
Abstract
Human and sheep parturition are more akin than currently recognised. In both glucocorticoids are key. The difference being mechanisms controlling glucocorticoid levels. Sheep have low cortisol during pregnancy which rise at term: humans control local glucocorticoid levels via the fetal adrenal and DHEA-sulphate. This increases 11β-HSD2 expression protecting the fetus from maternal cortisol by converting this to cortisone. During pregnancy DHEA inhibits placental and fetal membrane 11β-HSD1 expression. This plus hexose-6-phosphate dehydrogenase inhibition decreases 11β-HSD1 oxido-reductase/increases dehydrogenase action converting cortisol to cortisone via transcription factor C/EBPβ. This has a key role in progesterone synthesis blocking COX-2 transcription and prostaglandin synthesis. DHEA-induced protection reverses at parturition onset via pro-inflammatory cytokines increasing C/EBPα. This lowers progesterone and enhances local glucocorticoid production stimulating prostaglandins, oxytocin receptor production and cervical ripening. Lowered progesterone and increased TNFα/IL-1β markedly increase myometrial purinergic receptors promoting calcium entry, contraction and hence parturition.
Keywords
- cortisol
- DHEA
- 11β-HSD2 and 11β-HSD1
- fetal adrenal
- hexose-6-phosphate dehydrogenase
1. Introduction
It is clear that the factors controlling parturition are complex and poorly understood. Several studies suggest that the fetus is in control of the timing of the onset of labour. In sheep and cows activation at term of the fetal hypothalamic-pituitary-adrenal axis (HPA) plays a major role. Cortisol directly upregulates the placental enzyme 17α-hydroxylase/17,20-lyase (CYP17) that catalyses the conversion of pregnenolone to 17-OH-Pregnenolone and then to Dehydroepiandrosterone (DHEA). DHEA can then be further metabolised to oestrogen (oestriol from DHEA-S and oestradiol and oestrone from testosterone and androstenedione). It is suggested that the switch in the progesterone/oestrogen ratio then provides the stimulus for uterine prostaglandin production and hence labour. Oestrogens are thought to increase prostaglandin and oxytocin receptors, and upregulate myosin light chain kinase that enhances myometrial contraction. This pathway however was thought not apply in humans as human placentae were said to lack the CYP17 enzyme. More recent work has suggested that this is not true. Escobar and colleagues have shown that the human placenta does express CYP17 and can generate androgens
Many authors have suggested that the human placenta is an ‘incomplete steroidogenic organ’ and that oestrogen synthesis by the placenta requires the C19 oestrogen precursor DHEA-S that is produced by the fetal adrenal. It is suggested that DHEA-S is metabolised by the fetal liver to 16-hydroxy DHEA-S that then travels to the placenta where it is metabolised to oestrogen.
This chapter focuses on a possible novel role for DHEA and DHEA-S and suggests that these steroids are not simply produced as oestrogen precursors but regulate the local intracellular concentrations of glucocorticoid in critical sites and at key times in pregnancy.
2. The human fetal adrenal
Ishimoto and Jaffe have written an excellent review of the development and function of the human fetal adrenal cortex [3]. They summarise the evidence that there is early cortisol secretion by the fetal adrenal followed by suppressed synthesis until late gestation. Studies in infants with 21-hydroxylase deficiency (who cannot synthesise adequate amounts of cortisol) show that they have a compensatory rise in ACTH that produces fetal adrenal hyperplasia and associated adrenal androgen production (DHEA-S). Maternal administration of dexamethasone that crosses the placenta reverses the virilisation of the external genitalia if given before the 7th or 8th week of gestation.
One of the major problems in studying fetal adrenal function has been the major species differences so that rodents cannot be used as a model [4].
Goto [5] has suggested that in the normal fetus early cortisol synthesis has a negative feedback effect on ACTH secretion which lowers ACTH-induced androgen secretion and safeguards normal female sexual development.
Despite the lack of cortisol secretion by the fetal adrenal for the majority of pregnancy adrenal size suggests that this is not its function. By 20 weeks gestation the fetal adrenal is as big as the fetal kidney and by 30 weeks it achieves a relative size that is 10–20 fold that of the adult adrenal gland. This rapid growth is almost entirely due to enlargement of the fetal zone that is responsible for the production of DHEA-S. By 16–20 weeks the fetal zone (FZ) dominates the gland.
A key enzyme in the conversion of pregenolone to progesterone, 17-OH-pregenolone to 17-OH-progesterone and DHEA to androstenedione is HSD3B2 (3β-HSD)(Figure 1). Levels of this enzyme peak in the adrenal at 8–9 weeks post-conception in keeping with the early peak of cortisol synthesis. With the exception of the first trimester HSD3B2 is not expressed in the fetal zone (i.e., oestrogens and androgens would need to be produced peripherally). After 23 weeks gestation HSD3B2 is detectable in both the Definitive Zone and the Transitional Zone. Interestingly HSD3B2 is readily expressed
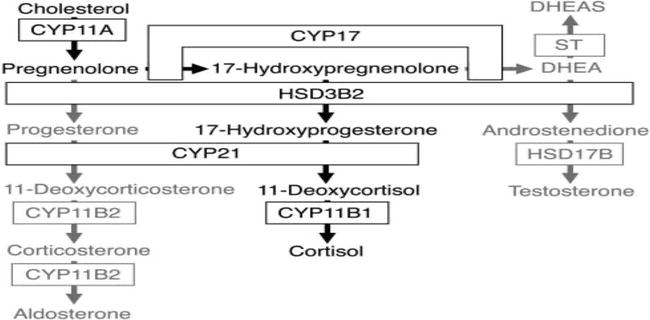
Figure 1.
Adrenal steroidogenesis.
The key steroid produced by the fetal adrenal is DHEA-S. The localization of the DHEA sulfotransferase (SULT2A1) is thus of importance. It is found in the Fetal Zone and the Transitional Zone but not the Definitive Zone (DZ). The DZ is the likely site of aldosterone synthesis late in gestation (i.e., becomes the zona glomerulosa) and the TZ cortisol, again late in gestation (becomes zona fasciculata).
DHEA-S production starts at about 8–10 weeks gestation and progressively increases, so at term the fetal adrenals produce about 200 mg per day.
Beverley Murphy reviewed the data on cord blood levels of cortisol during human gestation [6]. At about 15 weeks the level was 8 ng/ml (22 nmol/l) falling to 4 at 17–20 weeks. It then rose to 20 ng/ml at 35–37 weeks and 45 ng/ml at term. The cortisol rise appeared to be steepest immediately before the onset of labour and could not be attributed to the stress of labour.
All this raises intriguing questions—why does the fetal adrenal produce such large quantities of DHEA-S and why is it produced as a sulfate rather than as DHEA? Given the amount produced and its time course it would seem unlikely that it is simply a source of oestrogen. The hypothesis put forward here suggests an alternative role.
3. The importance of cortisol metabolism in controlling local glucocorticoid levels
Our previous studies indicated the critical importance of local metabolism of cortisol and its inter-conversion with the inactive steroid cortisone [7]. Failure to convert cortisol to cortisone results in the syndrome of apparent mineralocorticoid excess where the mineralocorticoid receptor is exposed to cortisol which produces marked sodium retention, hypertension and hypokalemia [8]. The enzyme responsible for this is 11β-Hydroxysteroid Dehydrogenase type 2 (11β-HSD2). In contrast to this unidirectional NAD-dependent enzyme the one responsible for the inter-conversion of cortisol and cortisone is 11β-HSD1.
3.1 Hexose-6-phosphate dehydrogenase and its interaction with DHEA
Elegant studies by Paul Stewart et al. showed that 11β-HSD1 lives in the endoplasmic reticulum and has a very close relationship with hexose-6-phosphate dehydrogenase (H6PDH) [9]. H6PDH converts NADP to NADPH. This cofactor is then used by 11β-HSD1 to drive the enzyme as an oxido-reductase converting cortisone to cortisol. In the absence of H6PDH 11β-HSD1 acts as a dehydrogenase and converts cortisol to cortisone. This role was specific for H6PDH. Glucose-6-phosphate dehydrogenase was not effective.
3.2 Placental hexose-6-phosphate dehydrogenase
If the mechanism controlling the activity of 11β-HSD1 in the placenta is similar to that in, for example, adipose tissue then the presence of H6PDH to generate NADPH would be key. This enzyme, H6PDH, is found in high concentration in the human placenta, is located in the luminal space of the placental microsomes and generates NADPH on the inner side of the endoplasmic reticulum membrane [10].
3.3 The control of the expression and the set point of 11β-HSD1
Given the key role of H6PDH,what controls H6PDH?. This paper suggests that this could relate to DHEA-S. As in the fetus DHEA-S is the most abundant steroid produced by the adult adrenal cortex (20 times the secretion rate of cortisol). DHEA-S is produced by catarrhine primates—the Old World monkeys and the apes. Rodents do not produce large amounts of DHEA and this needs to be remembered when looking at animal models.
Marks and Banks [11] found that DHEA and pregnenolone were non-competitive inhibitors of G6PD. A concentration of DHEA of 1 × 10−5 M produced 82% inhibition of G6PD in a crude preparation of human liver.
The specificity of this reaction was further examined by Raineri and Levy [12]. They found that a 17-keto group was required for inhibition. Polar substitution at C-3 decreased the inhibitory activity but this was least with a β-OH (DHEA is 3β-hydroxy-5β-androstan-17-one).
Ziboh et al. looked at DHEA inhibition of lipid synthesis and G6PD in rat skin [13]. They found that synthesis from acetate was inhibited 44–56% by 2.5 × 10−4 M DHEA. This could be reversed by adding NADPH. They made the important point that the plasma concentrations of DHEA are between 5 × 10−9 and 2 × 10−8 M (i.e., 2 to 3 orders of magnitude below the concentrations required to be effective in inhibiting lipid synthesis
Two papers have looked at the possibility that 11β-HSD1 is regulated by the pentose pathway flux; the first of these was by McCormick and colleagues [14]. They suggested that despite the fact that 11β-HSD1 is located in the endoplasmic reticulum that it is interconnected with the cytosolic pentose phosphate pathway, the primary producer of reduced pyridine nucleotides. They looked at this in intact rodent adipocytes. Inhibitors of NADPH generation via the pentose pathway (DHEA and norepinephrine) inhibited 11β-HSD1 oxido-reductase activity while decreasing cellular NADPH content. These compounds also augmented the reverse or dehydrogenase reaction (100 μΜ DHEA produced 187% increase in dehydrogenase activity). They then tested the compounds against isolated intact microsomes containing the tandem 11β-HSD1 and H6PDH unit. In this situation the inhibitors were not effective.
One of the surprising things about these results is that it is normally thought that the microsomal membrane is poorly permeable to pyridine nucleotides (i.e., that NADPH produced in the cytoplasm would not cross to enable 11β-HSD1 to act as a reductase). They incubated pyridine nucleotides with liver microsomes and found that NADPH very significantly increased 11β-HSD1 reductase activity. Interestingly NADP had no significant effect.
Another fascinating possibility needs consideration. It has been found that the sedoheptulose kinase CARKL directs macrophage polarisation through control of glucose metabolism [15]. Inhibition of CARKL by LPS, for example, results in a type 1 pro-inflammatory macrophage. This results in inhibition of the production of Fructose-6-phosphate (F-6-P) which normally contributes to glycolysis. Senesi et al. [16] showed that F-6-P contributes to glucocorticoid activation in the ER. It is transported into the ER and then isomerised to G-6-P which acts as the substrate for H6PDH, produces NADPH and activates the reductase action of 11β-HSD1.
A second key paper gives a different perspective. McNelis and colleagues [17] showed that DHEA had an anti-glucocorticoid action on human pre-adipocytes. Glucocorticoids stimulate human pre-adipocyte proliferation, differentiation and glucose uptake. Key to this is the local conversion of cortisone to cortisol by 11β-HSD1. The authors specifically looked at human cells given the relative lack of production of DHEA by rodent adrenals. They found that DHEA but not DHEA-S significantly inhibited pre-adipocyte proliferation and that this was due to reduced 11β-HSD1 oxido-reductase activity. This effect was significant at 1 μΜ DHEA and highly significant at 10 and 25 μΜ. At the highest dose (25 μΜ) there was a concurrent increase in dehydrogenase activity. DHEA also significantly increased insulin-independent glucose uptake.
The critically important part of the paper was the discovery that DHEA treatment inhibited the
The question is whether these two mechanisms (i.e., non-competitive inhibition of G6PD and decreased expression of H6PDH) are both relevant and, possibly, synergistic. Thus could DHEA inhibit the pentose pathway in the cytoplasm and thus reduce the availability of F-6-P for transfer into the ER? If DHEA can also inhibit the expression of H6PDH then this would further reduce the intra-ER level of NADPH and hence decrease 11β-HSD1 conversion of cortisone to cortisol and, with high levels of DHEA, enhance conversion of cortisol to cortisone. Given the likelihood that the sulfatase in the endoplasmic reticulum is active the basic model could be as follows:
DHEA-S enters the cell via a specific transporter (OATP-D) and then crosses into the smooth endoplasmic reticulum (ER) where it is converted to DHEA by the sulfatase enzyme.
DHEA then diffuses out into the cytoplasm and inhibits the pentose phosphate pathway by direct binding to G6PD. This then reduces the amount of F-6-P that is available to cross the ER membrane to be isomerized to G-6-P and thus limits the production of NADPH and hence drives dehydrogenase activity of 11β-HSD1 in the ER.
DHEA also inhibits the expression of H6PDH. This then also lowers the production of NADPH within the ER.
The Odermatt group reported [18, 19] reduced 11β-HSD1 expression following treatment with DHEA in 3T3-L1 adipocytes and in liver and adipose tissue of treated mice. They found that the CCAAT/enhancer binding protein (C/EBP) family of transcription factors (C/EBP-α, C/EBP-β, and C/EBP-δ) were involved in the DHEA-mediated effect on 11β-HSD1 expression. C/EBP-α and C/EBP-β were shown to bind directly to the 11β-HSD1 promoter and acted in concert to regulate gene expression in liver cells and adipocytes.
C/EBP-α, a potent activator of 11β-HSD1 gene transcription, was downregulated by DHEA treatment in the liver and adipose tissue of the treated mice in comparison to C/EBP-β and C/EBP-δ which were either elevated or unchanged. Thus a low C/EBP-α/C/EBP-β ratio was associated with decreased 11β-HSD1 transcription.
They then went on to investigate whether C/EBP might also modulate HSD11B2 gene expression. They found that DHEA up-regulated the expression and activity of 11β-HSD2 in rat renal cortical collecting duct cells. The main effect appeared to be via activation of gene transcription. In comparison to 11β-HSD1 11β-HSD2 was more stimulated by C/EBP-β and a low C/EBP-α/C/EBP-β ratio was linked to increased expression of 11β-HSD2. They suggested that DHEA modulated the expression of 11β-HSD2 in a phosphatidylinositol-3-kinase/AKT-dependent manner by increasing C/ΕΒP. In the context of this paper and parturition it is of interest that they investigated whether metabolism of DHEA to oestrogen or androgen was required for the DHEA effect on 11β-HSD2. Neither the oestrogen receptor antagonist tamoxifen nor the androgen receptor antagonist flutamide affected the DHEA-mediated upregulation of 11β-HSD2.
4. Transcription factors and 11β-HSD1/2 transcription
No such studies have been performed in relation to DHEA and the placental 11β-HSD2. It would of major physiological and potentially pathological interest if DHEA enhanced the transcription of placental 11β-HSD2 via this mechanism. DHEA did not affect 11β-HSD2 expression in Caco-2 and SW620 colon cells, suggesting tissue-specific regulation [19].
Chapman has reviewed the role and regulation of 11β-HSD1 in inflammation [20]. The enzyme is transcribed from three promoters, P1, P2 and P3. Liver and brain transcription is mainly from P2 and, as described above, is dependent on C/EBP as is the acute phase response. Mice lacking C/EBPα cannot mount such a response and die from neonatal hypoglycaemia. Pro-inflammatory cytokines interact with this mechanism. TNF-α increases the ratio of C/EBPα/C/EBPβ and hence enhances the transcription of 11β-HSD1. It is suggested that TNF promotes C/EBPβ phosphorylation and thus its export from the nucleus.
Working in a very different area Hardy et al. studied fibroblasts from patients with either rheumatoid arthritis or osteoarthritis [21]. These contained 11β-HSD1. Expression and activity of the enzyme increased following treatment with either IL-1β or TNFα (bone marrow: 37 and 8-fold respectively compared to vehicle; dermal fibroblasts 14-fold and 4-fold; synovial fibroblasts 31-fold and 7-fold; all p < 0.001 compared to vehicle). If this was happening in the amnion and 11β-HSD1 was acting as an oxido-reductase this could markedly increase the intracellular glucocorticoid levels.
This effect may well depend on the cell type used. Thus Ahasan et al. [22] have reported that TNFα and IL-1β increase the expression and activity of 11β-HSD1 in mesenchymal stromal cell types and tissues. This was different to what had been found in hepatocytes and blood cells. In addition combined treatment of glucocorticoids and pro-inflammatory cytokines
Li et al. [25] looked at the relation between cortisol and expression of 11β-HSD1 by IL1-β In cultured human chorionic trophoblast cells. They found that cortisol significantly increased 11β-HSD1 expression and reductase activity. They suggested that the chorion was the most abundant site of 11β-HSD1 within intrauterine tissues. They found that treatment of chorionic trophoblast cells with cortisol (1 μΜ) induced both cytosolic phospholipase A2 and cyclooxygenase 2 mRNA expression. They concluded that cortisol up-regulated 11β-HSD1 expression through induction of promoter activity, and the effect was enhanced by IL-1, suggesting that more biologically active glucocorticoids could be generated in the fetal membranes in the presence of infection.
5. The metabolism of DHEA
Also located within the endoplasmic reticulum (ER) in many tissues which use 11β-HSD1 to control local glucocorticoid levels is the CYP7B1 enzyme that 7-hydroxylates DHEA. 7OH-DHEA is converted to 7keto-DHEA by 11β-HSD1 [26]. This form of DHEA is more potent for promoting memory function in mice [27]. This 7-hydroxylase enzyme is found in the human placenta with high levels in decidual and trophoblastic cells. The function of this molecule is unknown.
There is a clear difference between the liver hydroxylation of DHEA and that of other tissues. Tissue extracts with the exception of the liver from mice homozygous for targeted disruption of CYP7B fail to generate hydroxylated products of DHEA. The liver has a separate enzyme CYP7A.
This could be very interesting in suggesting that the metabolic needs of the liver are rather different to tissues such as the skin and adipocyte.
Fetal 16-hydroxylation of DHEA and DHEA-S appears to be important [28]. IIt is suggested that steroid sulfates are directly metabolized without prior cleavage of the sulfate group in human fetal livers. The level of DHEA 16α-hydroxylase in human fetal liver is significantly higher than in adult liver. Estriol of fetal origin originates exclusively from the 16- hydroxylation of either DHEA or DHEA-S. The hydroxylated product undergoes aromatization in the syncytiotrophoblast and the estriol then transfers into the maternal circulation [29].
Due to the haemochorial nature of the placenta it is suggested that more than 90% of estriol generated in the syncytiotrophoblast enters the maternal circulation. Nothing is known about a possible interaction between 16OH-DHEA-S and 11β-HSD1 activity. This is important given that cord plasma 16OH-DHEA-S levels are approximately 2.5 times those of DHEA-S.
All this suggests that either the local production and metabolism of DHEA within the ER or its hepatic metabolism may be important. Could it be that metabolites of DHEA and not DHEA itself could affect H6PD expression and hence NADPH production? Thus local conversion of DHEA to 7OH-DHEA and its move to the cytoplasm could produce the non-competitive inhibition of G6PD. This would result in 11β-HSD1 acting as a dehydrogenase and hence converting 7OH-DHEA to 7-keto-DHEA, known to be a more potent neurosteroid and possibly an inhibitor of G6PD.
6. Placental sulfatase
One of the intriguing aspects of this hypothesis is that it potentially explains how it is that DHEA might be biologically relevant even though the circulating levels are two orders of magnitude below those that are necessary to affect NADPH production. It is the generation of DHEA within the ER by the action of steroid sulfatase on DHEA-S that produces very high intracellular DHEA levels. Thus DHEA-S acts as a pro-hormone that is then activated in cells with steroid sulfatase. This enzyme is within the ER and hence ideally placed to generate high levels of DHEA.
Sulfatase activity was first demonstrated in the human placenta by Pulkkinen in 1961 [30] and confirmed by Warren and colleagues [31].
If the hypothesis is correct then placental sulfatase deficiency is of particular interest. Rabe described 76 pregnancies with this [32]. The pregnancies are associated with low oestriol secretion and a prolonged half-life of DHEA-S after venous loading. Only 7 out of 76 pregnancies had an uncomplicated vaginal delivery. Eighteen required Caesarean section. All had a prolonged pregnancy with lack of cervical dilatation and absence of the normal process for the induction of labour. All 76 babies were male. Sulfatase deficiency is a sex-specific, X-linked placental enzyme defect.
7. Why does the placenta have both 11β-HSD1 and 11β-HSD2?
In 1997 we published evidence that cortisol infused into the maternal side of the human placenta was metabolised to cortisone (Figure 2) [33]. No conversion of cortisone to cortisol was detected. This study was not designed to examine whether this conversion was solely by 11β-HSD2 or whether 11β-HSD1 could be playing a role. A further more detailed study was published by Sun et al. [34]. They perfused cortisol into the maternal intervillous space of term placentae and found substantial cortisol in the fetal vein. The output of cortisol was increased when carbenoxolone, a non-specific inhibitor of 11β-HSD was added. Of particular interest was their finding that cortisol formation increased in a dose-dependent manner with infusion of cortisone. Cortisol was detected at a low level in both the maternal and fetal outflows. This suggests that
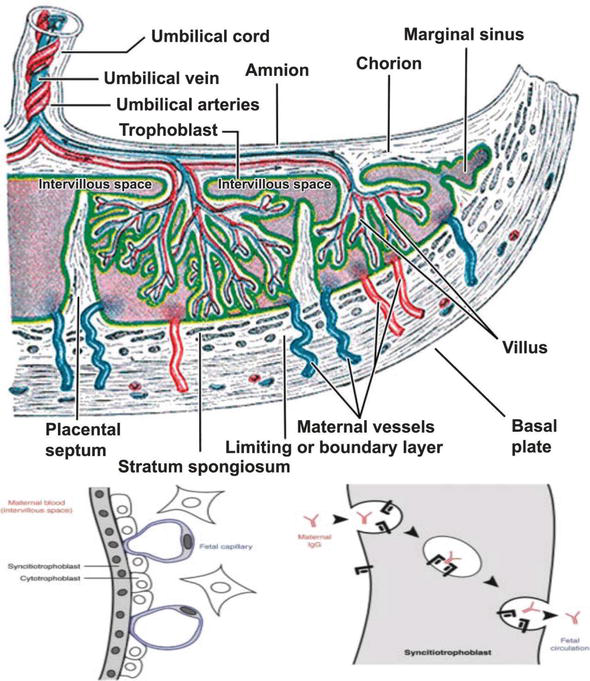
Figure 2.
Diagram of the human placenta. Maternal cortisol enters the inter-villous space and is then metabolised to cortisone by the 11β-HSD2 enzyme in the syncytiotrophoblast. The fetus is either then perfused with cortisone via the umbilical vein or with cortisol if the inactivation is ineffective or the 11β-HSD1 in the amnion, chorion and decidua acts as an oxido-reductase.
In the kidney the inactivation of cortisol to cortisone plays a critically important role in the protection of the mineralocorticoid receptor. The affinity of the glucocorticoid receptor for cortisol is 10–40 times lower than that of the MR. Thus in the kidney there is a need for an extremely effective protective mechanism. Presumably this is also necessary for the placenta in its role of protecting the fetus from maternal cortisol.
Sun et al. looked at the differential expression of 11β-HSD1 and 11β-HSD2 in human placental and fetal membranes [24]. They found that 11β-HSD2 was present in the placenta in the syncytiotrophoblast (Figure 2). By contrast 11β-HSD1 was found in the cytotrophoblast, amnion and chorion. 11β-HSD2 was thus well placed to protect the fetus from maternal cortisol. In the mother the circulating levels of free cortisol and cortisone are approximately equal [35].
Sun et al. [36] suggest that cortisol regeneration in the fetal membranes is not a coincidental but a required event for human parturition. In addition to the role of cortisol in stimulating prostaglandin synthesis (see below) they suggest that local cortisol production induces extracellular matrix (ECM) remodelling and that decreased collagen I, III and IV plays a key role in membrane rupture.
If therefore placental 11β-HSD1 acted normally throughout pregnancy as an oxido-reductase, as most of the literature suggests, it would convert maternal cortisone and cortisone derived from syncytiotrophoblast metabolism to cortisol. This makes little biological sense and is at variance with the studies quoted above which indicate that for the majority of human pregnancy circulating fetal cortisol levels are very low. This suggests that not only is placental 11β-HSD2 effective but that 11β-HSD1 is either down-regulated or acts as a dehydrogenase. This underlines the importance of the mechanism responsible for determining the set point of 11β-HSD1.
Murphy et al. [37] showed that placental 11β-HSD2 activity decreased significantly between 38 and 40 weeks. In contrast there were no changes in 11β-HSD2 mRNA or protein expression.
In comparison 11β-HSD1 mRNA abundance markedly increased with the onset of spontaneous labour as did fetal cortisol concentrations. The presence of the two enzymes in the placenta and 11β-HSD1 in the fetal membranes raises some fascinating questions. Could they function in different ways at different times during pregnancy? Thus placental 11β-HSD2 might be the main way by which the fetus is protected from maternal cortisol but 11β-HSD1 could play an additional local protective role as a dehydrogenase in the majority of the pregnancy (i.e., synergistic with 11β-HSD2). It could then be an oxido-reductase at term increasing
Sun and Myatt [23] looked at 11β-HSD1 expression in cultured human amnion cells, the major tissue producing prostaglandins. Term amnion fibroblasts had higher 11β-HSD1 expression than amnion epithelial cells. Dexamethasone dose-dependently increased 11β-HSD1 expression in amnion fibroblasts but not epithelial cells. This was blocked by the GR receptor antagonist RU486. The cytokines IL-1β and TNFα had little effect on 11β-HSD1 expression alone but in combination with dexamethasone significantly enhanced expression. These results suggest that the control of the amniotic glucocorticoid environment could play an important role in the genesis of parturition.
Konstantakou et al. reviewed the 11β-HSD dysregulation during pregnancy and beyond [38]. This focuses on the critical role played by glucocorticoids in developmental programming. Both animal and human studies have confirmed that 11β-HSD2 insufficiency is related to pregnancy complications (pre-eclampsia, intra uterine growth retardation (IUGR), pre-term birth, low birth weight and an increased risk of hypertension, heart disease and type 2 diabetes in adulthood). Konstantakou suggested that the main influence of 11β-HSD1 on fetal physiology is its contribution to fetal lung maturation. This is clearly important but If this was the case it would be difficult to understand why the enzyme is in the placenta and fetal membranes. An interesting study suggested that polymorphisms within the 11β-HSD1 gene conveyed an increased risk of pregnancy-induced hypertension and pre-eclampsia [39].
In summary, 11β-HSD2 with its low Km for cortisol (about 50 nM) (i.e., high affinity) is well placed in the syncytiotrophoblast to convert maternal cortisol to cortisone and thus protect the fetus. By contrast 11β-HSD1 with its higher Km (380 nM for cortisone) is localised to the cytotrophoblast, chorion and amnion and can control local glucocorticoid levels there.
8. Glucocorticoids and prostaglandin production
It might be thought that, given the classical anti-inflammatory effect of glucocorticoids, they would suppress the production of prostaglandins; the reverse appears to be the case. This paradox was noted by Casey et al. [40]. They found that PG production by human amnion and decidual cells in culture was not inhibited by dexamethasone. Indeed the concentration of PGE2 in the culture medium at day 3 after dexamethasone had been added was doubled. This was very different to the effect of dexamethasone on myometrial smooth muscle cells producing prostacyclin responsible for uterine quiescence. This prostaglandin was markedly inhibited at low dose dexamethasone.
Sun’s group delineated the paradoxical mechanism and has admitted that this is very complicated [36]. It involves the glucocorticoid induction of cytosolic phospholipase A2 and COX-2 with subsequent activation of multiple transcription factors.
Others confirmed the paradoxical effect in human amnion and showed that PG stimulation by glucocorticoids induces expression of COX-2 [23]. Amnion fibroblasts produced 50-fold more PGE2 per cell than epithelial cells. Dexamethasone (0.01–1 μM) increased PGE2 production in amnion fibroblasts but did not affect PG production in amnion epithelial cells. As indicated above it is the amniotic fibroblasts that contain 11β-HSD1.
Dexamethasone is obviously not an endogenous GR ligand. It is metabolised by 11β-HSD2 to 11-dehydro dexamethasone. 11β-HSD1 can convert this back to dexamethasone but is unable to carry out the reverse dehydrogenase reaction. It is thus important to confirm that cortisol has the same effects. This has been demonstrated by Guo et al. [41]. They also showed that the effect of cortisol on COX-2 in human amnion fibroblasts was modulated by progesterone.
9. Glucocorticoids and oxytocin
Another important possible interaction is between glucocorticoids and oxytocin in amniotic cells. Hinko and Soloff [42] found that oxytocin receptors (OTRs) in rabbit amnion increased 200 fold towards the end of gestation. To study this they first incubated the amnion cells with 20nM cortisol for 24 hours. This increased labelled oxytocin antagonist binding 16-18 fold [43].
Studies with human amniotic cells suggested that labour and the inflammatory cytokine IL-1β increased the expression of oxytocin receptors [44]. They used primary human amnion cell cultures from patients before and after the onset of labour. The expression of OTR mRNA was 16 fold higher in the post-labour cells than in the pre-labour. Treatment of pre-labour cells with IL-1β brought the level of OTR expression to that observed in post-labour cells. In post-labour cells IL1β produced no significant further increase in OTR.
Previous studies have shown that IL-1β concentrations within the uterus increase at the time of either term or pre-term labour [43].The same is true for several other so-called labour associated proteins including PTGS2 and IL-8.
9.1 DHEA and nuclear factor kappa B (NF-κB)
Labour, both term and pre-term, is associated with an increase in both the expression and activity of cyclo-oxygenase 2 (PTGS2) in the amnion, the key enzyme involved in the synthesis of prostaglandins [45]. It is regulated by NF-κB [44]. Both basal NF-κB DNA binding and transcriptional activity increase in human amnion at the onset of labour. PTGS2 expression was increased 40 fold and the oxytocin receptor 24 fold. In addition to increased oxytocin receptor mRNA, inflammatory cytokines such as IL-I are found within the amniotic fluid in relation to both term and pre-term labour often related to infection [43]. It is thus of interest that DHEA-S has been shown to markedly inhibit the expression of NF-κB in response to pro-inflammatory cytokines such as TNFα [46]. In the myometrium where abundant Progesterone Receptor B (PRB) is expressed prostaglandin synthesizing enzymes are among the NF-κB mediated pro-inflammatory genes that are inhibited by progesterone during gestation. Is the effect of DHEA-S on NF-κB an additional way in which DHEA can modulate the
NF-κB is essential for up-regulation of interleukin-8 expression in human amnion and cervical epithelial cells [47].
9.2 NF-κB and amniotic oxytocin receptors
Terzidou et al. [44] have shown that both labour and inflammation increase the expression of oxytocin receptors (OTRs) in the human amnion. Amniotic prostaglandin synthesis occurs mainly via cyclo-oxygenase 2 that is regulated by NF-κB. Labour, both pre-term and term, is associated with increased expression of this enzyme in the amnion and basal NF-κB DNA binding and transcriptional activity increase with the onset of labour. Using whole genome cDNA arrays they studied the range of genes that have increased expression in amnion that had high levels of NF-κB compared to amnion with low levels [47]. OTR expression showed the second highest difference with 24 fold increase in expression versus 40 fold for COX-2. A total of 919 genes were up-regulated including numerous inflammatory genes. They concluded that amnion activation is largely an inflammatory event that occurs in the amnion epithelial layer as a prelude to the onset of labour.
9.3 Interaction between the NF-κB and C/EBP transcription factor families
Given the very specific effects of pro-inflammatory cytokines such as TNF-α and DHEA on NF-κB and C/EBP transcription factors it is interesting to ask whether they interact. Stein et al. have shown that there is a clear functional and physical interaction [48]. This results in inhibition of promoters with kappa B enhancer motifs and in the synergistic stimulation of promoters with C/EBP binding sites.
10. What controls the production of DHEA-S by the fetal adrenal
If DHEA-S plays regulates placental 11β-HSD1 and 11β-HSD2 then what controls adrenal production of DHEA-S? Mesiano and Jaffe reviewed human fetal adrenal function in pregnancy and parturition [49]. The human placenta produces Corticotrophin Releasing Hormone (CRH) (41 amino acids: Chromosome 8) and the closely related urocortin (40 amino acids: chromosome 2). CRH directly and preferentially stimulates DHEA-S production by fetal adrenal cortical cells [50]. CRH was as effective as ACTH at stimulating DHEA-S production but 70% less potent than ACTH in stimulating cortisol. CRH production was up-regulated by cortisol.
There are two main types of CRH receptor—CRH-R1 and CRH-R2. Sirianni et al. showed that CRH and Urocortin act through type 1 CRH receptors to stimulate DHEA-S production in human fetal adrenal cells [51]. Urocortin mRNA is present in the cytotrophoblast and syncytiotrophoblast [52]. Unlike CRH, urocortin is not secreted into the maternal blood.
Maternal CRH levels rise exponentially as human pregnancy advances [53]. The CRH binding protein (CRH-BP) binds the hormone in an equimolar ratio and prevents the recognition of CRH by the CRH receptor. McLean and colleagues studied this in 485 pregnant women. CRH was detectable in the maternal circulation from early in the second trimester and then rose dramatically. Three hundred and eight women went into spontaneous labour at term: 24 had spontaneous pre-term delivery and 29 were delivered beyond term. The CRH plasma levels were significantly higher in those delivering pre-term and significantly lower in those delivering post-term. These differences were present from the earliest stage in pregnancy when CRH could be detected. CRH-BP was present greatly in excess of CRH throughout most of pregnancy but the exponential rise in CRH resulted in an equivalence point being reached 20 days before spontaneous term delivery. The CRH-BP levels then decreased significantly so that CRH was present in excess of CRH-BP at serial time points from 20 days before spontaneous delivery. The authors suggest that CRH-BP will block the bioactivity of CRH in the maternal circulation until the final 3 weeks before the onset of spontaneous labour. It is unclear what role CRH-BP might play in the fetal circulation but Perkins et al. have shown that CRH binding in fetal plasma is virtually the same as the maternal samples. If this affects the bioactivity of CRH does this prevent CRH from stimulating the synthesis and release of DHEA-S? Does this suggest that urocortin acting via the adrenal type 1 CRH receptor controls fetal DHEA-S levels?
It is suggested that humans are the only species to have high levels of placental CRH at term and no other species has been reported to have circulating CRH-BP.
Thus if DHEA-S by intra-placental conversion to DHEA controls both the conversion of cortisol to cortisone by 11β-HSD2, as might be the case given the studies of the Odermatt group on this enzyme in the kidney [54] and also the oxido-reductase activity of 11β-HSD1 the following could obtain:
Low DHEA-S results in decreased 11β-HSD2 expression and increased 11β-HSD1 conversion of cortisone to cortisol in the placenta.
The increase in placental cortisol then enhances the production of placental urocortin that in turn stimulates the fetal adrenal to produce DHEA-S—a classic feedback control system.
Given that the level of CRH rises exponentially during gestation and that the free level of this hormone may be linked to the timing of parturition this poses the question ‘What role does placental CRH play in the activation of the fetal hypothalamic pituitary adrenal axis?’. Does free CRH from the maternal circulation cross into the fetus? Is the free level such that it can not only activate the fetal HPA but also directly stimulate cortisol production by the fetal adrenal as shown by Sirianni et al. [51]. If this is so then there are several ways by which circulating cortisol levels in the fetus may rise just before term.
Decreased placental 11β-HSD2 activity just before term reduces the fetal protection from maternal cortisol. The transfer increases placental cortisol and hence placental CRH. This then stimulates both the fetal pituitary to secrete ACTH and the adrenal to produce DHEA-S. The ACTH then activates the transitional zone that synthesises cortisol. This further stimulates CRH and hence cortisol.
The reduction in maternal CRH-BP results in increased free CRH that crosses the placenta.
11. DHEA-S, cortisol and the onset of labour
Yoon et al. [55] showed that an increase in fetal plasma cortisol but not DHEA-S is followed by the onset of pre-term labour in patients with pre-term premature rupture of the membranes. In those who went into spontaneous labour within 7 days the mean fetal plasma cortisol was 8.35 μg/dl. In contrast those whose onset of labour was beyond 7 days had a mean cortisol of 4.75 μg/dl (p < 0.0001). In contrast the DHEA-S levels in the first group had a mean of 154 μg/dl versus 194 μg/dl in the second (p = 0.09). This work was confirmed by Oh et al. [56]. They found that fetal plasma cortisol was low until 36 weeks gestation and increased then up to term. Active labour was associated with an increase in plasma cortisol. Fetal plasma DHEA-S increased up to term but not during active labour. What they described as the stress index (cortisol/DHEA-S) increased with progressive gestation and with active labour at term. Others confirmed that infants delivered by Caesarean section had the lowest cortisol levels. Fetal plasma cortisol was significantly elevated in connection with spontaneous human parturition and highest in those born by instrumental delivery [57].
12. The switch from glucocorticoid protection to local glucocorticoid excess
This paper suggests that 11β-HSD enzymes play a key role in protecting the fetus from glucocorticoids. 11β-HSD2 does this by converting maternal cortisol to cortisone in the syncytiotrophoblast and hence together with the failure of cortisol production by the fetal adrenal produces low circulating cortisol levels in the fetus from about 8–10 weeks gestation until just before term. 11β-HSD1 is expressed at low level in the amnion, chorion and decidua and probably acts as a dehydrogenase converting cortisol to cortisone locally within these tissues. DHEA-S produced by the fetal adrenal is then converted to DHEA in the placenta and membranes by placental sulfatase. This local production of DHEA plays a key role in enhancing expression of 11β-HSD2 and decreasing the expression of 11β-HSD1 and H6PDH. Because of the avascular nature of the amnion it is likely that DHEA diffuses through the amnion cells to reach the fibroblasts containing 11β-HSD1.
Key to understanding parturition must very likely involve understanding how the fetus switches from low circulating and low intracellular cortisol levels to higher circulating and very high local glucocorticoid levels which switch on the increased production of oxytocin receptors, prostaglandin E2 and inhibit prostacyclin. The switch does not appear to be related to a decrease in the circulating levels of DHEA-S. This poses the question as how an increase in cortisol or other agents such as pro-inflammatory cytokines might block the inhibitory effect of DHEA.
It is clear that there are a number of key interactions between transcription factors, pro-inflammatory cytokines and glucocorticoids [58]. Grontved looked at the interaction between C/EBP and the Glucocorticoid Receptor (GR). As previously discussed DHEA has a specific effect on the transcription of 11β-HSD1. In particular it decreases the expression of C/EBPα and thus decreases the ratio of C/EBPα/C/EBPβ. What Grontved has shown is that C/EBPβ plays a key role in facilitating glucocorticoid receptor recruitment to DNA steroid response elements. The GR is exclusively associated with accessible chromatin and 62% of GR binding sites are occupied by C/EBPβ. Τhe paper shows that in the liver C/EBPβ frequently occupies the site prior to GR binding and sometimes joins GR at the site after activation. The authors found that accessibility of DNA is highly linked to occupancy of the DNA by C/EBPβ and suggest that C/EBPβ has a priming function for GR binding (Figure 3).
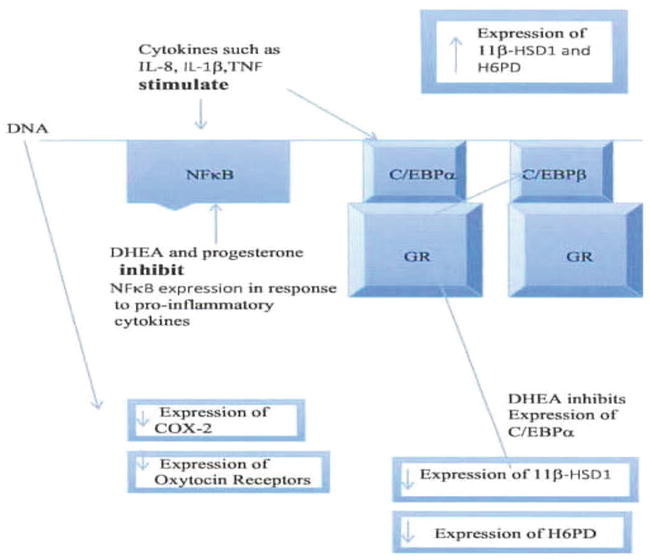
Figure 3.
Interaction of transcription factors with DHEA and pro-inflammatory cytokines with consequent effects on expression of key enzymes and receptors related to parturition.
Rudiger et al. [59] focus on the fact that the majority of glucocorticoid-modulated genes lack a DNA binding site for the glucocorticoid receptor (GR). They test the hypothesis that besides direct DNA binding of the GR, interaction with other transcription factors modulates the effect of GR binding. Using cultured human mesenchymal cells and leukocytes they found that glucocorticoids induced the formation of a complex between the GR and enhancer binding protein C/EBPα. This interaction inhibited glucocorticoid action on mesenchymal and lymphocytic cell proliferation.
13. A comparison of sheep and human parturition
Seminal papers by Liggins and colleagues showed that parturition in sheep is initiated by a sharp rise in the rate of secretion of cortisol by the fetal adrenal [60]. The rise in secretion was thought to be due to a combination of enhanced responsiveness to ACTH and partly to increased fetal ACTH levels. As discussed above cortisol acts in the sheep on placental enzymes active in the biosynthesis of oestrogens from progesterone. Thus placental secretion of oestrogen increases and that of progesterone decreases. They proposed that this change in the ratio of oestrogen: progesterone, particularly the rise in oestrogen, stimulated the release of prostaglandin F2α (PGF2α) from the maternal placenta and myometrium. PGF2α then enhanced the myometrial response to oxytocin and, after a latent period, stimulated contractions. They noted that the onset of parturition is normally associated with softening of the cervix, the mechanism of which is uncertain. The combination of uterine contractions in the presence of a distensible cervix leads to parturition.
In man as already discussed the process has long been thought to be different from that in sheep. In man the placenta was thought to lack the glucocorticoid responsive CYP17 enzyme. This has now been disputed. What this paper suggests is that the fundamental process is cortisol dependent in both species. What differs is the way in which the human fetal adrenal plays a crucial role in controlling the placental and tissue glucocorticoid levels. The effective loss of this control results in a marked increase in the intracellular and peri-cellular levels of cortisol in key tissues secondary to the decreased inactivation of maternal cortisol by placental 11β-HSD2 and the increased expression and oxido-reductase activity of 11β-HSD1 in the amnion, chorion and placenta. This initiates a cascade that stimulates
13.1 Pathologies that might affect the modulation of 11β-HSD1 by DHEA
At first sight it might be thought that placental sulfatase deficiency would be a good test of the hypothesis. One might imagine that in such pregnancies the fetus would not be able to produce high concentrations of DHEA within the placenta and thus could not reduce the oxido-reductase activity of 11β-HSD1. The fetus would then be exposed to a much higher
13.2 Pathologies that might affect the modulation of 11β-HSD2
Of particular interest is work showing that cholestatic pregnancy is associated with reduced placental 11β-HSD2 expression [62]. Morris et al. [63] showed that the bile acid chenodeoxycholic acid was a potent inhibitor of 11β-HSD1 dehydrogenase activity. Higher levels were required to inhibit 11β-HSD2. Of special importance was the finding that maternal cholestasis in pregnancy programs metabolic disease in the offspring (males had increased BMI and females increased waist and hip girth) [62]. This was very much in keeping with our studies in pregnant rats given dexamethasone in pregnancy [64]. Given the current global obesity pandemic it would be important to determine what role such neonatal programming is playing in the genesis of the disease.
14. Progesterone
Progesterone plays a key role not only in the maintenance of pregnancy but as a promoter of uterine quiescence. Is there any possibility that it could act in conjunction with DHEA?
In a surprising finding Nadjafi-Triebsch and colleagues discovered that males given DHEA had a rise in their progesterone levels. The mechanism for this was unclear.
Mizutani et al. [65] showed that C/EBPβ plays an important role in progesterone synthesis. These authors found that steroidogenic factor 1 (SF-1) induced the differentiation of mesenchymal stem cells into steroidogenic cells. They then looked at the mechanisms by which SF-1 modulated functions. A key component of the SF-1 nuclear protein complex was C/EBPβ. C/EBPβ is known to be essential for ovulation and luteinisation. C/EBPβ knock down in granulosa cells reduced cAMP- induced progesterone production. They concluded that C/EBPβ is an important mediator of progesterone production by working together with SF-1, especially under trophic hormone-stimulated conditions.
As discussed above DHEA has been shown to down-regulate C/EBPα and hence reduce the C/EBPα/C/EBPβ ratio. These transcription factors act either as homodimers or heterodimers. Does the decrease in C/EBPα lead to an increase in the C/EBPβ homodimer and hence enhance progesterone production as found in the Nadjafi-Triebsch paper?
15. Purinergic receptors in the myometrium
Calcium plays a key role in myometrial contraction and in the oxytocin response [66]. Like in other smooth muscle calcium binds to calmodulin and activates myosin light chain kinase. This can then phosphorylate myosin light chains at serine residue 19. This then allows the myosin crossbridge to bind to actin. The factors that control intracellular calcium in the myometrium are thus of special interest. We demonstrated that aldosterone stimulated the release of ATP from A6 cells [67] which acted on purinergic receptors (P2X4) on the releasing and adjacent cells and thus stimulated calcium entry into the cell. This produced cell contraction and opening of the sodium channel. This could be prevented by adding hexokinase to the cells.
Rat myometrial cells express both P2X4 and P2X7 receptors [68]. The expression of these receptors increased during the late stages of pregnancy. This increase was markedly enhanced in hormone-induced and inflammation-induced pre-term delivery models. Mifepristone increased P2X4 and P2X7 levels 2.1 and 4.1 fold: lipopolysaccharide by 7.4 and 18.6 times the control value.
16. Conclusions
Diczfalusy et al. [69] proposed that the role of the fetal zone of the human fetal adrenal was to produce large amounts of adrenal androgens which were then converted to oestrogens by the placenta. Despite the fact that the role of this oestrogenic milieu is still quite unclear this has been the suggested rationale for the enormous production of DHEA-S (Figure 4).
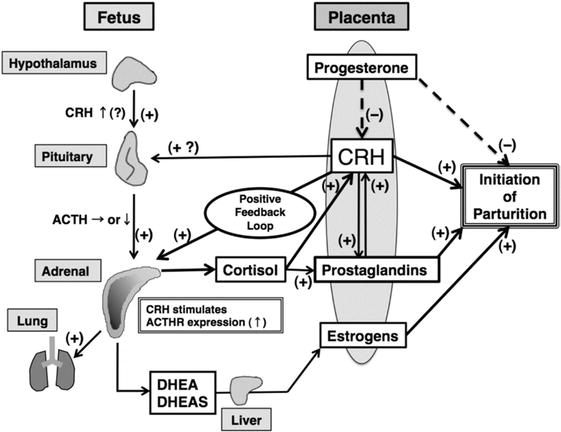
Figure 4.
Classical model of human feto-placental unit (Source: From Ishimoto and Jaffe [
It might seem strange that the FZ develops and DHEA-S secretion starts to increase at the end of the first trimester but the proposed roles of oestrogen are in parturition. One possibility is that cortisol and oestrogens both play a role in parturition that is synergistic (Figure 5).
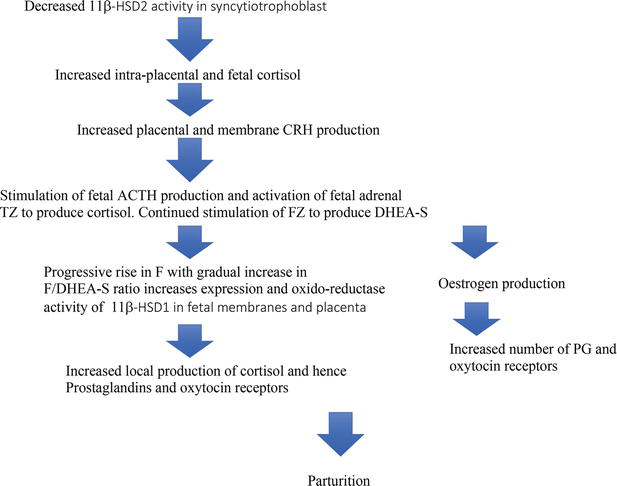
Figure 5.
Possible sequence of events leading to human parturition.
DHEA-S in addition to its function as a source of oestrogens has a vital function as the modulator of 11β-HSD1. Thus for the major part of pregnancy this results in this enzyme having both its expression reduced and its function controlled as a dehydrogenase converting cortisol to cortisone. This in conjunction with placental 11β-HSD2 results in a low glucocorticoid environment
The interaction between DHEA and progesterone leads to a different model of parturition (Figure 6).
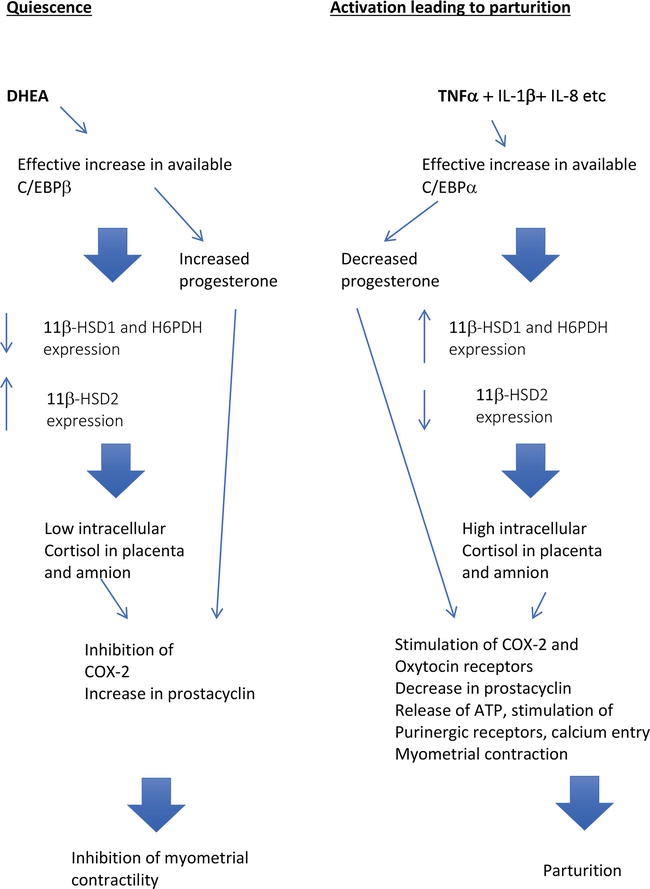
Figure 6.
Model 2 showing the interaction of DHEA with enhancer binding proteins and progesterone producing uterine quiescence and the activation steps leading to parturition.
In the first part of the model DHEA and progesterone are synergistic in producing quiescence during the majority of pregnancy. DHEA increases the C/EBPβ: C/EBPα ratio and thus the availability of C/EBPβ. This together with SF-1 stimulates the synthesis of progesterone which inhibits production of prostaglandins by COX-2. At the same time by the same mechanism DHEA decreases 11β-HSD1 and H6PDH expression resulting in very low amniotic and chorion intracellular cortisol levels. In the myometrium low levels of glucocorticoid enhance the production of prostacyclin and hence uterine quiescence.
In this model parturition is produced, as suggested by Lim et al. [47], by an increase in pro-inflammatory cytokines such as IL-8 TNFα and IL1-β (levels rise in normal pregnancy at term and are significantly higher with infection) that stimulate the production of C/EBPα. This then blocks the inhibitory effect of DHEA on the expression of 11β-HSD1 and H6PDH and also the stimulatory action on 11β-HSD2 expression. As a result there is decreased inactivation of maternal cortisol with increased availability of cortisol within the placenta. This cortisol is synergistic with TNFα and IL1-β in increasing 11β-HSD1 expression and oxido-reductase activity that enables the fetal membranes to convert cortisone to cortisol and thus produce a further major increase of cortisol within the cells. The increased production of C/EBPα induced by inflammatory cytokines inhibits prostaglandin synthesis which is dependent on C/EBPβ. This combination of an increase in local cortisol concentrations coupled with the decrease in progesterone within the cell results in COX-2 induction and hence prostaglandin production. The local glucocorticoid production also stimulates a marked increase in oxytocin receptors, inhibits uterine prostacyclin production and hence enhances myometrial contractility. Myometrial contraction is further enhanced by TNFα and IL1-β increasing the expression of P2X4 and P2X7 receptors in the myometrium. These may play an important role in the elevation of intracellular calcium induced by oxytocin.
References
- 1.
Escobar JC, Patel SS, Beshay VE, Suzuki T, Carr BR. The human placenta expresses CYP17 and generates androgens De Novo . The Journal of Clinical Endocrinology and Metabolism. 2011;96 (5):1385-1392 - 2.
Hong SH et al. Expression of steroidogenic enzymes in human placenta according to the gestational age. Molecular Medicine Reports. 2019; 49 (5):3903-3911 - 3.
Ishimoto H, Jaffe RB. Development and function of the human fetal adrenal cortex: A key component in the feto-placental unit. Endocrine Reviews. 2011; 32 (3):317-355 - 4.
Pignatti E, du Toit T, Flück CE. Development and function of the fetal adrenal. Reviews in Endocrine & Metabolic Disorders. 2023; 24 (1):5-21 - 5.
Goto M et al. In humans, early cortisol biosynthesis provides a mechanism to safeguard female sexual development. The Journal of Clinical Investigation. 2006; 116 (4):953-960 - 6.
Murphy BEP. Human fetal serum cortisol levels related to gestational age: Evidence of a midgestational fall and a steep late gestational rise, independent of sex or mode of delivery. American Journal of Obstetrics and Gynecology. 1982; 144 (3):276-282 - 7.
Edwards CR et al. Localisation of 11 beta-hydroxysteroid dehydrogenase—Tissue specific protector of the mineralocorticoid receptor. Lancet. 1988; 2 (8618):986-989 - 8.
Stewart PM, Corrie JE, Shackleton CH, Edwards CR. Syndrome of apparent mineralocorticoid excess. A defect in the cortisol-cortisone shuttle. The Journal of Clinical Investigation. 1988; 82 (1):340-349 - 9.
Bujalska IJ et al. Hexose-6-phosphate dehydrogenase confers oxo-reductase activity upon 11 beta-hydroxysteroid dehydrogenase type 1. Journal of Molecular Endocrinology. 2005; 34 (3):675-684 - 10.
Barash V, Erlich T, Bashan N. Microsomal hexose-6-phosphate and 6-phosphogluconate dehydrogenases in extrahepatic tissues: Human placenta and pig kidney cortex. Biochemistry International. 1990; 20 (2):267-274 - 11.
Marks PA, Banks J. Inhibition of mammalian glucose-6-phosphate dehydrogenase by steroids. Proceedings of the National Academy of Sciences of the United States of America. 1960; 46 (4):447-452 - 12.
Raineri R, Levy HR. On the specificity of steroid interaction with mammary glucose 6-phosphate dehydrogenase. Biochemistry. 1970; 9 (11):2233-2243 - 13.
Ziboh VA, Dreize MA, Hsia SL. Inhibition of lipid synthesis and glucose-6-phosphate dehydrogenase in rat skin by dehydroepiandrosterone. Journal of Lipid Research. 1970; 11 (4):346-354 - 14.
McCormick KL, Wang X, Mick GJ. Evidence that the 11 beta-hydroxysteroid dehydrogenase (11 beta-HSD1) is regulated by pentose pathway flux. Studies in rat adipocytes and microsomes. The Journal of Biological Chemistry. 2006; 281 (1):341-347 - 15.
Haschemi A et al. Article the sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metabolism. 2012; 15 (6):813-826 - 16.
Senesi S, Legeza B, Balazs Z, Csala M, Odermatt A. Contribution of fructose-6-phosphate to glucocorticoid activation in the endoplasmic reticulum: Possible implication in the metabolic syndrome. Endocrinology. 2010; 151 (October):4830-4839 - 17.
McNelis JC et al. Dehydroepiandrosterone exerts antiglucocorticoid action on human preadipocyte proliferation, differentiation, and glucose uptake. American Journal of Physiology. Endocrinology and Metabolism. 2013; 305 (9):E1134-E1144 - 18.
Apostolova G, et al. Dehydroepiandrosterone Inhibits the Amplification of Glucocorticoid Action in Adipose Tissue. American Journal of Physiology-Endocrinology and Metabolism. 2005; 288 :E957-E964 - 19.
Balazs Z, Schweizer RAS, Frey FJ, Rohner-Jeanrenaud F, Odermatt A. DHEA induces 11 -HSD2 by acting on CCAAT/enhancer-binding proteins. Journal of American Society of Nephrology. 2008; 19 (1):92-101 - 20.
Chapman KE, Coutinho AE, Gray M, Gilmour JS, Savill JS, Seckl JR. The role and regulation of 11β-hydroxysteroid dehydrogenase type 1 in the inflammatory response. Molecular and Cellular Endocrinology. 2009; 301 (1-2):123-131 - 21.
Hardy RS et al. Differential expression, function and response to inflammatory stimuli of 11beta-hydroxysteroid dehydrogenase type 1 in human fibroblasts: A mechanism for tissue-specific regulation of inflammation. Arthritis Research & Therapy. 2006; 8 (4):R108 - 22.
Ahasan MM et al. Inflammatory regulation of glucocorticoid metabolism in mesenchymal stromal cells. Arthritis and Rheumatism. 2012; 64 (7):2404-2413 - 23.
Sun K, Myatt L. Enhancement of glucocorticoid-induced 11β-hydroxysteroid dehydrogenase type 1 expression by proinflammatory cytokines in cultured human amnion fibroblasts. Endocrinology. 2003; 144 (12):5568-5577 - 24.
Sun K, Yang K, Challis JRG. Differential expression of 11β-hydroxysteroid dehydrogenase types 1 and 2 in human placenta and fetal membranes 1. The Journal of Clinical Endocrinology and Metabolism. 1997; 82 (1):300-305 - 25.
Li W, Gao L, Wang Y, Duan T, Myatt L, Sun K. Enhancement of cortisol-induced 11  -hydroxysteroid dehydrogenase type 1 expression by interleukin 1  in cultured human chorionic trophoblast cells. 2006; 147 (5):2490-2495 - 26.
Nashev LG et al. Hexose-6-phosphate dehydrogenase modulates 11beta-hydroxysteroid dehydrogenase type 1-dependent metabolism of 7-keto- and 7beta-hydroxy-neurosteroids. PLoS One. 2007; 2 (6):e561 - 27.
Yau JLW et al. Dehydroepiandrosterone 7- hydroxylase CYP7B: Predominant expression in primate hippocampus and reduced expression in Alzheimer’s disease. Neuroscience. 2003; 121 (2):307-314 - 28.
Kitada M, Kamatakis T, Itahashiq K, Rikihisa T, Kanakubo Y. P-450 HFLa, a form of cytochrome P-450 purified from human fetal livers, is the 16~hydroxylase of dehydroepiandrosterone 3=Sulfate*. The Journal of Biological Chemistry. 1987; 262 :13534-13537 - 29.
Falah N, Torday J, Quinney SK, Haas DM. Estriol review: Clinical applications and potential biomedical importance. Clinical Research Trials. 2015; 1 (2):29-33 - 30.
Pulkkinen M. Arylsulphatase and the hydrolysis of some steroid sulphates in developing organism and placenta. Acta Physiologica Scandinavica. Supplementum. 1961; 52 (180):1-92 - 31.
French AP, Warren JC. Sulfatase activity in the human placenta. Steroids. 1966; 8 (1):79-85 - 32.
Rabe T, Hösch R, Runnebaum B. Sulfatase deficiency in the human placenta: Clinical findings. Biological Research in Pregnancy and Perinatology. 1983; 4 (3):95-102 - 33.
Benediktsson R, Calder AA, Edwards CRW, Seckl JR. Placental 11β-hydroxysteroid dehydrogenase: A key regulator of fetal glucocorticoid exposure. Clinical Endocrinology. 1997; 46 (2):161-166 - 34.
Sun K, Adamson SL, Yang K, Challis JR. Interconversion of cortisol and cortisone by 11beta-hydroxysteroid dehydrogenases type 1 and 2 in the perfused human placenta. Placenta. 1999; 20 (1):13-19 - 35.
Walker BR, Connacher AA, Lindsay RM, Webb DJ, Edwards CR. Carbenoxolone increases hepatic insulin sensitivity in man: A novel role for 11-oxosteroid reductase in enhancing glucocorticoid receptor activation. The Journal of Clinical Endocrinology and Metabolism. 1995; 80 (11):3155-3159 - 36.
Wang W, Guo C, Sun K. Cortisol regeneration in the fetal membranes: A coincidental or requisite event in human parturition? Frontiers in Physiology. 2020; 11 (May):1-10 - 37.
Murphy VE et al. Alterations in human placental 11beta-hydroxysteroid dehydrogenase type 1 and 2 with gestational age and labour. Placenta. 2003; 24 (7):739-744 - 38.
Konstantakou P, Mastorakos G, Vrachnis N, Tomlinson JW, Valsamakis G. Dysregulation of 11beta-hydroxysteroid dehydrogenases: Implications during pregnancy and beyond. Journal of Maternal and Neonatal Medicine. 2017; 30 (3):284-293 - 39.
Shimodaira M et al. Glucocorticoid synthesis-related genes: HSD11B1 andHSD11B2 in hypertensive disorders in pregnancy. Gynecological Endocrinology. 2013;29 (7):657-661 - 40.
Casey ML, MacDonald PC, Mitchell MD. Despite a massive increase in cortisol secretion in women during parturition, there is an equally massive increase in prostaglandin synthesis. A paradox? The Journal of Clinical Investigation. 1985; 75 (6):1852-1857 - 41.
Guo CM, Zhu XO, Ni XT, Yang Z, Myatt L, Sun K. Expression of progesterone receptor a form and its role in the interaction of progesterone with cortisol on cyclooxygenase-2 expression in amnionic fibroblasts. The Journal of Clinical Endocrinology and Metabolism. 2009; 94 (12):5085-5092 - 42.
Hinko A, Soloff MS. Up-regulation of oxytocin receptors in rabbit amnion by glucocorticoids: Potentiation by cyclic adenosine 3′,5′-monophosphate. Endocrinology. 1993; 133 (4):1511-1519 - 43.
Romero R et al. Interleukin-1 alpha and interleukin-1 beta in preterm and term human parturition. American Journal of Reproductive Immunology. 27 (3-4):117-123 - 44.
Terzidou V, Blanks AM, Kim SH, Thornton S, Bennett PR. Labor and inflammation increase the expression of oxytocin receptor in human amnion. Biology of Reproduction. 2011; 84 (3):546-552 - 45.
Zakar T, Olson DM, Teixeira FJ, Hirst JJ. Regulation of prostaglandin endoperoxide H2 synthase in term human gestational tissues. Acta Physiologica Hungarica. 1996; 84 (2):109-118 - 46.
Altman R, Motton DD, Kota RS, Rutledge JC. Inhibition of vascular inflammation by dehydroepiandrosterone sulfate in human aortic endothelial cells: Roles of PPARα and NF-κB. Vascular Pharmacology. 2008; 48 (2-3):76-84 - 47.
Lim S et al. Nuclear factor kappa B activation occurs in the amnion prior to labour onset and modulates the expression of numerous labour associated genes. PLoS One. 2012; 7 (4):e34707 - 48.
Stein B, Cogswell PC, Baldwin AS. Functional and physical associations between NF-kappa B and C/EBP family members: A Rel domain-bZIP interaction. Molecular and Cellular Biology. 1993; 13 (7):3964-3974 - 49.
Mesiano S, Jaffe RB. Developmental and functional biology of the primate Fetal adrenal cortex 1. Endocrine Reviews. 1997; 18 (3):378-403 - 50.
Smith R, Mesiano S, Chan E-C, Brown S, Jaffe RB. Corticotropin-releasing hormone directly and preferentially stimulates dehydroepiandrosterone sulfate secretion by human fetal adrenal cortical cells. The Journal of Clinical Endocrinology and Metabolism. 1998; 83 (8):2916-2920 - 51.
Sirianni R, Mayhew BA, Carr BR, Parker CR, Rainey WE. Corticotropin-releasing hormone (CRH) and urocortin act through type 1 CRH receptors to stimulate dehydroepiandrosterone sulfate production in human fetal adrenal cells. The Journal of Clinical Endocrinology and Metabolism. 2005; 90 (9):5393-5400 - 52.
Watanabe F et al. Urocortin in human placenta and maternal plasma. Peptides. 1999; 20 (2):205-209 - 53.
McLean M, Smith R. Corticotropin-releasing hormone in human pregnancy and parturition. Trends in Endocrinology and Metabolism. 1999; 10 (5):174-178 - 54.
Balazs Z, Schweizer RAS, Frey FJ, Oise Rohner-Jeanrenaud F, Odermatt A. DHEA induces 11β-HSD2 by acting on CCAAT/enhancer-binding proteins. Journal of American Society Nephrology. 2008; 19 :92-101 - 55.
Yoon BH et al. An increase in fetal plasma cortisol but not dehydroepiandrosterone sulfate is followed by the onset of preterm labor in patients with preterm premature rupture of the membranes. American Journal of Obstetrics and Gynecology. 1998; 179 (5):1107-1114 - 56.
Oh S-Y, Romero R, Shim S-S, Park JS, Jun JK, Yoon BH. Fetal plasma cortisol and dehydroepiandrosterone sulfate concentrations in pregnancy and term parturition. Journal of Maternal and Neonatal Medicine. 2006; 19 (9):529-536 - 57.
Mears K, McAuliffe F, Grimes H, Morrison J. Fetal cortisol in relation to labour, intrapartum events and mode of delivery. Journal of Obstetetrics and Gynaecology (Lahore). 2004; 24 (2):129-132 - 58.
Grøntved L et al. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements transaction report. EMBO Journal. 2013; 32 :1568-1583 - 59.
Rudiger JJ. Interaction of C/EBPalpha and the glucocorticoid receptor in vivo and in nontransformed human cells. The FASEB Journal. 2002; 16 (2):177-184 - 60.
Liggins GC, Fairclough RJ, Grieves SA, Forster CS, Knox BS. Parturition in the sheep. Ciba Foundation Symposium. 1977; 47 :5-30 - 61.
Osathanondh R, Canick J, Ryan KJ, Tulchinsky D. Placental sulfatase deficiency: A case study. The Journal of Clinical Endocrinology and Metabolism. 1976; 43 (1):208-214 - 62.
Martineau M et al. Cholestatic pregnancy is associated with reduced placental 11βHSD2 expression. Placenta. 2014; 35 (1):37-43 - 63.
Morris DJ, Souness GW, Latif SA, Hardy MP, Brem AS. Effect of chenodeoxycholic acid on 11β-hydroxysteroid dehydrogenase in various target tissues. Metabolism. 2004; 53 (6):811-816 - 64.
Benediktsson R, Lindsay RS, Noble J, Seckl JR, Edwards CRW. Glucocorticoid exposure in utero: New model for adult hypertension. The Lancet. 1993; 341 (8841):339-341 - 65.
Mizutani T et al. C/EBPβ (CCAAT/enhancer-binding protein β) mediates progesterone production through transcriptional regulation in co-operation with SF-1 (steroidogenic factor-1). The Biochemical Journal. 2014; 460 (3) - 66.
Arrowsmith S, Wray S. Oxytocin: Its mechanism of action and receptor signalling in the myometrium. Journal of Neuroendocrinology. 2014; 26 (6):356-369 - 67.
Gorelik J et al. Aldosterone acts via an ATP autocrine/paracrine system: The Edelman ATP hypothesis revisited. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102 (42):15000-15005 - 68.
Urabe S, Miyoshi H, Fujiwara H, Yamaoka K, Kudo Y. Enhanced expression of P2X4 and P2X7 purinergic receptors in the myometrium of pregnant rats in preterm delivery models. Reproductive Sciences. 2009; 16 (12):1186-1192 - 69.
Diczfalusy E. A modified theory of steroid synthesis in the human foeto-placental unit. In: Endokrinologie der Entwicklung und Reifung. Berlin, Heidelberg: Springer; 1970. pp. 32-46