Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
Diabetes mellitus is the leading cause of chronic kidney disease (CKD) worldwide, and its rising prevalence explains the significant increase in the number of patients receiving renal replacement therapy. Additionally, CKD represents a major cause of morbidity and mortality in patients with diabetes. Hyperglycemia is the cardinal feature that leads to diabetic nephropathy. In fact, elevated glucose levels initiate several inflammatory and metabolic pathways that result in activation of the renin angiotensin aldosterone system (RAAS) and dysregulation of the tubuloglomerular feedback (TGF). For decades, the standard of care to prevent or delay the progression of diabetic kidney disease has included RAAS inhibitors and optimal blood pressure and glycemic control. Fortunately, newer medications have joined the armamentarium of drugs against diabetic nephropathy. Specifically, we will review sodium glucose cotransporter 2 inhibitors (SGLT2is) and the selective mineralocorticoid receptor antagonist (MRA), finerenone, which have proven to decrease the progression of diabetic kidney disease and cardiovascular risk.
The global burden of chronic kidney disease (CKD) was estimated to be 10–15% in 2017, which accounts for 850 million people living with this condition worldwide [1, 2]. In 2021, the Centers for Disease Control and Prevention (CDC) established that approximately one in seven adults have CKD in the United States [3]. The most common cause of CKD is in fact diabetes.
Another pressing statistic is the mortality attributed to kidney disease. In 2017, CKD ranked as the 12th leading cause of death [1]. However, besides its direct impact in morbidity and mortality, kidney disease is also an independent risk factor for cardiovascular events [4]. Globally, CKD accounted for 61.3 million disability-adjusted life-years (DALYs) with 41.6% of those being directly attributed to cardiovascular disease. DALYs mainly resulted from ischemic heart disease, stroke, and peripheral artery disease [1]. Further, a rising CKD population leads to a higher number of patients with end-stage kidney disease (ESKD). It has been predicted that the incidence of ESKD will increase from 11 to 18% between 2015 and 2030 [5]. These trends, together with higher cardiovascular disease, will intensify the burden on healthcare systems.
Diabetes stands as the primary cause of CKD contributing to approximately half of the cases [6]. Currently, diabetes affects roughly 460 million persons worldwide with a projected increase to around 580 and 700 million cases by 2030 and 2045 respectively [7, 8]. It was estimated that 10.5% of the US population had diabetes in 2018, with a projected increase to 14% by 2030 [9, 10].
Clustering of diabetes and diabetic kidney disease (DKD) has been observed among specific racial and ethnic groups. As such, African Americans, Native Americans, and Latinx have higher prevalence of diabetic nephropathy compared with Caucasians. This is explained not only by epigenetic factors associated with the environment and access to care but also by a genetic component [11]. Although, DKD does not represent a simple Mendelian inheritance, several genes have been identified as potential candidates that may differ between populations. This includes the glucose transporter 2, transforming growth factor β, APOL1, and endothelial nitric oxide synthase genes. Further, studies suggest a relationship between polymorphisms of the angiotensin converting enzyme (ACE) gene and DKD. Specifically, there is evidence that ACE gene may play a role in determining which patients may respond to renin angiotensin aldosterone system (RAAS) blockade therapy [12].
Even though the pathogenesis of DKD involves several hemodynamic and metabolic disorders, hyperglycemia is the cardinal feature of diabetic nephropathy. Elevated blood glucose leads to activation of many biochemical pathways resulting in increased oxidative stress, advanced glycation end-products (AGE), cytokine and growth factor release, and hormonal imbalance [12]. Collectively, these factors affect glomerular endothelial, mesangial, tubular and epithelial cells, or podocytes [11].
Renal hemodynamic changes happen with diabetes onset. Even in the absence of hypertension, animal models have shown an elevated intraglomerular pressure that is mediated by angiotensin II and endothelin [13]. The main mediators between the metabolic and hemodynamic pathways are the transforming growth factor beta (TGF-β) and vascular endothelial growth factor (VEGF) [11].
Angiotensin II is a potent hemodynamic and growth factor mediator. It increases sodium reabsorption and volume expansion in the proximal and distal nephron. In the proximal tubule, angiotensin II directly activates the sodium-hydrogen transporter, thereby increasing proximal sodium reabsorption. In addition, angiotensin II acts on the zona glomerulosa in the adrenal cortex and stimulates the release of aldosterone, which in turn increases distal sodium reabsorption and potassium excretion. Further, this vasoactive hormone causes systemic vasoconstriction through G-protein-coupled receptors. In the glomeruli, the afferent and efferent arteriole resistance increases in response to angiotensin II, although there is a greater effect in the efferent one [14]. Lastly, as a growth factor, this hormone affects the proximal tubular and the mesangial cells through direct and indirect pathways [15].
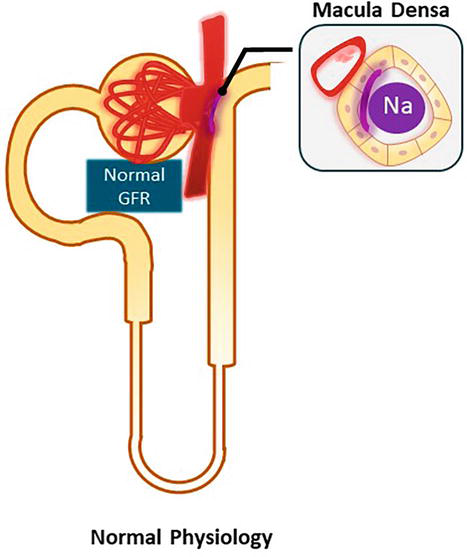
The role of angiotensin II in the tubuloglomerular feedback (TGF) is worth discussing separately. As shown in Figure 1, TGF is an autoregulatory mechanism within each single nephron, where specialized cells that belong to the juxtaglomerular apparatus (JGA) interact to maintain glomerular filtration rate (GFR). The JGA is formed by the macula densa, which is located between the ascending limb of the loop of Henle and the distal tubule, the juxtaglomerular cells of the afferent and efferent arterioles, and the mesangial cells. The macula densa senses distal tubular NaCl concentration. In states of reduced NaCl delivery, the macula densa releases renin resulting in efferent arteriole vasoconstriction [mediated by angiotensin II] and afferent arteriole vasodilation [mediated by a decrease in adenosine, which is a renal vasoconstrictor]. Therefore, in states of volume depletion with resulting decreased distal NaCl delivery, the net effect is an increase in single nephron GFR. In contrast, increased distal NaCl delivery decreases renin levels and stimulates the release of adenosine, which reduces single nephron GFR. Figure 1 shows the normal physiology of the tubuloglomerular feedback [12].
Figure 1.
Tubuloglomerular feedback. This autoregulatory mechanism is present within each single nephron. The macula densa, located in the distal nephron, senses tubular NaCl concentration and regulates the diameter of the renal arterioles to maintain GFR.
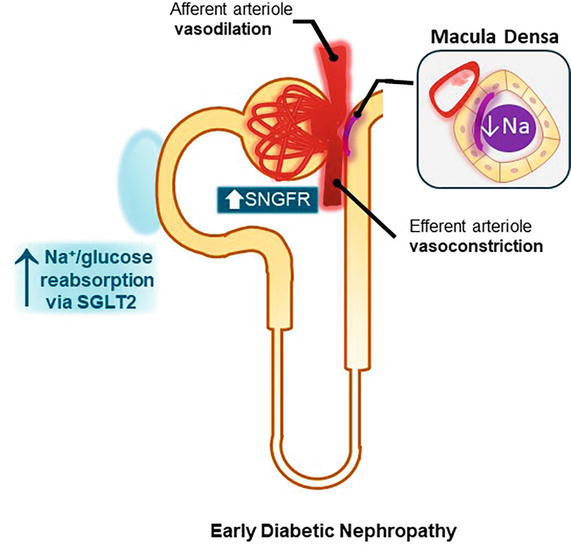
The diabetic milieu alters the tubuloglomerular feedback due to an increased activity of the sodium–glucose cotransporter 2 (SGLT2) in the proximal tubule. Proximal reabsorption of Na+ and glucose via sodium glucose cotransporter 2 leads to a decreased distal delivery of NaCl, which in turn increases single nephron GFR (SNGFR) and leads to hyperfiltration as shown in Figure 2 [12].
Figure 2.
Pathogenesis in diabetic nephropathy. In diabetes, there is an increased activity of the sodium glucose cotransporter 2 (SGLT2). This results in higher proximal NaCl reabsorption leading to a decreased distal delivery of NaCl. This is followed by a release of renin from the juxtaglomerular apparatus, RAAS activation with vasoconstriction of the efferent arteriole, and a decrease in adenosine that causes afferent arteriole vasodilation. The net effect is an increase in single nephron GFR (SNGFR).
DKD is characterized by different phases. Hyperfiltration is the initial stage and is followed by proteinuria in most cases and a subsequent decline in GFR secondary to a loss of nephron mass [16].
Hyperfiltration is defined as a GFR of at least two standard deviations above the mean GFR from its nondiabetic counterparts. It can be present in 10–67% of patients with type 1 and 6–73% of those with type 2 diabetes [11]. Hypertension and obesity frequently coexist with diabetes and contribute to glomerular hyperfiltration by causing glomerular enlargement [17, 18]. This results in increased renal plasma flow, glomerular capillary hyperperfusion, and higher glomerular transcapillary hydrostatic pressure gradient, which collectively lead to an increase in GFR.
The structural lesions of DKD start with thickening of the glomerular basement membrane followed by mesangial expansion and podocyte damage [19, 20, 21]. These changes lead to defects in selective glomerular capillary permeability, albuminuria, and protein extravasation into the mesangium. In fact, segmental mesangiolysis and Kimmelstiel-Wilson nodules are signs of progression of DKD [22, 23]. Interstitial fibrosis and glomerulosclerosis eventually take place manifesting as a decline in GFR [24, 25].
The presence of proteinuria in DKD is the single biggest predictor of CKD progression. Approximately 25% of patients with type 2 diabetes develop moderate albuminuria by 10 years [26]. Further, any degree of albuminuria also increases cardiovascular risk in this population [27, 28, 29]. It is important to note that roughly 20% of patients with diabetes develop DKD in the absence of albuminuria, and they have shown slower rates of CKD progression [30, 31, 32].
Diabetic nephropathy is usually identified after 5 years of diagnosis of type 1 diabetes. Because type 2 diabetes is recognized later in life and tends to be accompanied by elevated blood pressure and poor glycemic control, DKD may be evident at the time of diabetes diagnosis. Despite the differences in timing of recognition of kidney impairment, the clinical manifestations are similar in both types of diabetes [11]. Importantly, only 30–40% of patients with diabetes develop DKD, so nondiabetic glomerulopathies should also be considered in this population [33].
The natural history of DKD and the presence of other microvascular complications, such as retinopathy and/or neuropathy, can help determine the likelihood of kidney involvement. Among those, diabetic retinopathy has perhaps been the most widely used clinical feature. In fact, a population-based study in Wisconsin reported retinopathy in 98% of patients with type 1 diabetes mellitus (DM) compared with 78% of those with type 2 DM by 15 years [34, 35]. Further, in a meta-analysis of 2012 patients, the pooled positive predictive value of diabetic retinopathy for DKD was 0.72 (95% CI 0.68–0.75), while the negative predictive value was 0.69 (95% CI 0.67–0.72) [36]. In other words, diabetic retinopathy is concordant with diabetic nephropathy in almost all patients with type 1 diabetes, but it is less frequent in those with type 2 diabetes.
In regard to diabetic neuropathy, the correlation is less clear. The European Diabetes (EURODIAB) Prospective Complications Study showed that 23.5% of persons with type 1 DM had developed neuropathy by 7 years [37]. Other studies have reported that up to 50% of patients with types 1 and 2 diabetes have diabetic peripheral neuropathy [38].
However, it is important to highlight that the prevalence of nondiabetic kidney disease (NDKD) may vary between 10 and 85% depending on the study [39, 40, 41]. For example, a review of 233 kidney biopsies performed in persons with diabetes identified NDKD in 53% of the cases. The most common conditions observed included focal segmental glomerulosclerosis, minimal change disease, IgA nephropathy, and membranous glomerulonephritis [40].
Therefore, it is of utmost importance to identify when to pursue a kidney biopsy, which remains the gold standard for diagnosis. In general, a biopsy is indicated in the following clinical settings: nephrotic range proteinuria or GFR decline in the absence of diabetic retinopathy, onset of proteinuria less than 5 years from the diagnosis of type 1 diabetes, nephrotic range proteinuria with normal kidney function, active urinary sediment, such as microscopic hematuria, rapid decline in kidney function in patients with previously stable CKD [11].
The treatment of DKD is focused on four major pillars: cardiovascular risk reduction, blood pressure and glycemic control, and inhibition of the renin angiotensin aldosterone system (RAAS). Cardiovascular risk reduction is achieved by proper blood pressure and glycemic control, management of dyslipidemia, and smoking cessation [11].
5.1 Blood pressure control
Blood pressure goals in patients with diabetes have been highly debated. The Action in Diabetes and Vascular Disease: PreterAx and Diamicron-MR Controlled Evaluation (ADVANCE) trial showed a 21% risk reduction of kidney endpoints with systolic blood pressure levels below 135 mmHg compared with those values ~140 mm Hg [42]. Further, a review of studies showed that less GFR decline is achieved at a mean arterial pressure of 89 mm Hg (equivalent to a systolic blood pressure of 120 mm Hg and a diastolic of 75 mm Hg) [43].
The KDIGO guidelines recommend a blood pressure goal below 140/90 mmHg for DKD and a lower goal below 130/80 mmHg in the presence of albuminuria [44].
5.2 Glycemic control
Optimal glycemic control reduces CKD progression, cardiovascular events, and death. Most studies focus on targeting the hemoglobin A1c (HbA1c) since it is a surrogate of the 3-month average blood glucose. However, data have also shown that glucose variability predicts worse kidney and cardiovascular outcomes [45].
In patients with type 1 diabetes, the Diabetes Control and Complications Trial (DCCT) showed that intensive glycemic control (goal HbA1c < 6.05%) conferred a lower risk for the development of microalbuminuria by 39% and by 56% for overt proteinuria. Importantly, the median HbA1c concentration was 9.1% in the conventional group compared with 7.3% in the intensive group. Other microvascular complications were also lower in the intensive control arm [46].
In regard to patients with type 2 diabetes, the United Kingdom Prospective Diabetes Study (UKPDS) showed a 33% reduction in albuminuria in the group that achieved a median HbA1c of 7% compared with 7.9% in the conventional arm [47].
Further studies addressing whether lower A1c goals would have an additional benefit have shown mixed results. The ADVANCE, Action to Control Cardiovascular Risk in Diabetes (ACCORD), and VA Diabetes Trial (VADT) targeted and achieved HbA1c levels of ∼6.0% compared with a control arm of ∼7.0% [48, 49, 50]. ADVANCE trial showed positive kidney outcomes but no difference in cardiovascular events [48]. Contrarily, ACCORD and VADT established no cardiovascular or kidney benefit [49, 50].
Taking all this information together, the KDIGO guidelines recommend an individualized HbA1c target ranging from < 6.5 to < 8% [44].
5.3 Renin angiotensin aldosterone blockade
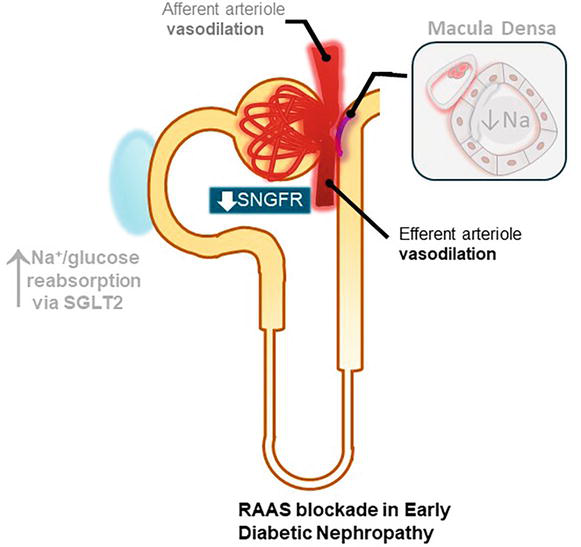
RAAS blockade has been the single most effective therapy for slowing the progression of DKD for decades, and it has been studied across all stages of diabetic nephropathy. As explained in the pathogenesis, RAAS is activated in diabetic nephropathy resulting in vasoconstriction of the efferent arteriole, which plays a role in increasing SNGFR. Figure 3 shows the nephroprotective effect of RAAS [12].
Figure 3.
Hyperfiltration in early diabetic nephropathy with concomitant RAAS blockade. Angiotensin II is a potent vasoconstrictor of both renal arterioles but predominantly of the efferent one. Therefore, Renin Angiotensin Aldosterone System (RAAS) blockade results in vasodilation of the efferent arteriole and decreases single nephron GFR (SNGFR).
In initial stages when albuminuria has not occurred, the use of RAAS inhibitors is controversial. In fact, studies conducted in patients with Type 1 diabetes have failed to show that RAAS blockade prevents the development of microalbuminuria, although this was a secondary endpoint [51, 52]. On the other hand, the Bergamo Nephrologic Diabetes Complications (BENEDICT) and the Randomized Olmesartan and Diabetes Microalbuminuria Prevention (ROADMAP) Trials showed that therapy prevented or delayed the development of microalbuminuria independent of blood pressure reduction [53, 54]. In 2016, a meta-analysis of 16921 patients further confirmed a potential role for RAAS blockade in the prevention of DKD, establishing a 16% risk reduction of development of microalbuminuria in patients with type 2 diabetes [55]. In regard to the cardiovascular risk associated to diabetes, the Health Outcomes and Prevention Evaluation (HOPE) study established that ACE inhibition decreased the risk of cardiovascular death, stroke, and myocardial infarction in patients with and without kidney disease [56]. Overall, RAAS blockade appears to play a role in prevention of DKD and cardiovascular protection irrespective of nephropathy. It is worth mentioning that current guidelines do not yet recommend it for the prevention of DKD [44].
Once hyperfiltration ensues and microalbuminuria develops, patients are at risk for transitioning from microalbuminuria to overt proteinuria. The Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria (IRMA-2) trial showed a dose-dependent effect favoring the use of irbesartan over placebo for the development of overt proteinuria, which was defined as ACR > 200 mg/day [57]. Similarly, in the Irbesartan Diabetic Nephropathy Trial (IDNT), RAAS blockade had superior kidney outcomes, independent of the effect on blood pressure, when compared with both the amlodipine and the placebo groups [58].
Maintaining RAAS blockade during the last stage of DKD where there is a progressive decline in GFR is still recommended. The Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan (RENAAL) study showed a 25% risk reduction of doubling of serum creatinine, 28% reduction of risk of ESKD, and 16% reduction in the composite endpoint, which was defined as doubling of serum creatinine or ESKD or death after 3.4 years of follow-up. Moreover, the therapy group showed 35% reduction of proteinuria assessed as secondary endpoint [59].
Another important clinical question is whether dual blockade of the RAAS provides additional benefit. Landmark trials including the ONTARGET, VA NEPHRON-D and the ALTITUDE presented safety concerns due to significant rates of hyperkalemia, kidney injury, or hypotension [60, 61, 62]. Collectively, these studies suggest avoiding dual blockade of the RAAS system in type 2 diabetes.
The twenty-first century began with this overwhelming evidence favoring the use of RAAS inhibitors across all clinical phenotypes of DKD. Unfortunately, the prevalence of RAAS blockade agents use decreased from 45% in 2006–2008 to 36% in 2012–2014 [63].
GLP-1 receptor agonists, also called incretins, are part of the arsenal of medications for the treatment of diabetes since 2005. However, only since 2019 an oral formulation has been available and approved by the FDA. GLP-1 receptor agonists initially gained popularity because of their ability to induce weight loss. They act by stimulating glucose-dependent insulin secretion and decrease glucagon secretion, gastric emptying, and appetite [64].
Another positive effect of GLP-1 receptor agonists is their cardiovascular protection. A meta-analysis of 56,004 patients with diabetes showed a significant 12% relative risk reduction in major adverse cardiovascular events [65]. In 2021, the Cardiovascular and Renal Outcomes with Efpeglenatide in Type 2 Diabetes (AMPLITUDE-O) trial showed a 27% risk reduction of incident major adverse cardiovascular events in the efpeglenatide group. Even though the kidney outcomes were a secondary endpoint, a composite kidney outcome of decrease in kidney function or macroalbuminuria was 32% lower in the medication arm [66].
The sodium glucose cotransporter 2 is responsible for the uptake of roughly 90% of glucose and majority of the sodium that is reabsorbed in the proximal tubule [12].
It has been shown that SGLT2is significantly reduce the renal threshold for glucose excretion from ~10 mmol/l (180 mg/dl) to ~ 1.2 mmol/L (21 mg/dL) in diabetic and to ~2 mmol/L (37 mg/dL) in nondiabetic subjects [67]. As a result, a better glycemic control addresses hyperglycemia, which is the main driving factor of the diabetic milieu. It is worth mentioning that glucose uptake by kidney cells is not insulin-dependent, so the effect of SGLT2is is present even in states of insulin deficiency [12].
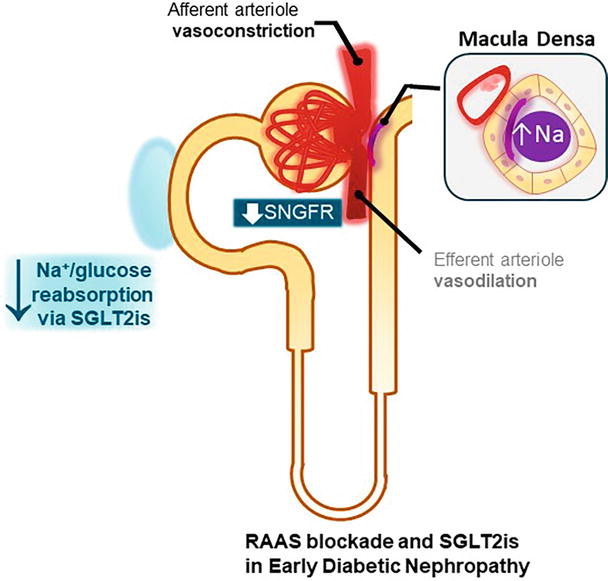
In addition to their effect on glucosuria, the benefits of the SGLT2is result from their natriuretic action and the restoration of the tubuloglomerular feedback. The increased distal delivery of sodium ameliorates the altered TGF characteristic of diabetic nephropathy by decreasing SNGFR, hyperfiltration, and intraglomerular pressure as shown in Figure 4 [68].
Figure 4.
Effect of RAAS blockade and SGLT2is in diabetic nephropathy. Sodium glucose cotransporter 2 inhibitors (SGLT2is) increase the distal delivery of NaCl to the macula densa, thereby restoring the tubuloglomerular feedback. Renin angiotensin aldosterone system (RAAS) blockade decreases single nephron GFR (SNGFR) through vasodilation of the efferent arteriole; however, the effect of SGLT2is lowers SNGFR further through vasoconstriction of the afferent arteriole.
Cardiovascular outcome trials with SGLT2is inhibitors, although not powered for secondary (renal) endpoints, showed beneficial renal effects by 40–70% [69, 70, 71, 72]. Notably, all these studies included patients with CKD, but most of them had early, if any, kidney disease. The Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes (EMPA-REG OUTCOME) trial included subjects with a mean eGFR of 74 mL/min/1.73 m2, and 60% of the patients enrolled did not have albuminuria [69, 70]. Further, the Canagliflozin Cardiovascular Assessment Study (CANVAS) observed patients with a mean eGFR of 76.5 mL/min/1.73 m2 and a median UACR of 12 mg/g [71]. Similarly, the Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes (DECLARE-TIMI 58) included patients with a mean eGFR of 85 mL/min/1.73 m2 while UACR data was not provided [72].
The Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy (CREDENCE) was the first study to establish primary renal endpoints. It included patients with stage 3 CKD with a mean eGFR of 56 mL/min/1.73 m2 and a median UACR of 927 mg/g. The CREDENCE trial showed a significant 30% risk reduction in the composite renal outcome without significant adverse events. This trial led to the FDA approval of canagliflozin for DKD [73].
In 2020, the Dapagliflozin in Patients with Chronic Kidney Disease (DAPA-CKD) trial showed a 39% risk reduction of the primary composite renal outcome in the dapagliflozin arm. Of note, the positive effect was observed across all subgroups including those with and without diabetes. The mean eGFR in this study was 43 mL/min/1.73 m2 with a median UACR of 965 mg/g [73].
Neither CREDENCE nor DAPA-CKD showed an increased risk of amputations, hyperkalemia, or hypoglycemia. However, CREDENCE showed a significantly higher risk of diabetic ketoacidosis in the canagliflozin group. This was not observed in the DAPA-CKD [73, 74]. In addition, patients treated with dapagliflozin developed more urinary tract infections and genital infections compared with placebo.
Randomized controlled trials with dedicated kidney outcomes have shown that SGLT2is are beneficial and safe in patients with CKD and proteinuria, regardless of diabetes status [73, 74]. Even though the majority of patients from the cardiovascular outcome trials included patients without proteinuria, subgroup analysis was not provided [69, 70, 71, 72]. Notably, the lowest GFR studied thus far is 20 mL/min/1.73 m2 in the EMPEROR- reduced trial, which also showed positive secondary kidney endpoints [75]. Finally, an ongoing clinical trial, The Study of Heart and Kidney Protection with Empagliflozin (EMPA-KIDNEY) with primary renal and cardiovascular outcomes also enrolled patients with an eGFR ≥ 20 mL/min/1.73 m2. [NCT03594110]
Table 1 summarizes trials of SGLT2is and selective mineralocorticoid receptor antagonist in DKD patients.
Trial
Patient population
Intervention
Primary endpoint
Follow-up (median, years)
Results
EMPA-REG OUTCOME (2015)
N = 7020 T2DM and CVD GFR> 30 No UACR criteria 60% had a UACR < 30
Empagliflozin vs Placebo
MACE
3.1
Primary outcome: HR 0.86, 95% CI: 0.74–0.99 Secondary (renal) outcome: Incident or worsening nephropathy 39% lower in the empagliflozin group HR 0.61; 95% CI: 0.53–0.70
CANVAS (2017)
N = 10,142 T2DM and CVD GFR ≥ 30 No UACR criteria 70% had a UACR < 30
Canagliflozin vs Placebo
MACE
2.4
Primary outcome: MACE: HR 0.86, 95% CI: 0.75–0.97
Secondary (renal) outcomes: Composite 40% reduction in GFR, renal replacement therapy or death from renal causes: HR 0.60; 95% CI, 0.47 to 0.77
Progression of albuminuria HR 0.73, 95% CI: 0.67–0.79
CREDENCE (2019)
N = 4401 T2DM GFR 30–90 UACR 300–5000 Maxim tolerated RAS blockade
Canagliflozin vs Placebo
Composite of ESKD, doubling of serum creatinine, or kidney/CV related death
2.6
Primary outcome: HR 0.70, 95% CI: 0.59–0.82
DECLARE-TIMI 58 (2019)
N = 17,160 T2DM and CVD GFR ≥ 60 No UACR criteria
Dapagliflozin vs Placebo
MACE; Composite CV death or hospitalization for HF
4.2
Primary outcome: MACE: HR 0.93, 95% CI: 0.84–1.03 Composite CV death or hospitalization for HF HR 0.83, 95% CI: 0.73 – 0.95
Secondary (renal) outcome: Composite of >40% decrease in GFR to < 60 ml/min, ESKD, or renal death: HR 0.53, 95% CI: 0.43–0.66
DAPA-CKD (2020)
N = 4304 T2DM: 67% GFR 25 to 75 UACR 200 to 5000
Dapagliflozin vs Placebo
Composite of sustained decline in GFR of at least 50%, ESKD, or death from kidney or cardiovascular causes
2.4
Primary outcome: HR 0.61, 95% CI: 0.51–0.72
Primary outcome according to subgroups: T2DM: HR 0.64 (95% CI: 0.52–0.79) No T2DM: HR 0.50 (95% CI: 0.35–0.72)
EMPEROR-REDUCED (2020)
N = 3730 HFrEF GFR ≥ 20 No UACR criteria
Empagliflozin vs Placebo
Composite of CV death or hospitalization for HF
1.3
Primary outcome: HR 0.75, 95% CI: 0.65–0.86
Secondary (renal) outcomes: Mean slope of change in GFR (hierarchical testing): HR 1.73 (95% CI: 1.10–2.37) Composite renal outcome: HR 0.50 (95% CI: 0.32–0.77)
FIDELIO-DKD (2020)
N = 5734 T2DM GFR 25 to 75 UACR 30 to 5000
Finerenone vs Placebo
Composite of kidney failure, a sustained GFR decrease of at least 40%, or death from renal causes
Recent chronic kidney disease randomized clinical trials of SGLT2is and selective mineralocorticoid receptor antagonist.
MACE: Major Advere Cardiovascular Events, T2DM: Type 2 Diabetes Mellitus, CVD: Cardio Vascular Disease, and HFrEF: Heart Failure with reduced Ejection Fraction, GFR: Glomerular Filtration Rate (mL/min/1.73 m2), UACR: Urine Albumin to Creatinine Ratio (mg/g).
Nonetheless, it is still recommended to avoid STGL2is in patients with recurrent genito-urinary tract infections, patients with urinary bladder catheterization or suprapubic catheters, or when there is a concern for volume depletion or prolonged fasting.
Aldosterone binds to the mineralocorticoid receptor (MR) of principal cells in the cortical collecting duct of the kidney and activates the epithelial sodium channel (ENaC), which will regulate salt excretion, extracellular volume, and blood pressure. MR is also present in nonepithelial cells such as cardiomyocytes, endothelial cells, vascular smooth muscle cells, mesangial cells, and podocytes. Importantly, cortisol also has high affinity to MR in nonepithelial cells due to the lack of the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11βHSD-2), which converts cortisol to cortisone, the nonactive form. Activation of the mineralocorticoid receptor stimulates the formation of reactive oxygen species, endothelial exocytosis, and adhesion. These sequences stimulate an inflammatory response in adipocytes that contributes to insulin resistance by increasing oxidative stress. Lastly, inflammation can eventually lead to fibrosis and remodeling in multiple organs including the heart, vasculature, and kidneys [76].
As such, a novel selective MR antagonist has also been added to the armamentarium against DKD. Finerenone and eplerenone are selective MR antagonists. However, finerenone is equally saturated in the kidneys and heart tissues, while eplerenone is more saturated in the kidneys. Spironolactone is a nonselective MR antagonist, which is also more saturated in the kidneys and can bind with the estrogen receptors in the mammary glands causing gynecomastia. By the end of 2020, the Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes (FIDELIO-DKD) trial showed promising renal outcome. Finerenone had a significant 18% reduction of a primary composite of kidney failure, sustained decreased of at least 40% in eGFR, and death from renal causes. Further, it showed a 31% greater reduction in albuminuria by month 4 compared with placebo [77].
In FIDELIO-DKD trial, the incidence of all serious adverse events was similar in both groups; however, the incidence of hyperkalemia leading to discontinuation of the trial regimen was higher in the finerenone group compared with placebo (2.3% vs. 0.9%, respectively) [77].
The positive kidney and cardiovascular outcomes seen in the FIDELIO-DKD persisted irrespective of SGLT2 inhibitor status [78].
The major therapeutic breakthroughs for DKD have led to updates in the current guidelines. The KDIGO 2020 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease recommends the use of SGLT2is in patients with Type 2 diabetes, CKD and GFR > 30 ml/min/1.73 m2. KDIGO also suggests initiating GLP-1 receptor agonists in patients with type 2 diabetes mellitus and CKD if glycemic targets are not met despite using metformin and SGLT2is or if unable to use those medications [44].
Similarly, the American Diabetes Association (ADA) recommends using GLP1-receptor agonists or SGLT2is in persons with type 2 diabetes and cardiovascular disease. However, when CKD or heart failure are predominant, SGLT2is are the preferred option [79].
We expect those guidelines to incorporate finerenone in the management of DKD given its beneficial role in CKD progression and cardiovascular outcomes in patients with diabetes mellitus Type 2.
Dr. Hanouneh is a speaker for AstraZeneca and Bayer.
Dr. Cervantes has no conflicts of interest.
References
1.Collaboration GCKD. Global, regional, and national burden of chronic kidney disease, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2020;395(10225):709-733
2.Jager KJ, Kovesdy C, Langham R, Rosenberg M, Jha V, Zoccali C. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Nephrology, Dialysis, Transplantation. 2019;34(11):1803-1805
3.Centers for Disease Control and Prevention. Chronic kidney disease in the United States. 2021. Available from: https://www.cdc.gov/kidneydisease/publications-resources/ckd-national-facts.html
4.Jankowski J, Floege J, Fliser D, Böhm M, Marx N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation. 2021;143(11):1157-1172
5.McCullough KP, Morgenstern H, Saran R, Herman WH, Robinson BM. Projecting ESRD Incidence and Prevalence in the United States through 2030. Journalof American Society of Nephrology. 2019;30(1):127-135
6.Xie Y, Bowe B, Mokdad AH, Xian H, Yan Y, Li T, et al. Analysis of the Global Burden of Disease study highlights the global, regional, and national trends of chronic kidney disease epidemiology from 1990 to 2016. Kidney International. 2018;94(3):567-581
7.Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9. Diabetes Research and Clinical Practice. 2019;157:107843
8.Lin X, Xu Y, Pan X, Xu J, Ding Y, Sun X, et al. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Scientific Reports. 2020;10(1):14790
9.Centers for Disease Control. Chronic Kidney Disease Surveillance System. 2021
10.Lin J, Thompson TJ, Cheng YJ, Zhuo X, Zhang P, Gregg E, et al. Projection of the future diabetes burden in the United States through 2060. Population Health Metrics. 2018;16(1):9
12.DeFronzo RA, Reeves WB, Awad AS. Pathophysiology of diabetic kidney disease: Impact of SGLT2 inhibitors. Nature Reviews in Nephrology. 2021;17(5):319-334
13.Cooper ME. Pathogenesis, prevention, and treatment of diabetic nephropathy. Lancet. 1998;352(9123):213-219
14.Denton KM, Anderson WP, Sinniah R. Effects of angiotensin II on regional afferent and efferent arteriole dimensions and the glomerular pole. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2000;279(2):R629-R638
15.Wolf G, Neilson EG. Angiotensin II as a renal growth factor. Journal of American Society of Nephrology. 1993;3(9):1531-1540
16.Mogensen CE. Prediction of clinical diabetic nephropathy in IDDM patients. Alternatives to microalbuminuria? Diabetes. 1990;39(7):761-767
17.Grabias BM, Konstantopoulos K. The physical basis of renal fibrosis: Effects of altered hydrodynamic forces on kidney homeostasis. American Journal of Physiology. Renal Physiology. 2014;306(5):F473-F485
18.Premaratne E, Verma S, Ekinci EI, Theverkalam G, Jerums G, MacIsaac RJ. The impact of hyperfiltration on the diabetic kidney. Diabetes & Metabolism. 2015;41(1):5-17
19.Caramori ML, Parks A, Mauer M. Renal lesions predict progression of diabetic nephropathy in type 1 diabetes. Journal of American Society ofNephrology. 2013;24(7):1175-1181
20.Fioretto P, Mauer M. Histopathology of diabetic nephropathy. Seminars in Nephrology. 2007;27(2):195-207
21.Tyagi I, Agrawal U, Amitabh V, Jain AK, Saxena S. Thickness of glomerular and tubular basement membranes in preclinical and clinical stages of diabetic nephropathy. Indian Journal of Nephrology. 2008;18(2):64-69
22.Saito Y, Kida H, Takeda S, Yoshimura M, Yokoyama H, Koshino Y, et al. Mesangiolysis in diabetic glomeruli: Its role in the formation of nodular lesions. Kidney International. 1988;34(3):389-396
23.Stout LC, Kumar S, Whorton EB. Focal mesangiolysis and the pathogenesis of the Kimmelstiel-Wilson nodule. Human Pathology. 1993;24(1):77-89
24.PFP R. Diabetic nephropathy. In: Brenner and Rector’s The Kidney. 10th ed. PA, USA: Elsevier
25.Tervaert TW, Mooyaart AL, Amann K, Cohen AH, Cook HT, Drachenberg CB, et al. Pathologic classification of diabetic nephropathy. Journal of American Society of Nephrology. 2010;21(4):556-563
27.de Boer IH, Gao X, Cleary PA, Bebu I, Lachin JM, Molitch ME, et al. Albuminuria changes and cardiovascular and renal outcomes in Type 1 Diabetes: The DCCT/EDIC Study. Clinical Journal of the American Society of Nephrology. 2016;11(11):1969-1977
28.Nelson RG, Bennett PH, Beck GJ, Tan M, Knowler WC, Mitch WE, et al. Development and progression of renal disease in Pima Indians with non-insulin-dependent diabetes mellitus. Diabetic Renal Disease Study Group. The New England Journal of Medicine. 1996;335(22):1636-1642
29.Adler AI, Stevens RJ, Manley SE, Bilous RW, Cull CA, Holman RR, et al. Development and progression of nephropathy in type 2 diabetes: The United Kingdom Prospective Diabetes Study [UKPDS 64]. Kidney International. 2003;63(1):225-232
30.Caramori ML, Fioretto P, Mauer M. Low glomerular filtration rate in normoalbuminuric type 1 diabetic patients: An indicator of more advanced glomerular lesions. Diabetes. 2003;52(4):1036-1040
31.Mottl AK, Kwon KS, Mauer M, Mayer-Davis EJ, Hogan SL, Kshirsagar AV. Normoalbuminuric diabetic kidney disease in the U.S. population. Journal of Diabetes and its Complications. 2013;27(2):123-127
32.Thomas MC, Macisaac RJ, Jerums G, Weekes A, Moran J, Shaw JE, et al. Nonalbuminuric renal impairment in type 2 diabetic patients and in the general population [national evaluation of the frequency of renal impairment cO-existing with NIDDM [NEFRON] 11]. Diabetes Care. 2009;32(8):1497-1502
33.Umanath K, Lewis JB. Update on diabetic nephropathy: Core Curriculum 2018. American Journal of Kidney Diseases. 2018;71(6):884-895
34.Klein R, Klein BE, Moss SE, Davis MD, DeMets DL. The Wisconsin epidemiologic study of diabetic retinopathy. III. Prevalence and risk of diabetic retinopathy when age at diagnosis is 30 or more years. Archives of Ophthalmology. 1984;102(4):527-532
35.Klein R, Zinman B, Gardiner R, Suissa S, Donnelly SM, Sinaiko AR, et al. The relationship of diabetic retinopathy to preclinical diabetic glomerulopathy lesions in type 1 diabetic patients: The Renin-Angiotensin System Study. Diabetes. 2005;54(2):527-533
36.He F, Xia X, Wu XF, Yu XQ , Huang FX. Diabetic retinopathy in predicting diabetic nephropathy in patients with type 2 diabetes and renal disease: A meta-analysis. Diabetologia. 2013;56(3):457-466
37.Tesfaye S, Chaturvedi N, Eaton SE, Ward JD, Manes C, Ionescu-Tirgoviste C, et al. Vascular risk factors and diabetic neuropathy. The New England Journal of Medicine. 2005;352(4):341-350
38.Tesfaye S, Selvarajah D. Advances in the epidemiology, pathogenesis and management of diabetic peripheral neuropathy. Diabetes/Metabolism Research and Reviews. 2012;28(Suppl 1):8-14
39.Zhuo L, Ren W, Li W, Zou G, Lu J. Evaluation of renal biopsies in type 2 diabetic patients with kidney disease: A clinicopathological study of 216 cases. International Urology and Nephrology. 2013;45(1):173-179
40.Pham TT, Sim JJ, Kujubu DA, Liu IL, Kumar VA. Prevalence of nondiabetic renal disease in diabetic patients. American Journal of Nephrology. 2007;27(3):322-328
41.Oh SW, Kim S, Na KY, Chae DW, Jin DC, Chin HJ. Clinical implications of pathologic diagnosis and classification for diabetic nephropathy. Diabetes Research and Clinical Practice. 2012;97(3):418-424
42.Patel A, MacMahon S, Chalmers J, Neal B, Woodward M, Billot L, et al. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus [the ADVANCE trial]: A randomised controlled trial. Lancet. 2007;370(9590):829-840
43.Yamout H, Lazich I, Bakris GL. Blood pressure, hypertension, RAAS blockade, and drug therapy in diabetic kidney disease. Advances in Chronic Kidney Disease. 2014;21(3):281-286
44.Group KDIGOKDW. KDIGO 2020 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney International. 2020;98(4S):S1-S115
45.Wadén J, Forsblom C, Thorn LM, Gordin D, Saraheimo M, Groop PH, et al. A1C variability predicts incident cardiovascular events, microalbuminuria, and overt diabetic nephropathy in patients with type 1 diabetes. Diabetes. 2009;58(11):2649-2655
46.Nathan DM, Genuth S, Lachin J, Cleary P, Crofford O, Davis M, et al. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The New England Journal of Medicine. 1993;329(14):977-986
47.Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes [UKPDS 33]. UK Prospective Diabetes Study [UKPDS] Group. Lancet. 1998;352(9131):837-853
48.Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. The New England Journal of Medicine. 2008;358(24):2560-2572
49.Gerstein HC, Miller ME, Byington RP, Goff DC, Bigger JT, Buse JB, et al. Effects of intensive glucose lowering in type 2 diabetes. The New England Journal of Medicine. 2008;358(24):2545-2559
50.Duckworth W, Abraira C, Moritz T, Reda D, Emanuele N, Reaven PD, et al. Glucose control and vascular complications in veterans with type 2 diabetes. The New England Journal of Medicine. 2009;360(2):129-139
51.Bilous R, Chaturvedi N, Sjølie AK, Fuller J, Klein R, Orchard T, et al. Effect of candesartan on microalbuminuria and albumin excretion rate in diabetes: Three randomized trials. Annals of Internal Medicine. 2009;151(1):11-20
52.Mauer M, Zinman B, Gardiner R, Suissa S, Sinaiko A, Strand T, et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. The New England Journal of Medicine. 2009;361(1):40-51
53.Ruggenenti P, Fassi A, Ilieva AP, Bruno S, Iliev IP, Brusegan V, et al. Preventing microalbuminuria in type 2 diabetes. The New England Journal of Medicine. 2004;351(19):1941-1951
54.Haller H, Ito S, Izzo JL, Januszewicz A, Katayama S, Menne J, et al. Olmesartan for the delay or prevention of microalbuminuria in type 2 diabetes. The New England Journal of Medicine. 2011;364(10):907-917
55.Persson F, Lindhardt M, Rossing P, Parving HH. Prevention of microalbuminuria using early intervention with renin-angiotensin system inhibitors in patients with type 2 diabetes: A systematic review. Journal of the Renin-Angiotensin-Aldosterone System. 2016;17(3)
56.Mann JF, Gerstein HC, Pogue J, Bosch J, Yusuf S. Renal insufficiency as a predictor of cardiovascular outcomes and the impact of ramipril: The HOPE randomized trial. Annals of Internal Medicine. 2001;134(8):629-636
57.Parving HH, Lehnert H, Bröchner-Mortensen J, Gomis R, Andersen S, Arner P, et al. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. The New England Journal of Medicine. 2001;345(12):870-878
58.Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. The New England Journal of Medicine. 2001;345(12):851-860
59.Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. The New England Journal of Medicine. 2001;345(12):861-869
60.Mann JF, Schmieder RE, McQueen M, Dyal L, Schumacher H, Pogue J, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk [the ONTARGET study]: A multicentre, randomised, double-blind, controlled trial. Lancet. 2008;372(9638):547-553
61.Parving HH, Brenner BM, McMurray JJ, de Zeeuw D, Haffner SM, Solomon SD, et al. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. The New England Journal of Medicine. 2012;367(23):2204-2213
62.Fried LF, Emanuele N, Zhang JH, Brophy M, Conner TA, Duckworth W, et al. Combined angiotensin inhibition for the treatment of diabetic nephropathy. The New England Journal of Medicine. 2013;369(20):1892-1903
63.Tummalapalli SL, Powe NR, Keyhani S. Trends in Quality of Care for Patients with CKD in the United States. Clinical Journal of the American Society of Nephrology. 2019;14(8):1142-1150
64.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132(6):2131-2157
65.Kristensen SL, Rørth R, Jhund PS, Docherty KF, Sattar N, Preiss D, et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. The Lancet Diabetes and Endocrinology. 2019;7(10):776-785
66.Gerstein HC, Sattar N, Rosenstock J, Ramasundarahettige C, Pratley R, Lopes RD, et al. Cardiovascular and renal outcomes with Efpeglenatide in Type 2 Diabetes. The New England Journal of Medicine. 2021;385(10):896-907
67.DeFronzo RA, Hompesch M, Kasichayanula S, Liu X, Hong Y, Pfister M, et al. Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care. 2013;36(10):3169-3176
68.Tsimihodimos V, Filippatos TD, Elisaf MS. SGLT2 inhibitors and the kidney: Effects and mechanisms. Diabetes and Metabolic Syndrome: Clinical Research and Reviews. 2018;12(6):1117-1123
69.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in Type 2 Diabetes. The New England Journal of Medicine. 2015;373(22):2117-2128
70.Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, et al. Empagliflozin and progression of kidney disease in Type 2 Diabetes. The New England Journal of Medicine. 2016;375(4):323-334
71.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in Type 2 Diabetes. The New England Journal of Medicine. 2017;377(7):644-657
72.Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in Type 2 Diabetes. The New England Journal of Medicine. 2019;380(4):347-357
73.Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canagliflozin and renal outcomes in Type 2 diabetes and nephropathy. The New England Journal of Medicine. 2019;380(24):2295-2306
74.Heerspink HJL, Stefánsson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, et al. Dapagliflozin in patients with chronic kidney disease. The New England Journal of Medicine. 2020;383(15):1436-1446
75.Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. The New England Journal of Medicine. 2020;383(15):1413-1424
76.Grune J, Beyhoff N, Smeir E, Chudek R, Blumrich A, Ban Z, et al. Selective mineralocorticoid receptor cofactor modulation as molecular basis for Finerenone’s antifibrotic activity. Hypertension. 2018;71(4):599-608
77.Bakris GL, Agarwal R, Anker SD, Pitt B, Ruilope LM, Rossing P, et al. Effect of finerenone on chronic kidney disease outcomes in Type 2 diabetes. The New England Journal of Medicine. 2020
78.Rossing P, Filippatos G, Agarwal R, Anker SD, Pitt B, Ruilope LM, et al. Finerenone in predominantly advanced CKD and Type 2 diabetes with or without sodium-glucose Cotransporter-2 inhibitor therapy. Kidney International Reports. 2022;7(1):36-45
79.Draznin B, Aroda VR, Bakris G, Benson G, Brown FM, Freeman R, et al. Pharmacologic approaches to glycemic treatment: Standards of Medical Care in Diabetes-2022. Diabetes Care. 2022;45(Supplement_1):S125-SS43
Written By
Carmen Elena Cervantes and Mohamad Hanouneh
Submitted: 14 March 2022Reviewed: 24 March 2022Published: 06 July 2022
 Open access peer-reviewed chapter
Open access peer-reviewed chapter