
Abstract
Zebrafish are becoming a popular experimental animal model for vision science and human-inherited retinal diseases. In this chapter, we describe application of zebrafish for the retinitis pigmentosa (RP) caused by digenic LDL receptor-related protein 5 (LRP5) and Eyes shut homolog (EYS). RP is the most common genetic disorder in inherited retinal diseases, and EYS is one of the major causes of RP. EYS orthologs are absent in rodents but present in zebrafish. Using this advantage, we generated and analyzed the digenic eys+/−; low-density lipoprotein (LDL)-related receptor-5 (lrp5)+/− zebrafish, the same form of gene defects emerged from a human case report as a candidate of RP. The analysis discovers that retinol binding protein 1a (rbp1a) gene is remarkably downregulated and that Lrp5 protein is a strong candidate for the receptor of all-trans-retinol in the visual cycle. Furthermore, in this review, we also discuss functional roles of EYS in vertebrates with an emphasis on its possible involvement in the retinal metabolism, the visual cycle, aiming at integrating our findings with recent advances in the research field.
Keywords
- retinitis pigmentosa
- Eys
- Lrp5
- Rbp1
- visual cycle
- familial exudative vitreoretinopathy
1. Introduction
Humans rely heavily on vision for a lot of information. Curing or slowing the progression of the vision loss contributes significantly to medicine, as vision loss impairs quality of life significantly. Vision is an intricate biological phenomenon that involves the retina and the visual field in the brain. Vision is initiated and processed by a network of retinal cells and molecular processes within the retina, which is located at the back of the eye. The first step of vision is to detect light, i.e., photons, and convert the information to chemical information in photoreceptors within the retina (for review, see [1, 2]).
In the photoreceptors, 11-
Both environmental and genetic factors can cause blindness. In this review, we focus primarily on genetic factors (i.e., IRDs). Among IRDs, retinitis pigmentosa (RP) and familial exudative vitreoretinopathy (FEVR) are included. In developed countries, IRD is going up in ranking as the cause of visual impairment. Although mouse genetics using the knocked-out (KO) approach is powerful to study whether a gene of interest is causative of an IRD and/or elucidating its pathogenesis and molecular functions, there are cases where the human phenotype is not recapitulated well [5, 6] or a gene of interest does not exist in mice [7, 8].
Researchers have been using zebrafish (
In this chapter, we aim to insightfully bridge the current knowledge gap between fish and mammals and get insight into the role of EYS in human vision that is key to photoreceptor degeneration by closely reviewing the accumulating results of
This review first focuses on the findings from the zebrafish low-density lipoprotein (LDL)-related receptor-5 (Lrp5) and Eys proteins and then possible EYS roles in vision. Recent advancements in genetic manipulation techniques, including CRISPR/Cas9, have enabled the creation of knockout zebrafish models more easily than before, offering a unique opportunity to investigate the functional consequences of disruption of gene(s) of interest. Although not the main topic of this review, we briefly introduce these techniques for readers who are not familiar with zebrafish.
Human
2. Inherited retinal diseases (IRDs)
IRD is a group of diseases characterized by a progressive degeneration of the retina as time goes, which can lead to progressive severe vision loss or blindness. IRD primarily affects the structure and/or function of the retina and alters them [12]. This process could be during the developing of the retina (this can be syndromic that may also be critically important for normal development in tissues other than the retina) or after the completion of development of the retina (i.e., maintenance and therefore, typically nonsyndromic) depending on types of IRDs. Because phenotypes can be changed by the types of mutations in a same gene, and sometimes it is difficult to distinguish phenotypes by those typical names, it may be preferable to use the term IRD in some cases, which would include all types of blindness.
There are several different types of IRDs where photoreceptors are especially affected, among the six major retinal neuronal cell types [13]. Leber congenital amaurosis (LCA) is a congenital retinal disease that results in severe vision loss present at birth [14, 15]. LCA can be caused by mutations in genes involved in a wide variety of photoreceptor functions such as centrosomal protein 290 (
Ciliopathies are a group of diseases that involve dysfunction of the cilium. Because the photoreceptor outer segment is one of the cilia particularly specialized for absorbing light, mutations in genes important for formation and/or maintenance of cilia can cause IRDs frequently accompanying a wide range of syndromic phenotypes such as obesity. Among them, Bardet-Biedl syndrome (BBS) is well known [18], and Meckel-Gruber syndrome (MKS) and Alström syndrome (ALMS) are also known [19, 20, 21].
Adjacent supporting cells, the retinal pigment epithelium (RPE), are crucial for maintenance of photoreceptors. Therefore, dysfunction of RPE cells can kill photoreceptors, and there are known IRDs such as RP, AMD, and CRD.
FEVR is also a group of IRDs. This disease is characterized by abnormal retinal angiogenesis and abnormal vascularization of the peripheral retina with subsequent retinal ischemia. Abnormality can be both hypovascularization and hypervascularization. Norrin cystine knot growth factor NDP (
One thing to note about FEVR is that the phenotype of mouse models of angiogenesis including knockout mice often does not recapitulate the symptoms of human patients; in other words, the phenotypes of humans and mice are often different [24], presumably due to the difference of capillary. In relation to zebrafish, confirmative studies have been conducted for
Usually, mutation(s) in a single gene account for IRD. On the other hand, it could be caused by multiple genes. For example, it is well known that digenic mutations in peripherin 2 (
3. Visual cycle
Visual cycle is a retinoid metabolism that regenerates photoisomerized retinal that allows rhodopsin or cone opsin can capture light multiple times for sustained vision [29, 30]. The visual cycle takes place mainly between photoreceptors and the RPE as well as between cones and Müller cells (called cone-specific visual cycle). In darkness, 11-
Upon absorption of light, 11-
Inside the RPE, retinol binding protein 1 (RBP1) plays a role [31, 32, 33]. RBP1 binds and transports atROL to endoplasmic reticulum (ER). atROL is esterified within this RBP1-atROL complex by lecithin retinol acyltransferase (LRAT). This all-trans-retinyl ester is converted to 11-
Retinoid metabolism crucial for sustained light absorption involves multicellular and multiple proteins. These proteins are essential and some of them show no phenotype in mice suggesting that visual cycle is conserved in vertebrates but there could be some variation among species. atROL could be carried by IRBP, however, how atROL is released from photoreceptors and is incorporated into the RPE has not been well known [34, 35].
4. LRP5 in humans and zebrafish and its involvement in IRDs
LRP5 protein is a member of the LDL receptor superfamily conserved in vertebrates. LRP5 protein was identified in 1998 in human and mouse cDNA libraries [36, 37, 38, 39]. LRP5 is best known as a co-receptor for Wnt ligands involved in the wingless (Wnt) signaling pathway on cell membranes, which is essential for the development of vascular endothelial cells, Müller cells, and retinal interneurons [40]. LRP5 is also reported to be involved in cholesterol and glucose uptake [41, 42]. Furthermore, the possible involvement of LRP5 in cell types involved in vitamin A (i.e., atROL) is suggested based on its expression [43].
Domain structures of human LRP5 and zebrafish Lrp5 are well conserved between humans and zebrafish. Human and zebrafish LRP5 proteins consist of a signal peptide sequence, low-density lipoprotein receptor YWTD (LY; also known as β-propeller), EGF, and LDLa domains in the N-terminal extracellular domains. These domains are followed by one transmembrane domain and low complexity region in the C-terminal intracellular region. LRP5 protein seems to act as a receptor of both protein and small molecules.
In addition to our group, there are at least two groups that use
In vision,
5. EYS in humans and zebrafish and its involvement in IRDs
Human eyes shut homolog (EYS) gene (Online Mendelian Inheritance in Man no. 612424), an ortholog of the Drosophila eyes shut/spacemaker (
The predominant human
This protein is present in invertebrates as well. Eys was originally identified in Drosophila that is a secreted protein filling a space in photoreceptors [63, 64]. This is in accordance with the other related proteins such as Pikachurin and Agrin that are secreted and functions outside cells [65]. In
Our group reported that this gene is most prevalent in the Japanese population [58], and five mutations that are most frequently observed in the Japanese population (JV1, c.4957dupA and p.(Ser1653Lysfs*2); JV2, c.8868C > A and p.(Tyr2956*); JV3, c.2528G > A and p.(Gly843Glu); JV4, c.6557G > A and p.(Gly2186Glu); JV5, c.6563 T > C and p.(Ile2188Thr)) [69]. These five mutations are located from N-terminal region to C-terminal region of EYS protein. Full-length Eys is highly expressed in zebrafish photoreceptors and this expression is specific or dominant [60], and full-length EYS is expressed is expressed in human photoreceptor-like cells derived from dermal fibroblasts (our unpublished observation). It would be important to note that the evidence suggests that full-length EYS protein is crucial for preventing photoreceptors from degeneration.
6. Available resources and genetic manipulation in zebrafish
In zebrafish, the eye starts to form around 20–24 hours post-fertilization (hpf) [70, 71]. Around this time point, zebrafish eyes are transparent allowing unique opportunities for eye formation or photoreceptor differentiation [72], which occurs around 48 hpf in the ventronasal patch [73].
We initially investigated zebrafish orthologs of human
One more important thing to note is that there are multiple strains actively used in zebrafish; from our experience using two different strains, AB and TL, we do not observe substantial differences in molecular cloning in both strains.
We used mutant zebrafish obtained from Zebrafish International Resource Center (ZIRC) where
We received 50–100 of 3–5 days post-fertilization (dpf) zebrafish embryos and raised the embryos to adults. We collected tails, extracted genomic DNAs, and genotyped to screen the obtained zebrafish and about 25% of them carried the described mutation in the
To avoid off-target effect potentially arisen from ENU-treatment, it would be ideal to cross-obtained mutant zebrafish with wild-type zebrafish for several generations to reduce the possibility of unexpected ENU-induced mutations being introduced at other loci. In our experience, this is probably sufficient for practical purposes, as the zebrafish obtained from these resource centers are of F2 generation, and ENU-induced mutations that induce point mutations are more random than CRISPR-induced off-target mutations.
Alternatively, because the CRISPR/Cas9 system has become accessible and much easier in zebrafish, researchers can generate knockout zebrafish using CRISPR/Cas9 in their own labs. We briefly explain our method here [75], which we believe is easiest. We obtain Cas9 protein (Alt-R® S.p. HiFi Cas9 Nuclease V3) [76], crRNA (Alt-R® CRISPR-Cas9 crRNA), and tracrRNA (Alt-R® CRISPR-Cas9 tracrRNA), the components necessary for CRISPR/Cas9, from Integrated DNA Technologies (IDT). We form gRNA by annealing the equimolar of custom-synthesized crRNA with universal tracrRNA using Nuclease Free Duplex Buffer (IDT), and mix the gRNA with the Cas9 protein to form ribonucleoprotein (RNP) complex. We inject the RNP complex into one-cell stage zebrafish eggs and raise them to adults and uses as founders (F0).
It is important to minimize the possibility of off-target effects when we select target sequence for knockout. We use CRISPRdirect (http://crispr.dbcls.jp/) [77]. We typically select sequences having minimal similarity to other locations in the reference sequence by choosing “Zebrafish (
To create knock-in zebrafish is attractive. For target size with smaller than 200 nt, there are reports [78]. However, accurate visualization of the localization of proteins tagged with fluorescent proteins such as EGFP and mCherry may still be particularly challenging, as the typical size of fluorescent proteins is ~750 nt.
We also have to be aware that there are some differences between humans and zebrafish. For example, in humans, rods are dominantly located in peripheral regions while rods are uniformly distributed in zebrafish. In addition, in zebrafish, there are four types of cones expressed and red and green cones form unique shape called double cone. Retinomoter movement reflecting circadian rhythm and/light is also very unique to fish, which is not necessarily disadvantage but needs to be note [79, 80]. One more important aspect is that photoreceptors have capability to regenerate throughout life in zebrafish [81, 82, 83]. This is advantageous for the study of regenerative medicine; this may make analysis a bit more difficult when we study retinal diseases because of continuous supply of new retinal cells.
7. Lrp5 protein and the visual cycle
EYS does not exist in rodents but does exist in zebrafish, which makes zebrafish valuable for EYS study. In humans, a case study was reported in 2017 describing digenic form of the heterozygous mutations in
We chose mutations in the C-terminal region of the Eys gene. This is because its C-terminal region is frequently mutated in the
In the digenic
Then we subjected the whole eyes to an unbiased global gene expression analysis, microarray analysis, at 3.5 mpf. At this stage, zebrafish are sexually mature young adults and we expected that this stage would allow us to identify gene(s) that are primarily affected by the heterozygous digenic mutations. Indeed, retinol-binding protein 1 a (
This discovery highly motivated us to further investigate the reason(s) this gene is specifically downregulated in the
We then investigated localization of Lrp5 protein in the eye. Lrp5 protein colocalizes with Rbp1a protein at the most concentrated regions in the microvilli of the RPE, which is very close to the inner segment (IS) of photoreceptors in the outer retina. This is strong supporting evidence that Lrp5 protein plays a role in the visual cycle as a receptor of atROL at the tip of the microvilli in the RPE. Using
The expression level of the
8. Perspective: potential involvement of EYS protein in the visual cycle and/or ciliary transport
Eys was originally found in
(1) Visual cycle: there are studies that support the idea that Eys protein is involved in the visual cycle. First, we have shown that Lrp5 is a strong candidate for the receptor of the atROL based on the possible genetic interaction between Eys and Lrp5. In this study, genetic studies suggest that the expression level of
Indeed, Eys and Lrp5 proteins are structurally related to Agrin (AGRN) and LDL receptor-related protein 4 (LRP4) proteins, respectively, and AGRN interacts with LRP4 in neuromuscular junction [84, 85]. Applying this to photoreceptors and the RPE, Eys and Lrp5 could interact with each other in the interphotoreceptor matrix between photoreceptors and the RPE. Based on the above evidence and suggestions from the previous study, we proposed in 2020 that Eys could be involved in the visual cycle (Figure 1).
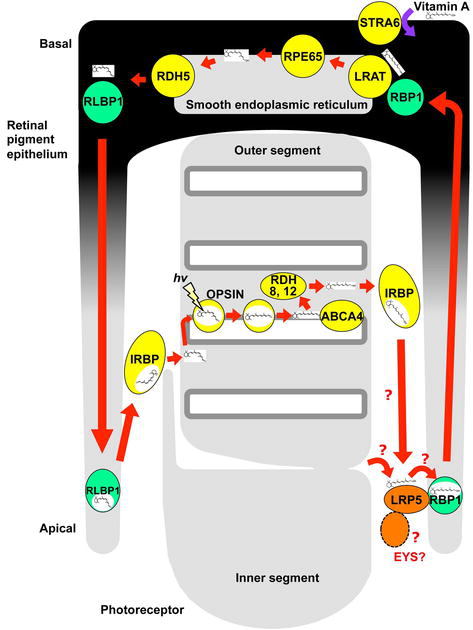
Figure 1.
A proposed model of LRP5 function in the visual cycle. LRP5 protein (orange in right bottom) is a strong candidate for the receptor of atROL in the visual cycle that forms a complex with RBP1 protein (the transporter of atROL; light green in right bottom) is involved in the uptake of atROL in the microvilli of the RPE (adopted from reference [
Studies in good line with our proposal are now emerging in both zebrafish and humans. First, human clinical trial published recently shows that administration of vitamin A worsened the photoreceptor function in the
(2) Ciliary transport/OS maintenance: the speculation that Eys protein is involved in ciliary transport may have arisen from two observations in the early studies. One is that, in Eys knockout zebrafish, ciliary pocket disappears in the cones [67], and opsins are mislocalized to IS to some extent in cones in terms of cell number [94, 97]. With respect to mislocalization of OS proteins, careful examination would be necessary because it is widely known that mislocalization of OS proteins to the IS and even non-OS proteins to the OS are rather a common phenomenon frequently observed in degenerative photoreceptors [98, 99, 100, 101, 102]. With this in mind, in the
9. Future direction: to investigate possible involvement of EYS protein in the visual cycle in terms of AOS and/or other aspects
Based on the current knowledge, the most clinically important aspect of the Eys protein is whether Eys is indeed involved in the visual cycle. If Eys is involved, the search for the existence of other proteins that might be involved in the visual cycle would be a meaningful topic from both clinical and biological perspectives.
In this regard, a possible interaction between Eys and the Prom1 protein has long been proposed based on studies in
Other possibly intriguing candidate proteins are Usher proteins. It is interesting that in humans, similar cone abnormality is observed in
In either case, AOS seems to be the crucial structural component for understanding EYS function and its possible involvement in the visual cycle. This is an interesting but unknown structure that has not been reported outside of fish, and even in fish it has been little studied. This may be an excellent opportunity to open up a new field of research. On the other hand, assuming that EYS functions in the visual cycle in humans based on previous studies, the presence of AOS seems natural, while it would be surprising if AOS is present in humans and other primates but has not been found before. Future studies on AOS are awaited.
It is a little bit surprising that photoreceptor degeneration is observed in zebrafish while this is not observed in medaka. It might be interesting that this is also observed when mutations are introduced in other loci in the medaka eys gene.
In addition, there are studies showing mislocalization of OS proteins in the IS in
In humans, cystoid macular edema (CME) is a well-known symptom frequently observed in RP patients [108, 109]. Involvement of WNT signaling is suggested in CME [110, 111], and LRP5 is one of the important WNT signaling molecules. If edema is observed in the outer retina rather than the inner retina, especially at early stages in the progression, it may suggest that both EYS and LRP5 play some roles in the retina.
There are a non-negligible number of studies claiming sector RP as one of possible phenotypes in
10. Conclusions
In this review, we summarized utilization of zebrafish in vision science and IRDs focusing on our recent findings in the digenic
Acknowledgments
We would like to thank the patients involved in the course of our study. This work was supported by JSPS KAKENHI Grant Numbers 17K16995 (to S.T.), 20K18364 (to S.T.), 15H04998 (to Y.S.), and 20H03845 (to Y.S.). The zebrafish mutant lines,
Conflict of interest
The authors declare there is no conflict of interest or no competing interest in this study.
References
- 1.
Kawamura S, Tachibanaki S. Rod and cone photoreceptors: Molecular basis of the difference in their physiology. Comparative Biochemistry and Physiology. Part A, Molecular & Integrative Physiology. 2008; 150 (4):369-377 - 2.
Kawamura S, Tachibanaki S. Molecular bases of rod and cone differences. Progress in Retinal and Eye Research. 2022; 90 :101040 - 3.
Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, et al. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin a. Science. 2007; 315 (5813):820-825 - 4.
Isken A, Golczak M, Oberhauser V, Hunzelmann S, Driever W, Imanishi Y, et al. RBP4 disrupts vitamin A uptake homeostasis in a STRA6-deficient animal model for Matthew-Wood syndrome. Cell Metabolism. 2008; 7 (3):258-268 - 5.
Gibbs D, Kitamoto J, Williams DS. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin VIIa, the Usher syndrome 1B protein. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100 (11):6481-6486 - 6.
Liu X, Bulgakov OV, Darrow KN, Pawlyk B, Adamian M, Liberman MC, et al. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104 (11):4413-4418 - 7.
Abd El-Aziz MM, Barragan I, O'Driscoll CA, Goodstadt L, Prigmore E, Borrego S, et al. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nature Genetics. 2008; 40 (11):1285-1287 - 8.
Collin RW, Littink KW, Klevering BJ, van den Born LI, Koenekoop RK, Zonneveld MN, et al. Identification of a 2 mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. American Journal of Human Genetics. 2008; 83 (5):594-603 - 9.
Nicolson T, Rusch A, Friedrich RW, Granato M, Ruppersberg JP, Nusslein-Volhard C. Genetic analysis of vertebrate sensory hair cell mechanosensation: The zebrafish circler mutants. Neuron. 1998; 20 (2):271-283 - 10.
Brockerhoff SE, Rieke F, Matthews HR, Taylor MR, Kennedy B, Ankoudinova I, et al. Light stimulates a transducin-independent increase of cytoplasmic Ca2+ and suppression of current in cones from the zebrafish mutant nof. The Journal of Neuroscience. 2003; 23 (2):470-480 - 11.
Wasfy MM, Matsui JI, Miller J, Dowling JE, Perkins BD. Myosin 7aa(−/−) mutant zebrafish show mild photoreceptor degeneration and reduced electroretinographic responses. Experimental Eye Research. 2014; 122 :65-76 - 12.
Peter VG, Kaminska K, Santos C, Quinodoz M, Cancellieri F, Cisarova K, et al. The first genetic landscape of inherited retinal dystrophies in Portuguese patients identifies recurrent homozygous mutations as a frequent cause of pathogenesis. PNAS Nexus. 2023; 2 (3):pgad043 - 13.
Livesey FJ, Cepko CL. Vertebrate neural cell-fate determination: Lessons from the retina. Nature Reviews. Neuroscience. 2001; 2 (2):109-118 - 14.
den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Progress in Retinal and Eye Research. 2008; 27 (4):391-419 - 15.
Daich Varela M, Cabral de Guimaraes TA, Georgiou M, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Current management and clinical trials. The British Journal of Ophthalmology. 2022; 106 (4):445-451 - 16.
Stradiotto E, Allegrini D, Fossati G, Raimondi R, Sorrentino T, Tripepi D, et al. Genetic aspects of age-related macular degeneration and their therapeutic potential. International Journal of Molecular Sciences. 2022; 23 (21):13280 - 17.
Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: Clinical features, molecular genetics, animal models and therapeutic options. The British Journal of Ophthalmology. 2017; 101 (1):25-30 - 18.
Hsu Y, Seo S, Sheffield VC. Photoreceptor cilia, in contrast to primary cilia, grant entry to a partially assembled BBSome. Human Molecular Genetics. 2021; 30 (1):87-102 - 19.
Marshall JD, Beck S, Maffei P, Naggert JK. Alstrom syndrome. European Journal of Human Genetics. 2007; 15 (12):1193-1202 - 20.
Hartill V, Szymanska K, Sharif SM, Wheway G, Johnson CA. Meckel-Gruber syndrome: An update on diagnosis, clinical management, and research advances. Frontiers in Pediatrics. 2017; 5 :244 - 21.
Parisi MA. Clinical and molecular features of Joubert syndrome and related disorders. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 2009; 151C (4):326-340 - 22.
Gilmour DF. Familial exudative vitreoretinopathy and related retinopathies. Eye (London, England). 2015; 29 (1):1-14 - 23.
Xiao H, Tong Y, Zhu Y, Peng M. Familial exudative vitreoretinopathy-related disease-causing genes and norrin/beta-catenin signal pathway: Structure, function, and mutation spectrums. Journal of Ophthalmology. 2019; 2019 :5782536 - 24.
Stahl A, Connor KM, Sapieha P, Chen J, Dennison RJ, Krah NM, et al. The mouse retina as an angiogenesis model. Investigative Ophthalmology & Visual Science. 2010; 51 (6):2813-2826 - 25.
Collin RW, Nikopoulos K, Dona M, Gilissen C, Hoischen A, Boonstra FN, et al. ZNF408 is mutated in familial exudative vitreoretinopathy and is crucial for the development of zebrafish retinal vasculature. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110 (24):9856-9861 - 26.
Caceres L, Prykhozhij SV, Cairns E, Gjerde H, Duff NM, Collett K, et al. Frizzled 4 regulates ventral blood vessel remodeling in the zebrafish retina. Developmental Dynamics. 2019; 248 (12):1243-1256 - 27.
Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science. 1994; 264 (5165):1604-1608 - 28.
Burkard M, Kohl S, Kratzig T, Tanimoto N, Brennenstuhl C, Bausch AE, et al. Accessory heterozygous mutations in cone photoreceptor CNGA3 exacerbate CNG channel-associated retinopathy. The Journal of Clinical Investigation. 2018; 128 (12):5663-5675 - 29.
Lamb TD. Evolution of phototransduction, vertebrate photoreceptors and retina. Progress in Retinal and Eye Research. 2013; 36 :52-119 - 30.
Sparrow JR, Hicks D, Hamel CP. The retinal pigment epithelium in health and disease. Current Molecular Medicine. 2010; 10 (9):802-823 - 31.
Bok D, Ong DE, Chytil F. Immunocytochemical localization of cellular retinol binding protein in the rat retina. Investigative Ophthalmology & Visual Science. 1984; 25 (8):877-883 - 32.
Bunt-Milam AH, Saari JC. Immunocytochemical localization of two retinoid-binding proteins in vertebrate retina. The Journal of Cell Biology. 1983; 97 (3):703-712 - 33.
Ghyselinck NB, Bavik C, Sapin V, Mark M, Bonnier D, Hindelang C, et al. Cellular retinol-binding protein I is essential for vitamin A homeostasis. The EMBO Journal. 1999; 18 (18):4903-4914 - 34.
Zeng S, Zhang T, Madigan MC, Fernando N, Aggio-Bruce R, Zhou F, et al. Interphotoreceptor retinoid-binding protein (IRBP) in retinal health and disease. Frontiers in Cellular Neuroscience. 2020; 14 :577935 - 35.
Cook JD, Ng SY, Lloyd M, Eddington S, Sun H, Nathans J, et al. Peropsin modulates transit of vitamin A from retina to retinal pigment epithelium. The Journal of Biological Chemistry. 2017; 292 (52):21407-21416 - 36.
Brown SD, Twells RC, Hey PJ, Cox RD, Levy ER, Soderman AR, et al. Isolation and characterization of LRP6, a novel member of the low density lipoprotein receptor gene family. Biochemical and Biophysical Research Communications. 1998; 248 (3):879-888 - 37.
Hey PJ, Twells RC, Phillips MS, Yusuke N, Brown SD, Kawaguchi Y, et al. Cloning of a novel member of the low-density lipoprotein receptor family. Gene. 1998; 216 (1):103-111 - 38.
Kim DH, Inagaki Y, Suzuki T, Ioka RX, Yoshioka SZ, Magoori K, et al. A new low density lipoprotein receptor related protein, LRP5, is expressed in hepatocytes and adrenal cortex, and recognizes apolipoprotein E. Journal of Biochemistry. 1998; 124 (6):1072-1076 - 39.
Dong Y, Lathrop W, Weaver D, Qiu Q , Cini J, Bertolini D, et al. Molecular cloning and characterization of LR3, a novel LDL receptor family protein with mitogenic activity. Biochemical and Biophysical Research Communications. 1998; 251 (3):784-790 - 40.
Xia CH, Yablonka-Reuveni Z, Gong X. LRP5 is required for vascular development in deeper layers of the retina. PLoS One. 2010; 5 (7):e11676 - 41.
Chin EN, Martin JA, Kim S, Fakhraldeen SA, Alexander CM. Lrp5 has a Wnt-independent role in glucose uptake and growth for mammary epithelial cells. Molecular and Cellular Biology. 2015; 36 (6):871-885 - 42.
Fujino T, Asaba H, Kang MJ, Ikeda Y, Sone H, Takada S, et al. Low-density lipoprotein receptor-related protein 5 (LRP5) is essential for normal cholesterol metabolism and glucose-induced insulin secretion. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100 (1):229-234 - 43.
Figueroa DJ, Hess JF, Ky B, Brown SD, Sandig V, Hermanowski-Vosatka A, et al. Expression of the type I diabetes-associated gene LRP5 in macrophages, vitamin A system cells, and the islets of Langerhans suggests multiple potential roles in diabetes. The Journal of Histochemistry and Cytochemistry. 2000; 48 (10):1357-1368 - 44.
Bek JW, Shochat C, De Clercq A, De Saffel H, Boel A, Metz J, et al. Lrp5 mutant and crispant Zebrafish faithfully model human osteoporosis, establishing the Zebrafish as a platform for CRISPR-based functional screening of osteoporosis candidate genes. Journal of Bone and Mineral Research. 2021; 36 (9):1749-1764 - 45.
Khrystoforova I, Shochat-Carvalho C, Harari R, Henke K, Woronowicz K, Harris MP, et al. Zebrafish mutants reveal unexpected role of Lrp5 in osteoclast regulation. Frontiers in Endocrinology. 2022; 13 :985304 - 46.
Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001; 107 (4):513-523 - 47.
Li Y, Peng J, Li J, Zhang Q , Li J, Zhang X, et al. The characteristics of digenic familial exudative vitreoretinopathy. Graefe's Archive for Clinical and Experimental Ophthalmology. 2018; 256 (11):2149-2156 - 48.
Yang H, Li S, Xiao X, Wang P, Guo X, Zhang Q. Identification of FZD4 and LRP5 mutations in 11 of 49 families with familial exudative vitreoretinopathy. Molecular Vision. 2012; 18 :2438-2446 - 49.
Li JK, Li Y, Zhang X, Chen CL, Rao YQ , Fei P, et al. Spectrum of variants in 389 Chinese probands with familial exudative vitreoretinopathy. Investigative Ophthalmology & Visual Science. 2018; 59 (13):5368-5381 - 50.
Zhao R, Wang S, Zhao P, Dai E, Zhang X, Peng L, et al. Heterozygote loss-of-function variants in the LRP5 gene cause familial exudative vitreoretinopathy. Clinical & Experimental Ophthalmology. 2022; 50 (4):441-448 - 51.
Ye X, Wang Y, Cahill H, Yu M, Badea TC, Smallwood PM, et al. Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell. 2009; 139 (2):285-298 - 52.
Ubels JL, Lin CM, Antonetti DA, Diaz-Coranguez M, Diegel CR, Williams BO. Structure and function of the retina of low-density lipoprotein receptor-related protein 5 (Lrp5)-deficient rats. Experimental Eye Research. 2022; 217 :108977 - 53.
Gao FJ, Zhang SH, Chen JY, Xu GZ, Wu JH. Digenic heterozygous mutations in EYS/LRP5 in a Chinese family with retinitis pigmentosa. International Journal of Ophthalmology. 2017; 10 (2):325-328 - 54.
Katagiri S, Akahori M, Hayashi T, Yoshitake K, Gekka T, Ikeo K, et al. Autosomal recessive cone-rod dystrophy associated with compound heterozygous mutations in the EYS gene. Documenta Ophthalmologica. 2014; 128 (3):211-217 - 55.
Pierrache LHM, Messchaert M, Thiadens A, Haer-Wigman L, de Jong-Hesse Y, van Zelst-Stams WAG, et al. Extending the spectrum of EYS-associated retinal disease to macular dystrophy. Investigative Ophthalmology & Visual Science. 2019; 60 (6):2049-2063 - 56.
Audo I, Sahel JA, Mohand- Said S, Lancelot ME, Antonio A, Moskova-Doumanova V, et al. EYS is a major gene for rod-cone dystrophies in France. Human Mutation. 2010; 31 (5):E1406-E1435 - 57.
Hosono K, Ishigami C, Takahashi M, Park DH, Hirami Y, Nakanishi H, et al. Two novel mutations in the EYS gene are possible major causes of autosomal recessive retinitis pigmentosa in the Japanese population. PLoS One. 2012; 7 (2):e31036 - 58.
Iwanami M, Oshikawa M, Nishida T, Nakadomari S, Kato S. High prevalence of mutations in the EYS gene in Japanese patients with autosomal recessive retinitis pigmentosa. Investigative Ophthalmology & Visual Science. 2012; 53 (2):1033-1040 - 59.
Suvannaboon R, Pawestri AR, Jinda W, Tuekprakhon A, Trinavarat A, Atchaneeyasakul LO. Genotypic and phenotypic profiles of EYS gene-related retinitis pigmentosa: A retrospective study. Scientific Reports. 2022; 12 (1):21494 - 60.
Takita S, Miyamoto-Matsui K, Seko Y. Intra- and interspecies comparison of EYS transcripts highlights its characteristics in the eye. The FASEB Journal. 2019; 33 (8):9422-9433 - 61.
Hohenester E. Laminin G-like domains: Dystroglycan-specific lectins. Current Opinion in Structural Biology. 2019; 56 :56-63 - 62.
Garcia-Delgado AB, Valdes- Sanchez L, Morillo-Sanchez MJ, Ponte-Zuniga B, Diaz-Corrales FJ, de la Cerda B. Dissecting the role of EYS in retinal degeneration: Clinical and molecular aspects and its implications for future therapy. Orphanet Journal of Rare Diseases. 2021; 16 (1):222 - 63.
Husain N, Pellikka M, Hong H, Klimentova T, Choe KM, Clandinin TR, et al. The agrin/perlecan-related protein eyes shut is essential for epithelial lumen formation in the Drosophila retina. Developmental Cell. 2006; 11 (4):483-493 - 64.
Zelhof AC, Hardy RW, Becker A, Zuker CS. Transforming the architecture of compound eyes. Nature. 2006; 443 (7112):696-699 - 65.
Furukawa T, Ueno A, Omori Y. Molecular mechanisms underlying selective synapse formation of vertebrate retinal photoreceptor cells. Cellular and Molecular Life Sciences. 2020; 77 (7):1251-1266 - 66.
Nie J, Mahato S, Mustill W, Tipping C, Bhattacharya SS, Zelhof AC. Cross species analysis of prominin reveals a conserved cellular role in invertebrate and vertebrate photoreceptor cells. Developmental Biology. 2012; 371 (2):312-320 - 67.
Yu M, Liu Y, Li J, Natale BN, Cao S, Wang D, et al. Eyes shut homolog is required for maintaining the ciliary pocket and survival of photoreceptors in zebrafish. Biology Open. 2016; 5 (11):1662-1673 - 68.
Alfano G, Kruczek PM, Shah AZ, Kramarz B, Jeffery G, Zelhof AC, et al. EYS is a protein associated with the ciliary axoneme in rods and cones. PLoS One. 2016; 11 (11):e0166397 - 69.
Iwanami M, Oishi A, Ogino K, Seko Y, Nishida-Shimizu T, Yoshimura N, et al. Five major sequence variants and copy number variants in the EYS gene account for one-third of Japanese patients with autosomal recessive and simplex retinitis pigmentosa. Molecular Vision. 2019; 25 :766-779 - 70.
Schmitt EA, Dowling JE. Early eye morphogenesis in the zebrafish, Brachydanio rerio . Journal of Comparative Neurology. 1994;344 (4):532-542 - 71.
Malicki J. Development of the retina. Methods in Cell Biology. 1999; 59 :273-299 - 72.
Godinho L, Mumm JS, Williams PR, Schroeter EH, Koerber A, Park SW, et al. Targeting of amacrine cell neurites to appropriate synaptic laminae in the developing zebrafish retina. Development. 2005; 132 (22):5069-5079 - 73.
Takita S, Wada Y, Kawamura S. Effects of NDRG1 family proteins on photoreceptor outer segment morphology in zebrafish. Scientific Reports. 2016; 6 :36590 - 74.
Parey E, Louis A, Montfort J, Guiguen Y, Roest Crollius H, Berthelot C. An atlas of fish genome evolution reveals delayed rediploidization following the teleost whole-genome duplication. Genome Research. 2022; 32 (9):1685-1697 - 75.
Takita S, Seko Y. eys+/− ;lrp5+/− Zebrafish reveals Lrp5 can be the receptor of retinol in the visual cycle. iScience. 2020;23 (12):101762 - 76.
Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nature Medicine. 2018; 24 (8):1216-1224 - 77.
Naito Y, Hino K, Bono H, Ui-Tei K. CRISPRdirect: Software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics. 2015; 31 (7):1120-1123 - 78.
Ranawakage DC, Okada K, Sugio K, Kawaguchi Y, Kuninobu-Bonkohara Y, Takada T, et al. Efficient CRISPR-Cas9-mediated knock-in of composite tags in zebrafish using long ssDNA as a donor. Frontiers in Cell and Development Biology. 2020; 8 :598634 - 79.
Burnside B. Light and circadian regulation of retinomotor movement. Progress in Brain Research. 2001; 131 :477-485 - 80.
Menger GJ, Koke JR, Cahill GM. Diurnal and circadian retinomotor movements in zebrafish. Visual Neuroscience. 2005; 22 (2):203-209 - 81.
Wan J, Goldman D. Retina regeneration in zebrafish. Current Opinion in Genetics & Development. 2016; 40 :41-47 - 82.
Lahne M, Nagashima M, Hyde DR, Hitchcock PF. Reprogramming muller glia to regenerate retinal neurons. Annual Review of Vision Science. 2020; 6 :171-193 - 83.
Raymond PA, Barthel LK, Bernardos RL, Perkowski JJ. Molecular characterization of retinal stem cells and their niches in adult zebrafish. BMC Developmental Biology. 2006; 6 :36 - 84.
Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, et al. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008; 135 (2):334-342 - 85.
Zhang W, Coldefy AS, Hubbard SR, Burden SJ. Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK). The Journal of Biological Chemistry. 2011; 286 (47):40624-40630 - 86.
Comander J, Weigel DiFranco C, Sanderson K, Place E, Maher M, Zampaglione E, et al. Natural history of retinitis pigmentosa based on genotype, vitamin A/E supplementation, and an electroretinogram biomarker. JCI Insight. 2023; 8 (15):e167546 - 87.
Fujinami-Yokokawa Y, Ninomiya H, Liu X, Yang L, Pontikos N, Yoshitake K, et al. Prediction of causative genes in inherited retinal disorder from fundus photography and autofluorescence imaging using deep learning techniques. The British Journal of Ophthalmology. 2021; 105 (9):1272-1279 - 88.
Burke TR, Duncker T, Woods RL, Greenberg JP, Zernant J, Tsang SH, et al. Quantitative fundus autofluorescence in recessive Stargardt disease. Investigative Ophthalmology & Visual Science. 2014; 55 (5):2841-2852 - 89.
Cremers FPM, Lee W, Collin RWJ, Allikmets R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Progress in Retinal and Eye Research. 2020; 79 :100861 - 90.
Schellens R, de Vrieze E, Graave P, Broekman S, Nagel-Wolfrum K, Peters T, et al. Zebrafish as a model to evaluate a CRISPR/Cas9-based exon excision approach as a future treatment option for EYS-associated retinitis pigmentosa. International Journal of Molecular Sciences. 2021; 22 (17):9154 - 91.
Yacob A, Wise C, Kunz YW. The accessory outer segment of rods and cones in the retina of the guppy, Poecilia reticulata P. (Teleostei). An electron microscopical study. Cell and Tissue Research. 1977;177 (2):181-193 - 92.
Hodel C, Niklaus S, Heidemann M, Klooster J, Kamermans M, Biehlmaier O, et al. Myosin VIIA is a marker for the cone accessory outer segment in zebrafish. Anatomical Record (Hoboken, N.J.). 2014; 297 (9):1777-1784 - 93.
Burnside B, Wang E, Pagh-Roehl K, Rey H. Retinomotor movements in isolated teleost retinal cone inner-outer segment preparations (CIS-COS): Effects of light, dark and dopamine. Experimental Eye Research. 1993; 57 (6):709-722 - 94.
Sato K, Liu Y, Yamashita T, Ohuchi H. The medaka mutant deficient in eyes shut homolog exhibits opsin transport defects and enhanced autophagy in retinal photoreceptors. Cell and Tissue Research. 2023; 391 (2):249-267 - 95.
Placidi G, Maltese PE, Savastano MC, D'Agostino E, Cestrone V, Bertelli M, et al. Retinitis pigmentosa associated with EYS gene mutations: Disease severity staging and central retina atrophy. Diagnostics (Basel). 2023; 13 (5):850 - 96.
Otsuka Y, Oishi A, Miyata M, Uji A, Oishi M, Hasegawa T, et al. Evaluation of outer nuclear layer overshadowed by retinal vessels in retinitis pigmentosa. Eye (London, England). 2022; 36 (5):1042-1049 - 97.
Nishiguchi KM, Miya F, Mori Y, Fujita K, Akiyama M, Kamatani T, et al. A hypomorphic variant in EYS detected by genome-wide association study contributes toward retinitis pigmentosa. Communications Biology. 2021; 4 (1):140 - 98.
Bales KL, Gross AK. Aberrant protein trafficking in retinal degenerations: The initial phase of retinal remodeling. Experimental Eye Research. 2016; 150 :71-80 - 99.
Wensel TG, Zhang Z, Anastassov IA, Gilliam JC, He F, Schmid MF, et al. Structural and molecular bases of rod photoreceptor morphogenesis and disease. Progress in Retinal and Eye Research. 2016; 55 :32-51 - 100.
Datta P, Allamargot C, Hudson JS, Andersen EK, Bhattarai S, Drack AV, et al. Accumulation of non-outer segment proteins in the outer segment underlies photoreceptor degeneration in Bardet-Biedl syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2015; 112 (32):E4400-E4409 - 101.
Radhakrishnan R, Dronamraju VR, Leung M, Gruesen A, Solanki AK, Walterhouse S, et al. The role of motor proteins in photoreceptor protein transport and visual function. Ophthalmic Genetics. 2022; 43 (3):285-300 - 102.
Radhakrishnan R, Leung M, Roehrich H, Walterhouse S, Kondkar AA, Fitzgibbon W, et al. Mice lacking the systemic vitamin A receptor RBPR2 show decreased ocular retinoids and loss of visual function. Nutrients. 2022; 14 (12):2371 - 103.
Sukumaran S, Perkins BD. Early defects in photoreceptor outer segment morphogenesis in zebrafish ift57, ift88 and ift172 intraflagellar transport mutants. Vision Research. 2009; 49 (4):479-489 - 104.
Zhu P, Xu J, Wang Y, Zhao C. Loss of Ift74 leads to slow photoreceptor degeneration and Ciliogenesis defects in zebrafish. International Journal of Molecular Sciences. 2021; 22 (17):9329 - 105.
Galli-Resta L, Placidi G, Campagna F, Ziccardi L, Piccardi M, Minnella A, et al. Central retina functional damage in usher syndrome type 2: 22 years of focal macular ERG analysis in a patient population from central and southern Italy. Investigative Ophthalmology & Visual Science. 2018; 59 (10):3827-3835 - 106.
Kelbsch C, Kempf M, Jung R, Kortum F, Reith M, Kuehlewein L, et al. Rod and cone function measured objectively by chromatic pupil campimetry show a different preservation between distinct genotypes in retinitis pigmentosa. Investigative Ophthalmology & Visual Science. 2023; 64 (11):18 - 107.
Lu Z, Hu X, Liu F, Soares DC, Liu X, Yu S, et al. Ablation of EYS in zebrafish causes mislocalisation of outer segment proteins, F-actin disruption and cone-rod dystrophy. Scientific Reports. 2017; 7 :46098 - 108.
Hajali M, Fishman GA. The prevalence of cystoid macular oedema on optical coherence tomography in retinitis pigmentosa patients without cystic changes on fundus examination. Eye (London, England). 2009; 23 (4):915-919 - 109.
Kitahata S, Gocho K, Motozawa N, Yokota S, Yamamoto M, Maeda A, et al. Evaluation of photoreceptor features in retinitis pigmentosa with cystoid macular edema by using an adaptive optics fundus camera. PLoS One. 2024; 19 (1):e0296493 - 110.
Zhang C, Lai MB, Pedler MG, Johnson V, Adams RH, Petrash JM, et al. Endothelial cell-specific inactivation of TSPAN12 (Tetraspanin 12) reveals pathological consequences of barrier defects in an otherwise intact vasculature. Arteriosclerosis, Thrombosis, and Vascular Biology. 2018; 38 (11):2691-2705 - 111.
Ji B, Lim D, Kim J, Kim HC, Chung H. Increased levels of Dickkopf 3 in the aqueous humor of patients with diabetic macular edema. Investigative Ophthalmology & Visual Science. 2016; 57 (4):2296-2304 - 112.
Marques JP, Porto FBO, Carvalho AL, Neves E, Chen R, Sampaio SAM, et al. EYS-associated sector retinitis pigmentosa. Graefe’s Archive for Clinical and Experimental Ophthalmology. 2022; 260 (4):1405-1413 - 113.
Chan CM, Tan TE, Jain K, Bylstra Y, Mathur RS, Tang RWC, et al. Retinitis pigmentosa associated with the EYS C2139Y variant: An important cause of blindness in east Asian populations. Retina. 2023; 43 (10):1788-1796 - 114.
Coussa RG, Basali D, Maeda A, DeBenedictis M, Traboulsi EI. Sector retinitis pigmentosa: Report of ten cases and a review of the literature. Molecular Vision. 2019; 25 :869-889 - 115.
Georgiou M, Grewal PS, Narayan A, Alser M, Ali N, Fujinami K, et al. Sector retinitis pigmentosa: Extending the molecular genetics basis and elucidating the natural history. American Journal of Ophthalmology. 2021; 221 :299-310