
Abstract
The epigenetic revolution has led to a paradigm shift in our understanding of gene regulation and function. Epigenetic modifications, including DNA methylation, posttranslational histone modifications, and regulatory noncoding RNAs, display unique features, such as reversibility and transgenerational inheritance. A great variety of environmental and lifestyle factors can cause changes in the epigenome. Epigenetic alterations can contribute to the underlying mechanisms of human diseases including cancer, cardiovascular, neurological, psychiatric, autoimmune, metabolic and inherited. The chapter focuses on the fine interplay between environmental stress, the epigenetic adaptive responses, and how the inability to adapt may trigger disease outcomes. A model of the epigenetic disease is postulated, epigenetic disease adaptational model (EDAM), according to which the epigenetic disease develops as a failure to adapt to environmental stressors. This may occur in at least two possible scenarios: (1) when the epigenetic adaptational programs are not adequate to stress nature, duration, intensity and/or stage of action and (2) when the epigenetic adaptational programs are not adequate to the situation. In the second scenario, the stressful situation is wrongly considered the most feasible situation, and the stressful conditions are taken as “norm.” The proposed model highlights important topics for future research in the field of epigenetics and disease.
Keywords
- environmental stress
- epigenetic modifications
- gene expression
- adaptive plasticity
- epigenetic disease
- epigenetic disease adaptational model (EDAM)
1. Introduction
The epigenetic revolution that has happened during the last decades has challenged our understanding of gene regulation and expression and has outlined the emergence of a new post-genomic era in biology. The long-term dogmatic views on heredity, exclusively relying on the genetic code inheritance, underwent significant scrutiny and questioning. The realization that the primary DNA sequence alone provides incomplete knowledge on the functioning of the genome has shifted the focus toward understanding the epigenetic processes, which modulate gene function by modifications in DNA or the associated proteins. This has been driven by advances in technology and methodologies such as DNA methylation profiling and chromatin immunoprecipitation that have allowed for precise mapping of epigenetic marks on a genome-wide scale. The definition of “epigenetics,” originally introduced by Conrad Waddington in the early 1940s [1], as “the branch of biology, which studies the causal interactions between genes and their products, which bring the phenotype into being,” has been notably evolved over time and now refers to “the study of changes in gene function that are heritable and that do not entail a change in DNA sequence” [2]. Epigenetic modifications (“epi” standing for “over” and “above”) occur independent of the genetic sequence and provide an additional level of hereditary information denoted as “epigenetic code” [3]. The changes in the epigenome include three main classes: methylation in DNA, posttranslational histone modifications, and noncoding RNAs. Epigenetic changes, compared to genetic, display several unique characteristics including higher frequency, occurrence in specific genome regions, and potential reversibility. Evidence that comes from epigenetic research demonstrated that the genome is highly dynamic and responsive to environmental shifts. A great variety of environmental and lifestyle factors were shown to cause changes in the gene expression patterns mediated by epigenetics. With reference to this, epigenetic changes could be regarded as sensitive indicators of the interactions between the organism and the environment. Moreover, early embryogenesis is most vulnerable to epigenetic stress responses, at least some of which may increase the risk of malfunctioning and disease pathology in later life. Studies have shown transmission of environmentally induced epigenetic changes to the offspring in at least several species [4, 5, 6]. Though the evolutionarily significance of transgenerational epigenetics is still debatable, its significance for the organisms themselves and their near descendants seems quite feasible. In addition to their impact on various cellular processes, including shaping of cellular identity, development, and aging, epigenetic modifications were attributed a role in disease onset as well. Changes in the epigenome were related to many human disorders, such as cancer, cardiovascular diseases, neurological and psychiatric disorders, autoimmune diseases, metabolic diseases, and inherited disorders. This opened up possibilities for development of targeted interventions and therapeutic approaches, which can modulate gene expression patterns. Overall, the epigenetic revolution has led to a paradigm shift in our understanding of gene function by highlighting the dynamic nature and plasticity of how genes are regulated.
In this chapter, we provide an overview of the role of epigenetics in gene regulation, the impact of environmental and lifestyle factors on the epigenome, and the association between epigenetic modifications and various diseases. We propose the Epigenetic Disease Adaptational Model (EDAM) as a conceptual framework to elucidate how the inability to effectively adapt to environmental stressors may contribute to the development of epigenetic diseases. This model aims to provide insights into the complex interplay between the environment and the epigenome highlighting new possibilities for therapeutic approaches.
2. Types of epigenetic modifications
Three major mechanisms governing epigenetic regulation could be distinguished: methylation in DNA, posttranslational histone modifications (PTMs), and noncoding RNAs (ncRNAs). They modulate gene expression by causing changes in DNA-histones interactions responsible for the rearrangement of chromatin from a more open and transcriptionally accessible state (euchromatin) to a condensed chromatin structure that is transcriptionally less active (heterochromatin) and vice versa (Figure 1).
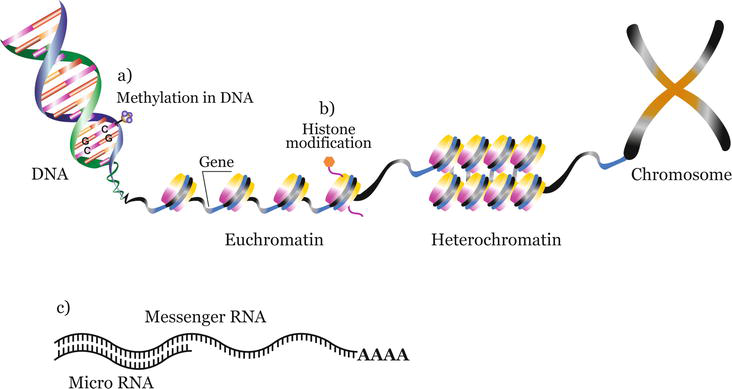
Figure 1.
Types of epigenetic modifications. (a) Methylation in DNA—Covalent attachment of a methyl group to the C5 position of cytosine residue in CpG dinucleotide sequences, (b) histone modifications—Posttranslational covalent addition of a chemical group (methylation, phosphorylation, acetylation, ubiquitylation, sumoylation), and (c) microRNAs (miRNAs)—Small noncoding RNAs which regulate post-transcriptional gene expression by binding to target mRNAs resulting in gene silencing. Epigenetic modifications regulate the dynamic transitions between transcriptionally active (euchromatin) or transcriptionally inactive (heterochromatin) states.
2.1 Methylation in DNA
Methylation in DNA is considered a relatively long term and stable epigenetic mark and can contribute to maintenance of cellular functions and phenotype. It refers to covalent attachment of a methyl group to the C5 position of cytosine residue in CpG dinucleotide sequences preferentially located at the 5′ promoter region of human genes forming CpG islands. Methylation at cytosines, other than those in CpGs, occurs in undifferentiated cells and is crucial for gene regulation in embryonic stem cells [7]. CpG methylation plays an essential role in transcriptional gene silencing by restricting the expression of tissue-specific genes during development and differentiation, and is also involved in X chromosome inactivation in females, DNA imprinting, and transcriptional repression of highly repeated genome sequences. The donor methyl groups are supplied by S-adenosylmethionine (SAM) to form 5-methylcytosine (m5C). Methylation in CpGs suppresses transcription by either directly blocking DNA recognition and access at specific transcriptional factor binding sites or by recruitment of methyl-binding domain proteins (MBDs). For example, methyl-CpG binding protein 2 (MeCP2), a member of the MBD family, binds to methyl CpG recruiting histone-modifying proteins, such as histone deacetylases (HDAC). This leads to histone deacetylation, promoting chromatin condensation, and subsequent transcription repression [8]. CpG methylation is a post-replicative process and is mediated by DNA methyltransferase enzymes (DNMTs). DNA methyltransferase 3 alpha (DNMT3A) and DNA methyltransferase 3 beta (DNMT3B) are involved in
2.2 Posttranslational histone modifications (PTMs)
Within chromatin, DNA undergoes intricate packaging, forming a highly condensed structure wrapped around histone octamers. This arrangement results in the formation of nucleosomes, which gives the characteristic “beads on a string” appearance and can effectively regulate DNA accessibility. Each histone octamer is composed of a tetramer of two copies of histone 2A (H2A) and two copies of histone 2B (H2B), flanked by dimers of histone 3 (H3) and histone 4 (H4). The histone proteins feature a globular C-terminal domain and an extended N-terminal tail, which are susceptible to various PTMs. These include methylation of lysine or arginine residues, phosphorylation of serine or threonine, acetylation and deacetylation of lysines, and ubiquitylation and sumoylation of lysines. Acetylation and methylation of lysine residues on H3 and H4 are the most extensively studied PTMs. Histone acetylation is governed by the “charge neutralization model,” wherein the positive charge of lysine residues on H3/H4 promotes tight DNA packaging with histones. Acetylation disrupts the tight configuration, allowing access for transcription factors, and thus facilitating transcription. Enzymes, such as histone acetyltransferases (HATs) and histone deacetylases (HDACs), regulate the addition and removal of acetyl groups. Unlike acetylation, histone methylation may have a different effect on targeted residues. For example, methylation at H3K4/36/79 generally activate transcription, while methylation at H3K9/27 and H4K20 is considered repressive [9]. Various histone methyltransferases (HMTs) catalyze these modifications, while removal of methyl groups from those marks is catalyzed by histone demethylases (HDMTs). Variations in the combinations of histone modifications, referred to as a “histone code,” may regulate chromatin structure and transcriptional status [10]. The concept of a “histone code” significantly expands the informational capacity of the genetic code. Different histone variations can act synergistically or antagonistically in order to modulate the affinities of chromatin-associated proteins. This, in turn, regulates the dynamic transitions between transcriptionally active or transcriptionally inactive chromatin states. Overall, histone modifications play an important regulatory role bringing about different downstream events through the dynamic regulation of chromatin structure.
2.3 Noncoding RNAs (ncRNAs)
Noncoding RNAs have a regulatory function and are mainly categorized into long ncRNAs (lncRNAs) and small ncRNAs (sncRNAs). LncRNAs are a diverse family that consists of long transcripts from different genomic regions and are shown to play an essential role in X-chromosome inactivation and genomic imprinting. Their gene silencing effect is partly due to their recruitment of remodeling complexes, which foster histone methylation [11]. SncRNAs are also implicated in modulation of gene transcriptional silencing. These include microRNAs (miRNAs), small inhibitory RNAs (siRNAs), and piwi-interacting RNA (piRNA). While all classes mediate epigenetic modifications, piRNAs notably repress transposon expression through
3. Environmental and lifestyle factors and the epigenome
The proper function of the organisms is a result of the delicate balance between the maintenance of the conserved nucleotide sequence, which facilitates the basic biological functions, and the dynamic changeability in the epigenome, which regulates gene expression, as a response to the constantly changing environment. This flexibility allows organisms to adapt to environmental shifts, and potentially “learn” from experience. The environmental impact on the regulation of gene expression plays a crucial role in reprogramming in early embryogenesis and development. During these stages, different sets of genes are epigenetically activated or deactivated, thus facilitating proper functioning of the organism through orchestrated events responsive to the environmental input. However, environmental factors, which affect gene expression networks, may disrupt the normal regulatory processes, potentially leaving long-lasting effects, and may even contribute to diseased outcomes. Many environmental and lifestyle factors were shown to epigenetically alter gene regulation and expression (Figure 2), and some of their known effects are discussed below.
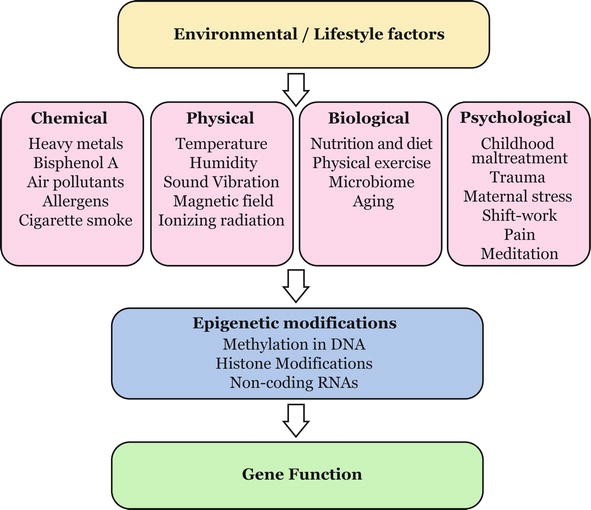
Figure 2.
Environment-epigenome interactions. Environmental and lifestyle factors, including chemical, physical, biological, and phycological, may cause changes in the epigenome, and thus can affect gene regulation and function by activating or silencing of genes.
3.1 Chemical factors
3.1.1 Heavy metals
Heavy metals, such as arsenic [15], cadmium [16], nickel [17], and lead [18], are common environmental pollutants that are able to impact DNA methylation, histone acetylation, and miRNA expression. Exposure to heavy metals in humans has been linked to a number of diseases, such as neurological disorders, cardiovascular conditions, central nervous system neuropathies, autoimmune diseases, and cancer and was shown to cause genotoxic and immunotoxic effects.
3.1.2 Bisphenol A (BPA)
Adverse consequences to the epigenome have also been demonstrated for another environmental chemical, bisphenol A (BPA), a common industrial plasticizer [19]. In mice, periconceptional exposure to BPA was shown to decrease the level of methylation in the IAP retrotransposon located upstream of the
3.1.3 Air pollutants
Exposure to ambient air pollutants correlates with global genomic DNA methylation [22], increased H3K4 demethylation and H3K9 acetylation [23], modified expression of miR-21 and miR-222 [24], and has been linked to cardiovascular and respiratory diseases.
3.1.4 Allergens
Allergen exposure was reported to lead to deacetylation of histone proteins, hypermethylation of DNA, and changes in the levels of miRNA [25]. A genome-wide analysis demonstrated clear differential DNA methylation patterns in patients with seasonal allergic rhinitis [26].
3.1.5 Cigarette smoke
Exposure to cigarette smoke is a well-established risk factor for chronic obstructive pulmonary disease, cardiovascular diseases, and cancer. Alteration in DNA methylation at multiple CpG sites induced by cigarette smoke was reported in different populations, including Europeans [27], African Americans [28], and Chinese [29]. Moreover, whole-genome bisulfite sequencing has revealed differential CpGs methylation in cord blood of newborns exposed to prenatal maternal smoking [30]. Additionally, cigarette smoke exposure causes dose- and time-dependent histone alterations
3.2 Physical factors
3.2.1 Temperature and humidity
Temperature extremes increase the risk for heatstroke, hyperthermia, or hypothermia and can worsen chronic conditions such as cardiovascular disease, respiratory disease, and diabetes. Studies have demonstrated an association between ambient temperature and relative humidity, LINE-1 and Alu methylation levels, and differential global DNA methylation
3.2.2 Sound vibration
Recently, it became apparent that sound vibration can induce physiological changes in plants mediated by epigenetic mechanisms as increased H3K27me3 in the promoter regions of defense-related genes in
3.2.3 Low-frequency magnetic fields
A few publications provided evidence that extremely low-frequency magnetic field exposure is related to increased global DNA methylation and aberrant site-specific DNA methylation [35], increased H3K9 acetylation [36], and altered miRNA expression [37].
3.2.4 Ionizing radiation
Ionizing radiation was also shown to cause epigenetic modifications, including global and gene-specific DNA methylation, histone modifications, and modulation of miRNA expression [38].
3.3 Biological factors
3.3.1 Nutrition and diet
Nutrition has been attributed epigenetic roles both in physiological and pathologic processes [39]. Folate deficiency, during conception and periconception, is related with several birth defects, including neural tube defects and congenital heart defects; however, application of folic acid periconceptionally can reduce birth defect frequency. The underlying mechanisms involve changes in the methylation of insulin-like growth factor 2 gene differentially methylation region (IGF2 DMR) [40]. Dietary choline, supplied prenatally or postnatally, leads to long-term enhancement of memory in rodents while choline deprivation during pregnancy decreases memory capacity in the offspring. A decrease in global DNA methylation and gene-specific DNA methylation of Cdkn3, which corresponded to increased expression of the encoded Kap protein, was reported in fetal brain from mothers fed a choline-deficient diet in mice [41]. Additionally, choline deficiency decreases H3K9me1 and H3K9me2 in murine fetal hippocampus and in cultured neural progenitor cells [42]. Decreased mean PPARα promoter methylation was found in the offspring of rats fed with a protein-restricted diet during pregnancy [43]. Alcohol consumption has been proven to cause global DNA hypomethylation and increased H3K43me in autopsy brain samples of patients with alcohol dependence [44]. Exposure to ethanol
3.3.2 Physical exercise
In addition to nutrition, physical exercise can significantly influence gene expression causing changes into the epigenome. A large number of differentially expressed genes were identified involved in metabolisms and mitochondrial biogenesis in skeletal muscle during recovery from endurance exercise [46]. The underlying mechanisms include increase in global leukocyte methylation [47] and global H3K36 acetylation [48] showing that exercise can induce chromatin remodeling associated with enhanced transcription.
3.3.3 Microbiota
Variations in the gut microbiota are related to many pathological conditions including intestinal and metabolic disorders, which can be mediated by microbiota-sensitive epigenetic mechanisms [49].
3.3.4 Aging
A role of epigenetic mechanisms in the process of aging and aging-related diseases was also established. Numerous studies on human and mouse tissues or cell cultures revealed that global DNA methylation generally decreases with age [50]. In addition, global changes in H3K9me3, H4K20me3, H3K27me3, and H3K9ac, and up- and downregulation of miRNAs in older individuals were also established [50]. Certain epigenetic modifications accumulate or change predictably with age providing a molecular signature that can be used to measure the aging process at a cellular level referred to as epigenetic clocks [51]. Epigenetic clocks are designed to estimate an individual’s biological age rather than their chronological age. One of the most well-known epigenetic clocks is the Horvath Clock, which utilizes DNA methylation patterns at 353 specific CpG sites across the genome.
3.4 Psychological factors
3.4.1 Childhood maltreatment
A hundred of empirical studies explored the relation between childhood maltreatment and DNA methylation of candidate genes both in children and adults [52]. The vast majority of these investigate the methylation status of genes involved in the regulation of glucocorticoid signaling showing hypermethylation of NR3C1, hypomethylation of FKBP5, and differentially methylated CpG sites in children with a history of maltreatment [52].
3.4.2 Trauma
Exposure to trauma can also alter stress response mechanisms. In post-traumatic stress disorder (PTSD), dysregulation of hypothalamic-pituitary-adrenal (HPA) axis has been observed, which alters the response to cortisol feedback
3.4.3 Maternal stress
Maternal psychosocial stress experiences during pregnancy can permanently alter the function of the HPA axis causing site-specific CpG methylation in exposed individuals [54].
3.4.4 Shift-working
Shift-working occupations have been linked to increased risk of age-related diseases and were associated with epigenetic age [55]. The authors found that working more than 10 years of night shift work was significantly associated with epigenetic age acceleration. Additionally, differentially methylated CpG sites were associated with shift working, including the ZFHX3 gene, which is involved in circadian rhythm [55].
3.4.5 Chronic pain
Chronic pain can induce epigenetic modifications as well, including reduction in H3K27me3 levels at the Mcp-3 gene promoter [56], decrease in global DNA methylation [57], and increase in miR-155 and miR-223 expression [58] in prefrontal cortex in murine models.
3.4.6 Meditation
A few studies indicated that meditation can induce changes in the epigenome such as decrease in expression levels of HDAC2, HDAC3, and HDAC9 and alterations in global modification of H4ac and H3K4me3 predicting a better cortisol recovery after a test of acute psychosocial stress [59]. In meditators, the epigenetic aging rate was significantly decreased, which correlated with the number of years of practice [60].
4. Epigenetics and disease
Epigenetics was attributed two primary roles: one in normal development, guiding major biological processes such as reprogramming in early embryogenesis, and the other in disease, where epigenetic changes can contribute to certain pathological phenotypes. A great variety of epigenetic alterations have been found in a vast number of human diseases prompting for abnormal transcription activation or repression. Among these, cancer epigenome is most well-studied. Various epigenetic changes were reported in cancers of the breast, lungs, prostate, colorectum, ovaries, bladder, pancreas, gastric cancer, leukemia, and others [61, 62, 63, 64]. The epigenetic component in tumorigenesis is characterized by abnormal patterns both in global DNA methylation and in local CpG methylation of target genes, disrupted patterns of PTMs, changes in chromatin composition and remodeling, and altered expression state of chromatin-modifying enzymes. A growing number of studies have attributed roles of miRNAs in almost all cancer types as well. Modifications in the epigenome take part in both cancer initiation and progression
Epigenetic modifications have also been associated with cardiovascular diseases such as coronary heart disease, acute myocardial infarction, heart failure, vascular calcification, and hypertension [65]. Aberrant DNA methylation patterns, histone modifications, and noncoding RNA regulation were shown to impact the function of cardiovascular disease-related genes and their expression levels, thus affecting cardiovascular disease progression.
A growing body of evidence attributes a role of epigenetics in a number of neurological (Alzheimer’s, Parkinson’s, and Huntington’s diseases) and psychiatric disorders (depression, schizophrenia, bipolar disorder, autism spectrum disorders, and substance use disorders) and their co-occurrence, including dysregulation in DNA methylation and histone modifications resulting in disease phenotypes [66, 67].
All three types of epigenetic alterations, DNA methylation, histone modifications, and noncoding RNAs, are shown to be involved in the etiology of human autoimmune diseases, including systemic lupus erythematosus, rheumatoid arthritis, systemic sclerosis, Sjogren’s syndrome, and autoimmune thyroid diseases [68].
A profound epigenetic component is described in the underlying mechanisms of metabolic diseases, such as obesity, type 2 diabetes, nonalcoholic fatty liver disease, osteoporosis, gout, and hyper- and hypothyroidism [69].
In a number of human disorders of genomic imprinting, such as Prader-Willi syndrome, Angelman syndrome, Beckwith–Wiedemann syndrome (BWS), pseudohypoparathyroidism (PHP), and Silver–Russell syndrome (SRS), “epimutations” leading to loss of gene expression were found, adding to the knowledge of how epigenetic defects lead to disease phenotypes [70].
The relationship between the genome and epigenome has extended our comprehension of the underlying molecular events that lead to human diseases. These could be either inherited or
The recognition of the role of epigenetics in the pathogenesis of diseases may justify why many therapeutic approaches in the past failed to achieve the anticipated outcomes. A deeper comprehension of the epigenetic mechanisms underlying human disorders is needed, and these hold promise for new therapeutic advancements. A breakthrough in the field of epigenetics and diseases is the approval of several epigenetic therapies (epidrugs) for cancer treatment [71]. The first approved DNMT inhibitor (DNMTi) primarily treats myelodysplastic syndrome by inhibiting DNA methylation, particularly targeting DNMT1 enzymes. The majority of approved compounds are HDAC inhibitors (HDACi), showing promise in selectivity and toxicity. Emerging therapies include miRNA and multidrug combinations which enhance cancer treatment and reduce drug resistance. Epigenetic therapies are continually assessed for cytotoxicity, pharmacological parameters, and mechanism of action in preclinical studies and in clinical trials.
5. Adaptation, homeostasis and allostasis
Over the course of millions of years, organisms have evolved characteristics, which allow them to fit exquisitely and operate optimally in their specific surroundings. However, these same traits may not be advantageous or beneficial in a different environmental context, and in some cases could even be detrimental to fitness. In view of the fact that no environment remains constant, even the most well-fitted organisms need be able to adapt to changes in order to survive. As a consequence, they have developed mechanisms designed to facilitate adaptation to changing environments. These allow them to cope with environmental challenges fostering resilience while faced with deviations from their usual operating conditions. In regard to this, the following types of responses to environmental changes may be undertaken by the organisms in pursuit of adaptation and survival:
Homeostasis: With homeostasis, the internal stability and balance within an organism are maintained relatively steady, regardless of external environmental fluctuations [72]. Homeostasis ensures that essential biological processes occur within a narrow range of optimal conditions.
Allostasis: The term “allostasis” was introduced by Sterling and Eyer [73] and literally means “maintaining stability through change.” Allostasis refers to the adaptive process in response to acute environmental challenges [74]. While allostasis may appear similar to homeostasis, it emphasizes the flexible adaptation to dynamic environments or stressful circumstances. Unlike homeostasis, which seeks to maintain stability within a narrow range and minimize variability, allostasis recognizes that the optimal internal state may vary depending on situational demands. In allostasis, having greater variability is considered advantageous because it signifies the capacity of the internal settings to meet specific challenges posed by the environment in order to adapt and support the body’s overall system [75]. When allostatic adaptive systems are activated and deactivated efficiently, “the body is able to cope effectively with challenges that it might not otherwise survive” [76].
Allostatic load: There are situations however where allostatic systems may be excessively stimulated or not functioning properly, and this state is referred to as “allostatic load” or the “cost of adaptation” [77]. “Allostatic load” describes the price that the body pays when it is compelled to adapt to challenging stressful conditions both physical and psychosocial. It indicates an excessive amount of stress or the inefficient functioning of the stress response systems, which should be activated during stress and then deactivated once the stressor subsides. When individuals experience repeated or chronic stressors, their bodies continuously activate the stress response. While the stress response is essential for coping with acute stressors, prolonged or repeated activation lacking adequate recovery can result in an accumulation of physiological dysregulation. Over extended periods of time, allostatic load can lead to the development of diseases [78]. McEwen distinguishes between the following types of allostatic load: repeated activation of allostatic systems that gradually lead to allostasis over time, failure to shut off allostatic activity after repeated stress resulting in failure to adapt, and insufficient primary adaptation mechanisms leading to the activation of compensatory mechanisms [77].
6. Epigenetic disease adaptational model (EDAM)
We will now explore the epigenetic disease in terms of environmental stress, epigenetic modification, and adaptative efficiency. By epigenetic disease, we will consider any pathological phenotype that is due to a change in gene expression in response to a stressful factor or a situation as a result of an epigenetic modification(s), which is either ontogenetic (occurring during the lifespan of an individual) or inheritable (epimutations), or both. Though genetic disease factors will not be considered here, in order to achieve simplicity, we will keep in mind that most diseases have a complex and multifactorial nature, and develop as a result of the interaction between both genetic and epigenetic determinants, that is. there is hardly a disease that is purely genetic or epigenetic. Moreover, genetic factors are involved not only in disease predisposition and progression but also may contribute to resilience, modulation of disease manifestation, immune response, disease recovery, or chronification. By epigenetic stress, referred below as “stress,” we will assume any exogenous or endogenous, environmental or lifestyle factor of either physical, chemical, biological, or psychological nature that causes a shift in the homeostatic equilibrium
Adaptation to the stress is successful and healthy equilibrium with environment is maintained or achieved. This outcome will be attained when the epigenetic adaptational programs initiated during the stress responseare adequate to the stress nature, duration, intensity, and/or period of action. In such a case, when the stress is turned off, the epigenetic adaptational programs will no longer be needed and will stop operating. Homeostasis will be sustained or allostasis will be reached (Figure 3a and b).Adaptation to stress is not successful and epigenetic disease programs are triggered. In this case, the epigenetic adaptational programsare not adequate to the stress nature, duration, intensity, and/or period of action. Upon exposure to highly intense and continuous or repeated stress, notably in vulnerable developmental periods, in order to meet the challenges, the epigenetic adaptational programs triggered will continue operating in an attempt to maintain or achieve equilibrium. The failure of the organisms to adapt will be manifested in an epigenetic disease phenotype (Figure 3c). The epigenetic disease programs will, therefore, represent either inefficient epigenetic adaptational programs or additionally activated compensatory mechanisms, or both.Adaptation to stress is not anymore needed. In circumstances such as these, the epigenetic adaptational program will not be adequate to thesituation . This may occur in case the stress is already “turned off” but the epigenetic adaptational programs are still ongoing. The stressful situation is misjudged or the threat is being overrated. In such a case, the stressful situation is wrongly considered asthe most probable situation, and the stressful conditions are taken as the “normal ” (in the sense of “usual” or “becoming a standard”) conditions. The still ongoing epigenetic adaptational programs are then outdated and needing a revision. An epigenetic disease phenotype will be manifested and/or sustained (Figure 3d).
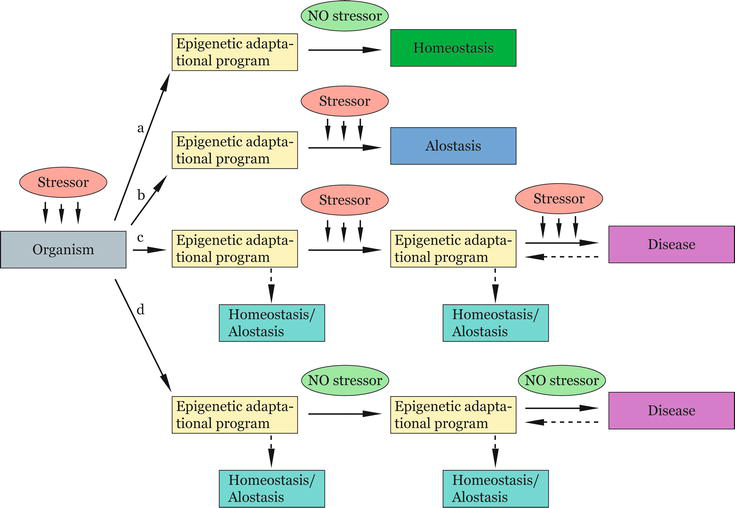
Figure 3.
Epigenetic disease adaptational model (EDAM). Upon stress exposure, epigenetic adaptational programs are triggered, which may have the following outcomes: (a) the epigenetic adaptational programs are adequate to the stressor/situation, and homeostasis is sustained, (b) the epigenetic adaptational programs are adequate to the stressor/situation and equilibrium is achieved
7. Illustrating the EDAM model
Let us take two examples to illustrate the above postulations. Studies on Dutch Hunger survivors showed that maternal undernutrition has important effects on birth weights in newborns and health in later life, which are dependent on its timing during gestation [79]. Upon exposure to famine during the first trimester of pregnancy, the newborns were more likely to have normal birth weight though in later life they tended to display glucose intolerance and higher obesity rates than the general population, together with other health issues including coronary heart disease [80]. Moreover, all first-born offspring of such mothers (that were exposed to famine intrauterine) showed an increase in the average birth weight even though they themselves were never exposed to malnutrition during early development [81]. Studies on DNA methylation patterns in Dutch Hunger survivors have revealed alterations in key genes involved in metabolism [82, 83]. While correlation alone does not directly establish causation, the observed data are consistent with the assumption that undernutrition during early developmental stages alters the epigenetic code and expression profiles of key metabolic genes.
We can speculate that such dramatic metabolic stress, as is the huge reduction in nutrient supply during early gestation, can induce substantial alterations in the epigenetic processes within fetal cells. In response to the decreased nutrient supply, fetal cells will undergo metabolic adaptations in an attempt to sustain optimal fetal growth. The cells will modulate their gene expression patterns in order to counterbalance the nutritional deficiency. The epigenetic programming of fetal cells will allow optimal utilization of the limited nutritional resources. These alterations will be imprinted in the epigenome, setting the stage for future gene expression. Their biological purpose will be to attempt at adapting the developing organism to the stressful situation by maintaining optimal equilibrium with an environment that has been dramatically changed. Importantly, this programming will continue operating even after the cessation of the environmental stressor that prompted it. Moreover, these adaptational programs may even be passed into the offspring who may never in their lives encounter a similar stressful situation. Though transgenerational epigenetic inheritance presumably enables the transmission of beneficial survival solutions, it appears that at least some of these may not be relevant to the environmental context of next generations.
Notably, offspring whose mothers experienced malnutrition during the critical early stages of pregnancy, when the proper execution of the developmental programs is most crucial, exhibit an elevated susceptibility to adult obesity. During the initial stages of development, the fetus is particularly sensitive to stress-related responses controlled by epigenetic processes. In the early embryogenesis, significant changes occur in epigenetic markers due to the high level of cell division. It is likely that this vulnerability to stress responses during the early development may attempt to enable the most advantageous fitness to a surrounding considered most feasible in later life.
Another example comes from the studies on the long-term effects of early traumatic experience. Children who have experienced adversity or neglect in childhood are more likely to develop adult psychopathology, such as mood disorders and schizophrenia [84]. Individuals exposed to childhood trauma exhibit longstanding modifications of the HPA axis, a major neuroendocrine axis regulating homeostasis in mammals, and elevated cortisol metabolism. Upon stress, the HPA axis is activated, and the resultant increase in cortisol prepares the body to cope with the stressor. Under normal conditions, this system is controlled by a negative feedback loop, which restricts the stress response activity. This is dependent upon the expression of the glucocorticoid receptor (GR) gene, NR3C1, which is controlled by the level of promoter DNA methylation. In case the GR promoter is hypomethylated, the receptor is overexpressed and small cortisol amounts are needed to restrict stress response. Persons with a history of childhood abuse display increased methylation in the GR promoter [85], which prevents the NGFI-A transcription factor binding and causes low GR expression levels. The negative feedback loop is then compromised resulting in high stress hormone levels.
The maintenance of high cortisol levels in persons with early traumatic experience may be viewed as an adaptive mechanism utilized to enable readily available stress responses. It seems that this is achieved through epigenetic modulation of stress response genes. Even though these epigenetic adaptational programs have been triggered far away in time, they are still ongoing regardless of whether the stress is still present or not. We can assume that the traumatic situation is considered the
8. Testing the EDAM model
The difficulty to test this model comes from the fact that most diseases are caused by multiple genetic and environmental factors. Furthermore, their effect is a result of factors interaction rather than of an individual influence. Different factors may act synergistically or antagonistically. Finally, we should take into consideration the possibility of occurrence of epigenetic changes, other than being adaptive, and the probable incidence of mutations in genes sequences, which can also contribute to the disease outcome. Nevertheless, a starting point for exploring the model can be the screening for epigenetic marks that can be related to a particular environmental or lifestyle factor, their possible significance for adaptation, and a further follow-up of how these are changed or sustained during pathology and recovery.
9. Conclusion
Organisms function in a fine balance with the environment, constantly adapting to changes, in order to maintain homeostasis or achieve allostasis. Upon stress, significant capacity for lasting adaptive plasticity is accomplished through epigenetic processes including methylation in DNA, PTMs, and ncRNAs. Cells undergo adaptations in an attempt to sustain optimal cellular functions. They modulate their gene expression patterns in order to counterbalance the stressor. The proposed EDAM model relies on the fine interplay between environmental stress, of either chemical, physical, biological or psychological nature, the response to it mediated through the epigenome, and how the failure to adapt may lead to epigenetic diseases. This may occur in at least two possible scenarios: first, when the epigenetic adaptational programs are not adequate to the stress nature, duration, intensity, and/or stage of action, and second, when the epigenetic adaptational programs are not adequate to the situation. In the last scenario, the stressful situation is wrongly considered the most feasible situation and the stressful conditions are taken as the “normal” conditions. Even though these adaptive programs may have been efficient in a past experience, their exact reproduction may be dysfunctional if not consistent with the current circumstances.
At least several questions rise from the proposed perspective: Are these not anymore advantageous programs editable? Have organisms evolved mechanisms to update the operating epigenetic adaptational programs with regard to the present context? In which cases is the stressful situation further considered a “norm” and how to counteract this? Are there set points at which: the epigenetic adaptational program turns into a disease program; the disease program can be brought back to health; and the disease program cannot be anymore reversed? What regulates the balance between these? How to monitor, and what strategies to use in order to manage the epigenetic burden that comes from the environment? How to target the modulation of the epigenome toward healthier states during life span and across generations?
The EDAM model adds a new perspective to understanding how failure to adapt to environmental stressors may lead to epigenetic diseases. Deeper understanding of the epigenetic bases of diseases has numerous potential applications. Some of these include (1) disease diagnosis and prognosis—epigenetic markers can serve as diagnostic tools for identifying diseases and predicting the course of the condition, (2) personalized treatment—allows for the development of targeted therapies based on an individual’s unique epigenetic profile, (3) therapeutic interventions—development of new epidrugs aimed at reversing or modifying specific epigenetic modifications, and (4) environmental and lifestyle interventions—possibility of modifying epigenetic marks through changes in lifestyle, diet, or exposure to specific environmental factors.
Acknowledgments
The author is grateful to the National Science Fund of Bulgaria, Grant KP-06-H25/5 for the financial support of studies in the field of Epigenetics.
Conflict of interest
The author declares no conflict of interest.
References
- 1.
Waddington CH. The epigenotype. Endeavour. 1942; 1 :18-20 - 2.
Wu C-t, Morris JR. Genes, genetics, and epigenetics: A correspondence. Science. 2001; 293 :1103-1105. DOI: 10.1126/science.293.5532.1103 - 3.
Turner B. Defining an epigenetic code. Nature Cell Biology. 2007; 9 :2-6. DOI: 10.1038/ncb0107-2 - 4.
Painter R, Osmond C, Gluckman P, Hanson M, Phillips D, Roseboom T. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG: An International Journal of Obstetrics and Gynaecology. 2008; 115 :1243-1249. DOI: 10.1111/j.1471-0528.2008.01822.x - 5.
Veenendaal MV, Painter RC, de Rooij SR, Bossuyt PM, van der Post JA, Gluckman PD, et al. Transgenerational effects of prenatal exposure to the 1944-45 Dutch famine. BJOG: An International Journal of Obstetrics and Gynaecology. 2013; 120 :548-554. DOI: 10.1111/1471-0528.12136 - 6.
Moore R, Kaletsky R, Murphy C. Piwi/PRG-1 Argonaute and TGF-ẞ mediate transgenerational learned pathogenic avoidance. Cell. 2019; 177 :1827-1841. DOI: 10.1016/j. cell.2019.05.024 - 7.
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009; 462 :315-322. DOI: 10.1038/nature08514 - 8.
Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998; 393 :386-389. DOI: 10.1038/30764 - 9.
Wiles ET, Selker EU. H3K27 methylation: A promiscuous repressive chromatin mark. Current Opinion in Genetics & Development. 2017; 43 :31-37. DOI: 10.1016/j.gde.2016.11.001 - 10.
Jenuwein T, Allis CD. Translating the histone code. Science. 2001; 293 :1074-1080. DOI: 10.1126/science.1063127 - 11.
Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: Insights into functions. Nature Reviews Genetics. 2009; 10 :155-159. DOI: 10.1038/nrg2521 - 12.
Ohnishi Y, Totoki Y, Toyoda A, Watanabe T, Yamamoto Y, Tokunaga K, et al. Small RNA class transition from siRNA/piRNA to miRNA during pre-implantation mouse development. Nucleic Acids Research. 2010; 38 (15):5141-5151. DOI: 10.1093/nar/gkq229 - 13.
Morris KV. RNA-directed transcriptional gene silencing and activation in human cells. Oligonucleotides. 2009; 19 :299-306. DOI: 10.1089/oli.2009.0212 - 14.
Siciliano V, Garzilli I, Fracassi C, Criscuolo S, Ventre S, di Bernardo D. MiRNAs confer phenotypic robustness to gene networks by suppressing biological noise. Nature Communications. 2013; 4 :2364. DOI: 10.1038/ncomms3364 - 15.
Reichard JF, Schnekenburger M, Puga A. Long term low-dose arsenic exposure induces loss of DNA methylation. Biochemical and Biophysical Research Communications. 2007; 352 :188-192. DOI: 10.1016/j.bbrc.2006.11.001 - 16.
Takiguchi M, Achanzar WE, Qu W, Li G, Waalkes MP. Effects of cadmium on DNA-(cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Experimental Cell Research. 2003; 286 :355-365. DOI: 10.1016/s0014-4827(03)00062-4 - 17.
Lee YW, Klein CB, Kargacin B, Salnikow K, Kitahara J, Dowjat K, et al. Carcinogenic nickel silences gene expression by chromatin condensation and DNA methylation: A new model for epigenetic carcinogens. Molecular and Cellular Biology. 1995; 15 :2547-2557. DOI: 10.1128/MCB.15.5.2547 - 18.
Wright RO, Schwartz J, Wright RJ, Bollati V, Tarantini L, Park SK, et al. Biomarkers of lead exposure and DNA methylation within retrotransposons. Environmental Health Perspectives. 2010; 118 :790-795. DOI: 10.1289/ehp.0901429 - 19.
Singh S, Li SS. Epigenetic effects of environmental chemicals bisphenol A and phthalates. International Journal of Molecular Sciences. 2012; 13 :10143-10153. DOI: 10.3390/ijms130810143 - 20.
Morgan HD, Sutherland HE, Martin DIK, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nature Genetics. 1999; 23 :314-318. DOI: 10.1038/15490 - 21.
Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104 :13056-13061. DOI: 10.1073/pnas.0703739104 - 22.
Tarantini L, Bonzini M, Apostoli P, Pegoraro V, Bollati V, Marinelli B, et al. Effects of particulate matter on genomic DNA methylation content and iNOS promoter methylation. Environmental Health Perspectives. 2009; 117 (2):217-222. DOI: 10.1289/ehp.11898 - 23.
Cantone L, Nordio F, Hou L, Apostoli P, Bonzini M, Tarantini L, et al. Inhalable metal-rich air particles and histone H3K4 dimethylation and H3K9 acetylation in a cross-sectional study of steel workers. Environmental Health Perspectives. 2011; 119 (7):964-969. DOI: 10.1289/ehp.1002955 - 24.
Bollati V, Marinelli B, Apostoli P, Bonzini M, Nordio F, Hoxha M, et al. Exposure to metal-rich particulate matter modifies the expression of candidate microRNAs in peripheral blood leukocytes. Environmental Health Perspectives. 2010; 118 (6):763-768. DOI: 10.1289/ehp.0901300 - 25.
Yang J, Zhong W, Xue K, Wang Z. Epigenetic changes: An emerging potential pharmacological target in allergic rhinitis. International Immunopharmacology. 2019; 71 :76-83. DOI: 10.1016/j.intimp.2019.03.004 - 26.
Nestor CE, Barrenäs F, Wang H, Lentini A, Zhang H, Bruhn S, et al. DNA methylation changes separate allergic patients from healthy controls and may reflect altered CD4+ T-cell population structure. PLoS Genetics. 2014; 10 (1):e1004059. DOI: 10.1371/journal.pgen.1004059 - 27.
Guida F, Sandanger TM, Castagné R, Campanella G, Polidoro S, Palli D, et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Human Molecular Genetics. 2015; 24 (8):2349-2359. DOI: 10.1093/hmg/ddu751 - 28.
Dogan MV, Shields B, Cutrona C, Gao L, Gibbons FX, Simons R, et al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics. 2014; 15 :151. DOI: 10.1186/1471-2164-15-151 - 29.
Zhu X, Li J, Deng S, Yu K, Liu X, Deng Q , et al. Genome-wide analysis of DNA methylation and cigarette smoking in a Chinese population. Environmental Health Perspectives. 2016; 124 (7):966-973. DOI: 10.1289/ehp.1509834 - 30.
Howe CG, Zhou M, Wang X, Pittman GS, Thompson IJ, Campbell MR, et al. Associations between maternal tobacco smoke exposure and the cord blood CD4+ DNA methylome. Environmental Health Perspectives. 2019; 127 (4):047009. DOI: 10.1289/EHP3398 - 31.
Liu F, Killian JK, Yang M, Walker RL, Hong JA, Zhang M, et al. Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene. 2010; 29 (25):3650-3664. DOI: 10.1038/onc.2010.129 - 32.
Schembri F, Sridhar S, Perdomo C, Gustafson AM, Zhang X, Ergun A, et al. MicroRNAs as modulators of smoking-induced gene expression changes in human airway epithelium. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106 (7):2319-2324. DOI: 10.1073/pnas.0806383106 - 33.
Bind MA, Zanobetti A, Gasparrini A, Peters A, Coull B, Baccarelli A, et al. Effects of temperature and relative humidity on DNA methylation. Epidemiology. 2014; 25 (4):561-569. DOI: 10.1097/EDE.0000000000000120 - 34.
Jung J, Kim SK, Jung SH, Jeong MJ, Ryu CM. Sound vibration-triggered epigenetic modulation induces plant root immunity against Ralstonia solanacearum. Frontiers in Microbiology. 2020; 11 :1978. DOI: 10.3389/fmicb.2020.01978 - 35.
Liu Y, Liu WB, Liu KJ, Ao L, Li J, Zhong JL, et al. Effect of 50 Hz extremely low-frequency electromagnetic fields on the DNA methylation and DNA methyltransferases in mouse spermatocyte-derived cell line GC-2. BioMed Research International. 2015; 2015 :237183. DOI: 10.1155/2015/237183 - 36.
Leone L, Fusco S, Mastrodonato A, Piacentini R, Barbati SA, Zaffina S, et al. Epigenetic modulation of adult hippocampal neurogenesis by extremely low-frequency electromagnetic fields. Molecular Neurobiology. 2014; 49 :472-1486. DOI: 10.1007/s12035-014-8650-8 - 37.
Erdal ME, Yılmaz SG, Gürgül S, Uzun C, Derici D, Erdal N. miRNA expression profile is altered differentially in the rat brain compared to blood after experimental exposure to 50 Hz and 1 mT electromagnetic field. Progress in Biophysics and Molecular Biology. 2018; 132 :35-42. DOI: 10.1016/j.pbiomolbio.2017.08.001 - 38.
Belli M, Tabocchini MA. Ionizing radiation-induced epigenetic modifications and their relevance to radiation protection. International Journal of Molecular Sciences. 2020; 21 (17):5993. DOI: 10.3390/ijms21175993 - 39.
Choi SW, Friso S. Epigenetics: A new bridge between nutrition and health. Advances in Nutrition. 2010; 1 :8-16. DOI: 10.3945/an.110.1004 - 40.
Steegers-Theunissen RP, Obermann-Borst SA, Kremer D, Lindemans J, Siebel C, Steegers EA, et al. Periconceptional maternal folic acid use of 400 microg per day is related to increased methylation of the IGF2 gene in the very young child. PLoS One. 2009; 4 :e7845. DOI: 10.1371/journal.pone.0007845 - 41.
Niculescu MD, Craciunescu CN, Zeisel SH. Dietary choline deficiency alters global and gene-specific DNA methylation in the developing hippocampus of mouse fetal brains. The FASEB Journal. 2006; 20 :43-49. DOI: 10.1096/fj.05-4707com - 42.
Mehedint MG, Niculescu MD, Craciunescu CN, Zeisel SH. Choline deficiency alters global histone methylation and epigenetic marking at the Re1 site of the calbindin 1 gene. The FASEB Journal. 2010; 24 :184-195. DOI: 10.1096/fj.09-140145 - 43.
Lillycrop KA, Phillips ES, Torrens C, Hanson MA, Jackson AA, Burdge GC. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR alpha promoter of the offspring. The British Journal of Nutrition. 2008; 100 (2):278-282. DOI: 10.1017/S0007114507894438 - 44.
Ponomarev I, Wang S, Zhang L, Adron Harris R, Dayne MR. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. The Journal of Neuroscience. 2012; 32 (5):1884-1897. DOI: 0.1523/JNEUROSCI.3136-11.2012 - 45.
Kaminen-Ahola N, Ahola A, Maga M, Mallitt KA, Fahey P, Cox TC, et al. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genetics. 2010; 6 (1):e1000811. DOI: 10.1371/journal.pgen.1000811 - 46.
Mahoney DJ, Parise G, Melov S, Safdar A, Tarnopolsky MA. Analysis of global mRNA expression in human skeletal muscle during recovery from endurance exercise. The FASEB Journal. 2005; 19 (11):1498-1500. DOI: 10.1096/fj.04-3149fje - 47.
Zhang FF, Cardarelli R, Carroll J, Zhang S, Fulda KG, Gonzalez K, et al. Physical activity and global genomic DNA methylation in a cancer-free population. Epigenetics. 2011; 3 :293-299. DOI: 10.4161/epi.6.3.14378 - 48.
McGee SL, Fairlie E, Garnham AP, Hargreaves M. Exercise-induced histone modifications in human skeletal muscle. The Journal of Physiology. 2009; 587 :5951-5958. DOI: 10.1113/jphysiol.2009 - 49.
Woo V, Alenghat T. Epigenetic regulation by gut microbiota. Gut Microbes. 2022; 14 (1):2022407. DOI: 10.1080/19490976.2021.2022407 - 50.
Wang K, Liu H, Hu Q , Wang L, Liu J, Zheng Z, et al. Epigenetic regulation of aging: Implications for interventions of aging and diseases. Signal Transduction and Targeted Therapy. 2022; 7 (1):374. DOI: 10.1038/s41392-022-01211-8 - 51.
Oblak L, van der Zaag J, Higgins-Chen AT, Levine ME, Boks MP. A systematic review of biological, social and environmental factors associated with epigenetic clock acceleration. Ageing Research Reviews. 2021; 69 :101348. DOI: 10.1016/j.arr.2021.101348 - 52.
Parade SH, Huffhines L, Daniels TE, Stroud LR, Nugent NR, Tyrka AR. A systematic review of childhood maltreatment and DNA methylation: Candidate gene and epigenome-wide approaches. Translational Psychiatry. 2021; 11 (1):134. DOI: 10.1038/s41398-021-01207-y - 53.
Howie H, Rijal CM, Ressler KJ. A review of epigenetic contributions to post-traumatic stress disorder. Dialogues in Clinical Neuroscience. 2019; 21 (4):417-428. DOI: 10.31887/DCNS.2019.21.4/kressler - 54.
Palma-Gudiel H, Córdova-Palomera A, Eixarch E, Deuschle M, Fañanás L. Maternal psychosocial stress during pregnancy alters the epigenetic signature of the glucocorticoid receptor gene promoter in their offspring: A meta-analysis. Epigenetics. 2015; 10 (10):893-902. DOI: 10.1080/15592294.2015.1088630 - 55.
White AJ, Kresovich JK, Xu Z, Sandler DP, Taylor JA. Shift work, DNA methylation and epigenetic age. International Journal of Epidemiology. 2019; 48 (5):1536-1544. DOI: 10.1093/ije/dyz027 - 56.
Imai S, Saeki M, Yanase M, Horiuchi H, Abe M, Narita M, et al. Change in microRNAs associated with neuronal adaptive responses in the nucleus accumbens under neuropathic pain. The Journal of Neuroscience. 2011; 31 (43):15294-15299. DOI: 10.1523/JNEUROSCI.0921-11.2011 - 57.
Tajerian M, Alvarado S, Millecamps M, Vachon P, Crosby C, Bushnell MC, et al. Peripheral nerve injury is associated with chronic, reversible changes in global DNA methylation in the mouse prefrontal cortex. PLoS One. 2013; 8 (1):e55259. DOI: 10.1371/journal.pone.0055259 - 58.
Descalzi G, Ikegami D, Ushijima T, Nestler EJ, Zachariou V, Narita M. Epigenetic mechanisms of chronic pain. Trends in Neurosciences. 2015; 38 (4):237-246. DOI: 10.1016/j.tins.2015.02.001 - 59.
Kaliman P. Epigenetics and meditation. Current Opinion in Psychology. 2019; 28 :76-80. DOI: 10.1016/j.copsyc.2018.11.010 - 60.
Chaix R, Alvarez-López MJ, Fagny M, Lemee L, Regnault B, Davidson RJ, et al. Epigenetic clock analysis in long-term meditators. Psychoneuroendocrinology. 2017; 85 :210-214. DOI: 10.1016/j.psyneuen.2017.08.016 - 61.
Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends in Genetics. 2000; 16 :168-174. DOI: 10.1016/s0168-9525(99)01971-x - 62.
Baylin SB, Jones PA. Epigenetic determinants of Cancer. Cold Spring Harbor Perspectives in Biology. 2016; 8 (9):a019505. DOI: 10.1101/cshperspect.a019505 - 63.
Portela A, Esteller M. Epigenetic modifications and human disease. Nature Biotechnology. 2010; 28 :1057-1068. DOI: 10.1038/nbt.1685 - 64.
Lu Y, Chan YT, Tan HY, Li S, Wang N, Feng Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Molecular Cancer. 2020; 19 (1):79. DOI: 10.1186/s12943-020-01197-3 - 65.
Shi Y, Zhang H, Huang S, Yin L, Wang F, Luo P, et al. Epigenetic regulation in cardiovascular disease: Mechanisms and advances in clinical trials. Signal Transduction and Targeted Therapy. 2022; 7 (1):200. DOI: 10.1038/s41392-022-01055-2 - 66.
Moosavi A, Motevalizadeh AA. Role of epigenetics in biology and human diseases. Iranian Biomedical Journal. 2016; 20 (5):246-258. DOI: 10.22045/ibj.2016.01 - 67.
Grezenko H, Ekhator C, Nwabugwu NU, Ganga H, Affaf M, Abdelaziz AM, et al. Epigenetics in neurological and psychiatric disorders: A comprehensive review of current understanding and future perspectives. Cureus. 2023; 15 (8):e43960. DOI: 10.7759/cureus.43960 - 68.
Mazzone R, Zwergel C, Artico M, Taurone S, Ralli M, Greco A, et al. The emerging role of epigenetics in human autoimmune disorders. Clinical Epigenetics. 2019; 11 (1):34. DOI: 10.1186/s13148-019-0632-2 - 69.
Wu YL, Lin ZJ, Li CC, Lin X, Shan SK, Guo B, et al. Epigenetic regulation in metabolic diseases: Mechanisms and advances in clinical study. Signal Transduction and Targeted Therapy. 2023; 8 :98. DOI: 10.1038/s41392-023-01333-7 - 70.
Zoghbi HY, Beaudet al. Epigenetics and human disease. Cold Spring Harbor Perspectives in Biology. 2016; 8 (2):a019497. DOI: 10.1101/cshperspect.a019497 - 71.
Miranda Furtado CL, Dos Santos Luciano MC, Silva Santos RD, Furtado GP, Moraes MO, Pessoa C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics. 2019; 14 (12):1164-1176. DOI: 10.1080/15592294.2019.1640546 - 72.
Cannon WB. The Wisdom of the Body. New York: W.W. Norton; 1932 - 73.
Sterling P, Eyer J. Allostasis: A new paradigm to explain arousal pathology. In: Fisher S, Reason J, editors. Handbook of Life Stress, Cognition, and Health. New York: John Wiley & Sons; 1988. pp. 629-649 - 74.
McEwen BS. Sex, stress and the hippocampus: Allostasis, allostatic load and the aging process. Neurobiology of Aging. 2002; 23 (5):921-939. DOI: 10.1016/s0197-4580(02)00027-1 - 75.
Carlson ED, Chamberlain RM. Allostatic load and health disparities: A theoretical orientation. Research in Nursing & Health. 2005; 28 (4):306-315. DOI: 10.1002/nur.20084 - 76.
McEwen BS. Protective and damaging effects of stress mediators: Central role of the brain. Dialogues in Clinical Neuroscience. 2006; 8 (4):367-381. DOI: 10.31887/DCNS.2006.8.4/bmcewen - 77.
Mc Ewen BS. Stress, adaptation, and disease. Allostasis and allostatic load. Annals of the New York Academy of Sciences. 1998; 840 :33-44. DOI: 10.1111/j.1749-6632.1998.tb09546.x - 78.
McEwen BS, Stellar E. Stress and the individual. Mechanisms leading to disease. Archives of Internal Medicine. 1993; 153 (18):2093-2101 - 79.
Lumey LH, Stein AD, Ravelli AC. Timing of prenatal starvation in women and birth weight in their first and second born offspring: The Dutch famine birth cohort study. European Journal of Obstetrics, Gynecology, and Reproductive Biology. 1995; 61 (1):23-30. DOI: 10.1016/0028-2243(95)02149-m - 80.
Roseboom T, de Rooij S, Painter R. The Dutch famine and its long-term consequences for adult health. Early Human Development. 2006; 82 (8):485-491. DOI: 10.1016/j.earlhumdev.2006.07.001 - 81.
Lumey LH. Reproductive outcomes in women prenatally exposed to undernutrition: A review of findings from the Dutch famine birth cohort. The Proceedings of the Nutrition Society. 1998; 57 (1):129-135. DOI: 10.1079/pns19980019 - 82.
Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, et al. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Human Molecular Genetics. 2009; 18 (21):4046-4053. DOI: 10.1093/hmg/ddp353 - 83.
Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105 (44):17046-17049. DOI: 10.1073/pnas.0806560105 - 84.
Devi F, Shahwan S, Teh WL, Sambasivam R, Zhang YJ, Lau YW, et al. The prevalence of childhood trauma in psychiatric outpatients. Annals of General Psychiatry. 2019; 18 :15. DOI: 10.1186/s12991-019-0239-1 - 85.
Shields AE, Wise LA, Ruiz-Narvaez EA, Seddighzadeh B, Byun HM, Cozier YC, et al. Childhood abuse, promoter methylation of leukocyte NR3C1 and the potential modifying effect of emotional support. Epigenomics. 2016; 8 (11):1507-1517. DOI: 10.2217/epi-2016-0074