airway epithelial cells induces production of fibrogenic growth factors and apoptotic cell death: A possible mechanism for bronchiolitis obliterans syndrome. Human Immunology. 2003;64(5):521-529
72.Sawyer GJ, Dalchau R, Fabre JW. Indirect T cell allorecognition: A cyclosporin A resistant pathway for T cell help for antibody production to donor MHC antigens. Transplant Immunology. 1993;1(1):77-81
Written By
Andrés Jaramillo and Katrin Hacke
Submitted: 11 January 2023Reviewed: 12 January 2023Published: 27 February 2023
 Open access peer-reviewed chapter
Open access peer-reviewed chapter