
Abstract
This chapter discusses the significant impact of N-heterocyclic carbene (NHC) ligands on the optical properties of metal complexes. It highlights the key role of NHC ligands in shifting the d−d transitions of these complexes to shorter wavelength regions, rendering them luminescent. The study focuses on NHC-based metal complexes involving copper, silver, platinum, iridium, and coinage metals, showcasing their luminescent properties and potential applications in organic light-emitting diodes (OLEDs). However, it also underscores a challenge: the quantum yields of these metal complexes may not be sufficient for practical applications. To address this issue, this chapter proposes modifying the NHC ligand with electron-rich substituents, aiming to reduce the HOMO-LUMO gap and shift d-d transitions into the UV region. Additionally, it introduces cyclic (alkyl)(amino)carbene (CAACs) as promising ligands for tuning the photophysical properties of metal complexes. CAACs are noted for their ability to decrease the HOMO-LUMO gap compared to NHCs and their electrophilic nature. Luminescent CAAC metal complexes are found to exhibit sub-microsecond lifetimes, making them suitable candidates for OLED applications. Ultimately, the research challenges researchers to better understand the structure, bonding, and properties of these complexes to create stable and highly luminescent materials using abundant and cost-effective metals like iron, copper, zinc, and silver.
Keywords
- N-heterocyclic carbene (NHC) ligands
- optical properties of metal complexes
- luminescent metal complexes
- cyclic (alkyl)(amino)carbene (CAACs) ligands
- structure-property of NH-Carbenes
1. Introduction
Carbenes are an intriguing class of carbon-containing compounds with a neutral divalent carbon atom with six electrons on its valence shell (Figure 1). However, unbound carbenes are extremely unstable due to their incomplete electron octet and coordinative unsaturation. Therefore, they have traditionally been considered extremely reactive, transient intermediates in chemical transformations like cyclopropanation. [1]
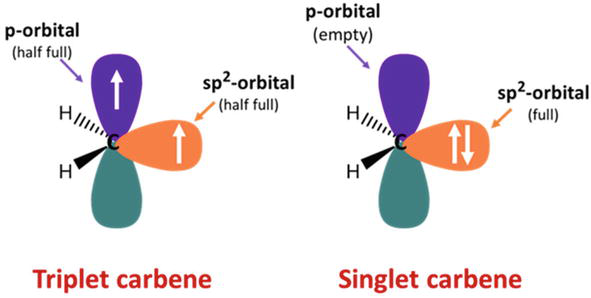
Figure 1.
Schematic representation of triplet and singlet carbene.
The exploration of carbenes has remained a significant and enduring focus of interest for researchers over the years, sparking numerous theoretical and experimental inquiries [2, 3, 4, 5, 6, 7, 8, 9, 10]. Among the most prominent questions that have arisen are those concerning the electronic configuration of carbenes – whether they exist in a singlet or triplet state – and the precise geometry around the carbenic carbon atom. Ongoing research endeavors, spanning from the realms of physical chemistry to organic chemistry, have provided a more systematic understanding of the nature of carbenes and their applications [3].
Initially conceived as intermediates in the context of organic transformations, these intriguing chemical entities later emerged as a central subject of study within the field of coordination chemistry [4, 11, 12]. The stability of carbenes became a crucial consideration for their utilization in coordination chemistry [5]. Consequently, the quest for isolating and definitively characterizing free, uncoordinated carbenes became a notable challenge in the late 1990s. In 1991, a milestone achievement was made when an isolable and “bottleable” carbene was successfully incorporated into a nitrogen heterocycle, giving rise to what is now known as N-heterocyclic carbene (NHC) [6].
1.1 Definition and a brief history of NHCs
NHCs are electron-rich nucleophilic heterocyclic species containing a carbene carbon and at least one directly bonded nitrogen atom within the ring structure NHCs. Compared with most carbenic systems, which are inherently unstable and exist only as transient intermediates in organic processes (e.g., cyclopropanations and C▬H insertions) without any additional stabilizing features. NHCs are stable compounds that can often be isolated and stored. NHCs, nowadays, find many applications across the life sciences, ranging from catalysis to materials science and biomedical applications [7].
The preparation of carbon having six valance electrons was a synthetic challenge for a long time, as there were many testified failed attempts to prepare them. [8] In the 1960s, Wanzlick tried to isolate carbenes but was unsuccessful. Later he proposed that a carbene center at the imidazole ring’s 2-position would be stable due to the electron-donating effects of nearby nitrogen atoms provided the conceptual foundation for the evolution of these species’ chemistry. [9] Bertrand et al. in 1988 proposed for the first time that the presence of heteroatoms, such as silicon or phosphorus, close to the carbon center increased the stability of carbenic species. [10] In 1991, Arduengo established the integration of the carbenic carbon atom into a nitrogen heterocycle decorated with bulky substituents. [6, 13] The inaugural achievement in isolating stable crystalline N-heterocyclic carbene (NHC) involved a meticulously orchestrated process. This milestone was realized through the deprotonation of bis-(1-adamantyl) imidazolium chloride using sodium hydride within the medium of tetrahydrofuran. The success of this reaction was facilitated by the presence of a catalytic quantity of dimethyl sulfoxide, as depicted in Figure 2 [6].
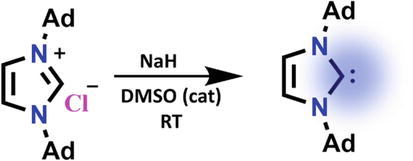
Figure 2.
Preparation of N-heterocyclic carbene by the deprotonation of bis-(1-adamantyl) imidazolium chloride.
With the discovery of the first NHCs in 1991, this field has expanded rapidly (as shown in Figure 3) in understanding the subtle behavior of NHCs and their fundamental applications in natural sciences. It is fascinating to observe that new applications for NHCs are continuously being discovered at a remarkable speed. [1, 15] An example of advancements in catalytic activity involves using NHCs as ligands in transition metal species. This approach goes beyond merely replacing phosphines, aiming to achieve increased stability and turnover frequencies. [16] In Figure 3, the studies carried out on NHCs from 1991 to 2021 are summarized very well.
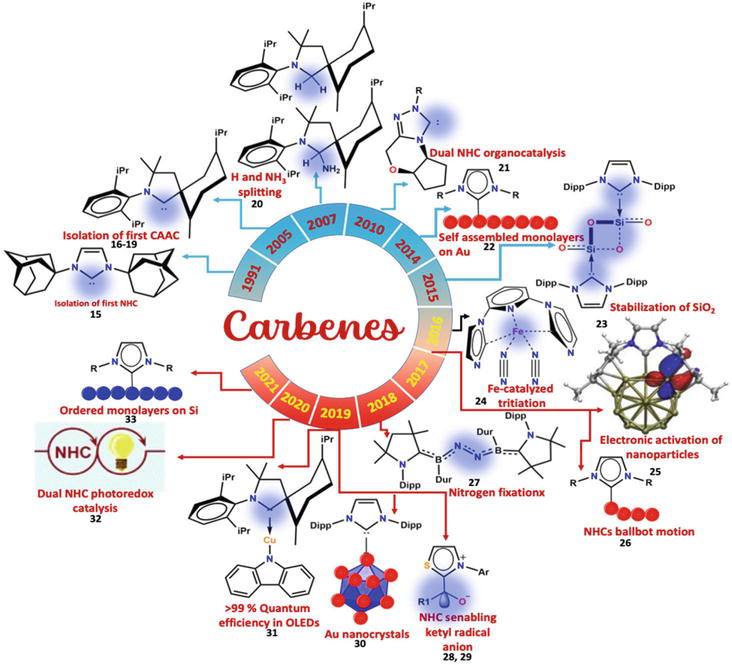
Figure 3.
Selected high impact contribution in the field of carbenes during last three decades. Ar = aryl; OLEDs = organic light-emitting diodes; NHC = N-heterocyclic carbene; Dur = 2,3,5,6-tetramethylphenyl; Dipp = 2,6-diisopropylphenyl; CAAC = cyclic (alkyl)(amino)carbene. Part of the figure was adapted with permission from ref. [
1.2 Structure and properties of NHCs
NHCs have proven cutting-edge classes of compounds with a significant influence on chemical science. [17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35] NHCs are known for their intrinsic reactivity; NHCs are not different from them. However, the presence of a cyclic scaffold and the heteroatom increased the level of stability in contrast to other congeners. [14] This chapter briefly describes their primary characteristics, structural variation, metal complexation, and photophysical behavior. Although heteroatoms, particularly nitrogen atoms, can enhance the stability of carbenes, NHCs possess various structural characteristics that contribute to their stability in terms of both kinetics and thermodynamics, as well as their unique reactivity binding patterns. [36] Roughly some of the core features of the NHC structure can contribute: (1) presence and location of heteroatoms (nitrogen and others); (2) substituents at proximal positions; (3) backbone structure (e.g. unsaturation) and substituents; (4) ring size. A representation of the general structures of NHCs, firstly reported compound 1,3-di(adamantyl)imidazol-2-ylidene (IAd) presented in Figure 2 [37].
The accessible lone pairs can overlap with the vacant p-orbital (LUMO, “electron donating”) while the neighboring nitrogen atoms can “withdraw electron density” from the non-bonding lone pair at the carbenic carbon (HOMO, “-electron withdrawing”) due to their high electronegativity (Figure 4) [38]. The reactive center is crucially protected from possible dimerization or other breakdown routes by the steric bulk on the N-substituents (such as adamantyl in Arduengo
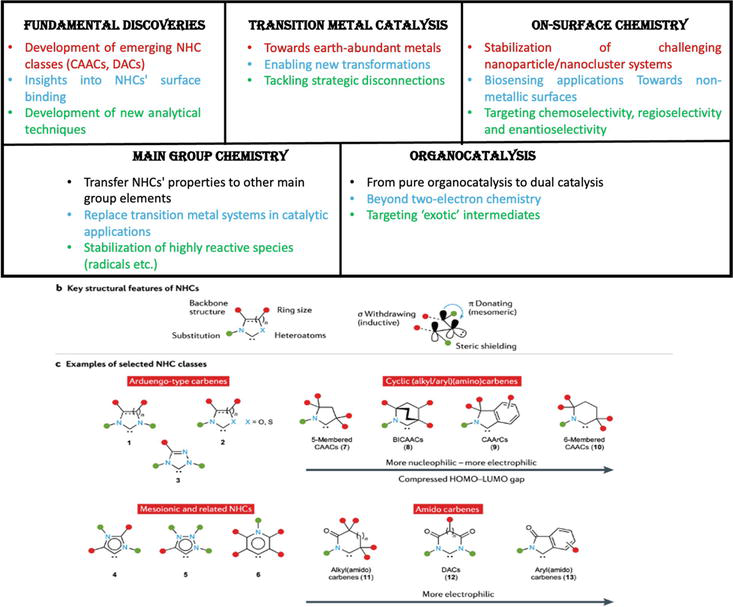
Figure 4.
Recent key advances, structural features, and selected N-heterocyclic carbenes classes. (a) Summary of the most significant N-heterocyclic carbene (NHC)-related discoveries during the past decades. (b) Crucial NHC structural characteristics. (c) Overview of a few established and new classes of NHCs.
1.3 Generation of carbenes and N-heterocyclic carbenes (NHCs)
Elimination and fragmentation processes are typically used to create carbene and NHC compounds. When groups linked to the carbon atom are broken as a result of photolysis, thermolysis, or a reaction with metals, carbenes are produced as intermediate product.
1.3.1 Generation of carbenes
1.3.1.1 α-elimination
The hydrolysis of chloroform in basic medium was probably the first reaction in which intermediate NHC formation was suggested [41]. In the early 1950s, Hine and coworkers investigated the mechanism of this reaction, ushering in the current age of carbene chemistry. [42, 43, 44] The acidic proton of chloroform separates in the basic environment, forming the trichloromethyl anion. When the chloride anion is taken out of the carbanion, dichlorocarbene is created. Carbon monoxide is produced
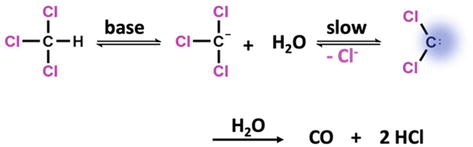
Figure 5.
The generation of intermediate carbenes through α-elimination reactions in chloroform.
Cyclopropane derivative can be prepared by the addition of NHC to the double bond electron system using aprotic solvents and strong bases, as presented in Figure 6. Before the reaction, the solvent used must be dried very well. Because the water in the environment can easily react with the NHC formed, and it also reacts with the base in the environment and completely prevents the formation of NHC.
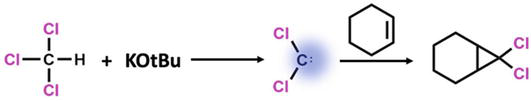
Figure 6.
Addition of carbenes to a double bond system to yield cyclopropane derivatives.
1.3.1.2 Three-membered carbon ring for carbenes
Due to high steric strain, three-membered rings frequently breakdown into carbene intermediates when heated or exposed to radiation. The photolysis of 1,1-dichloro-2-phenylcyclopropane produces CCl2, while oxirane photolytic decomposition gives arylcarbenes, as illustrated in Figure 7. For substituted arylcarbenes, such as diphenylcarbene [45], phenylmethylcarbene [46], cyanophenylcarbene [47, 48], and methoxycarbonylphenylcarbene [49], substituted phenyloxiranes serve as handy precursors. With unsymmetrical precursors, selective cleavage was seen; it appears that the thermodynamically more stable isomer is favorable.
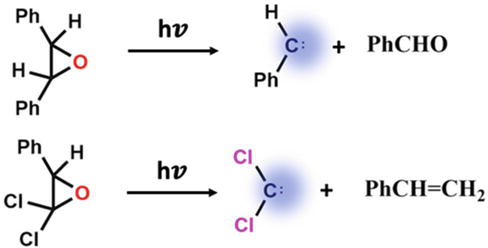
Figure 7.
Generation of carbenes from cyclopropanes and epoxides.
1.3.1.3 Simmon-Smith reaction
Methylene carbene cannot be produced using the α-elimination method. There are techniques for producing methylene carbene adducts, though. The reaction of alkenes with diiodomethane in the presence of zinc to yield cyclopropane compounds is the most frequently employed method (Figure 8) these reactions are popularly known as Simmon-Smith reaction [50, 51, 52].
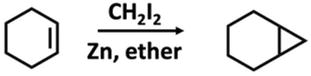
Figure 8.
Simmon-Smith reaction.
1.3.1.4 Strained alkenes
The π-bond is weakened by a greatly diminished p-p overlap and distortion from planarity when the alkene is very sterically hindered. As a result, the energy of the ground state is increased, making it feasible for heating to cause dissociation to NHCs. Tetranaphth-1-ylethene’s reversible dissociation into bis(naphth-1-yl) carbene at 250°C [53] is a well-known example of this mechanism (Figure 9).
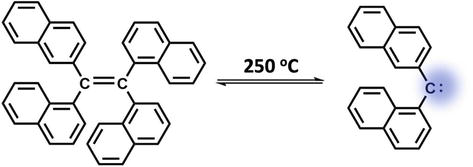
Figure 9.
Thermal dissociation of tetranaphth-1-ylethene
1.3.2 Generation of N-heterocyclic carbenes
1.3.2.1 Cyclization by introduction of the precarbenic atom moiety
The incorporation of the C1 precarbenic unit stands out as the most widely employed strategy for NHC precursors. Renowned for its high efficiency and flexibility, this method allows for the integration of diverse substituents. Depending on the desired core of the N-heterocyclic carbene precursor, various reagents can be utilized to introduce the carbenic carbon moiety. This section provides an overview of the different possibilities, accompanied by representative examples [14].
1.3.2.1.1 Trialkyl orthoformate (HC(OR)3) as the precarbenic unit
In 1991, Saba and Kaloustian reported that the treatment of
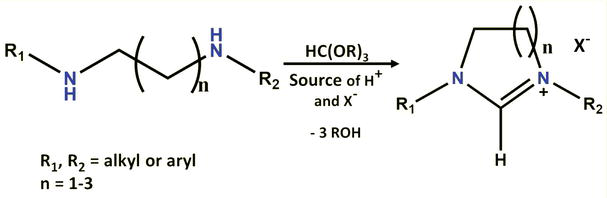
Figure 10.
General route to the cyclic formamidinium salt by condensation of a diamine with trialkyl orthoformate.
This reaction serves as the final step in a significant number of NHC-precursor syntheses due to its typically high yield and ease of execution. The resulting salts often precipitate, facilitating recovery through simple filtration. Essentially, any tethered secondary diamine can be transformed into the corresponding cyclic formamidinium salt, with the main challenge lying in finding a suitable synthesis for the starting diamine (Figure 10). Depending on the characteristics of the target NHC precursor (such as the nature of the linking backbone, nitrogen substituents, presence of chirality, or annelated cycles), several synthetic pathways to substituted diamines have been developed. These can be categorized into six major strategies: (a) formation of a bisimine and reduction; (b) bisacylation of two amines and reduction; (c) monoalkylation and monoacylation of two amines [55, 56, 57, 58, 59].
1.3.2.1.1.1 Condensation-reduction route
In a typical synthetic procedure, a diimine is initially formed through the condensation of an aryl or alkylamine with glyoxal. This diimine is then reduced to the corresponding diamine, which is isolated either as the free base or as the dihydrochloride salt at this stage. The cyclization step involves the use of triethyl orthoformate as the C1-building block, along with one equivalent of acid if starting from the free diamine, leading to the formation of the corresponding imidazolinium salt (Figure 11). This synthetic route remains the most widely adopted standard protocol for obtaining common imidazolinium salts, proving applicable to a broad range of primary amines, as demonstrated by selected examples in Figure 11 [60].
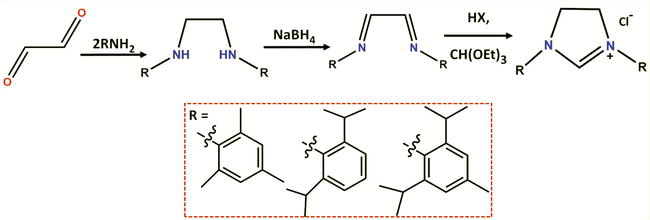
Figure 11.
Synthesis of the symmetrical imidazolinium chlorides 1a-q in a three-step sequence: Glyoxal condensation with an arylamine, reduction of the resulting diimine, and cyclization with triethyl orthoformate.
Hoveyda and co-workers successfully employed the method for synthesizing the unsymmetrical (and chiral) imidazolinium salt 4 from (S)-NOBIN in a two-step sequence, using Boc-protected amino aldehyde as the key coupling partner (Figure 12). The deprotection of the Boc group took place during the formation of the dihydrochloride salt of the diamine. However, it is worth noting that compound had to be synthesized first from mesitylamine in a three-step route, contributing to an overall lengthy synthetic procedure [61].
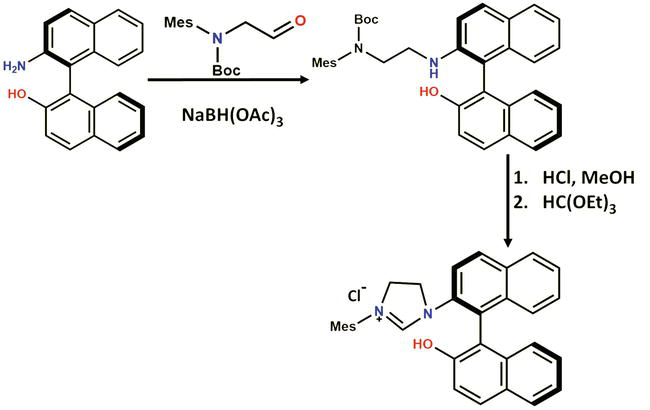
Figure 12.
Synthesis of the unsymmetrical, chiral imidazolinium chloride 4 according to Hoveyda.
Lassaletta and co-workers extended this method to the cyclization of bis-hydrazine, resulting in the formation of 1,3-bis(
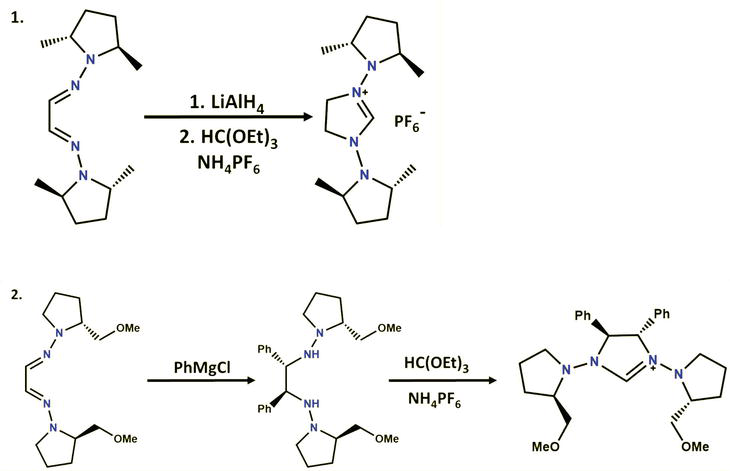
Figure 13.
Synthesis of the 1,3-Bis(N,N-dialkylamino)- imidazolinium salts 6a and 6b derived from chiral N,N-Dialkylhydrazines.
1.3.2.1.1.2 Bisacylation/reduction route
The use of oxalyl chloride or derivatives, such as monoethyl oxalyl chloride, for synthesizing
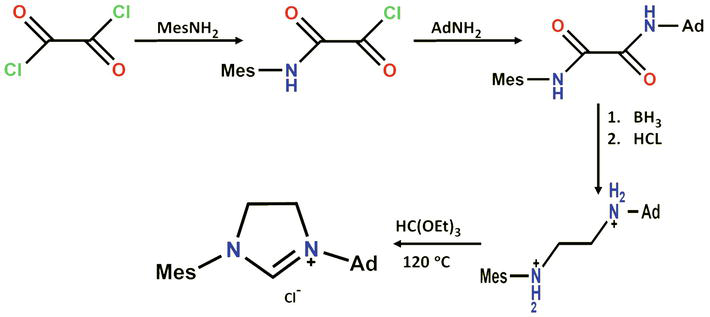
Figure 14.
Synthesis of 1-Adamantyl-3-mesitylimidazolinium chloride, according to Mol and co-workers.
2. s, p, d and f block metal complexes
2.1 Complexes with s-block elements
Complexes of s-block metals in their zero oxidation state offer chemists the opportunity to handle these elements in soluble and distinct molecular forms, typically in their electron-rich electronic state. Beryllium (Be) is one of them which very rarely investigated chemical element of the first octal row of the periodic table, except inert neon. The reason behind this is the high toxicity of Be, which makes the experiment
Beryllium chemistry is a productive field that utilizes the superior lewis acidity of beryllium compounds compared to other main-group elements. Beryllium oxide (BeO) is the strongest neutral main group Lewis acid known to date. Dehnicke et al. conducted a systematic study on the stable complexes that Beryllium dihalides BeX2 (X = F, Cl) form with lewis bases. In its zero oxidation state, beryllium atom showcase a significant lewis acidity. In 2013, studies predicted tri-coordinated beryllium complexes with CO, PR3, or NHC that would lead to an electron octet at beryllium, which could be potentially synthesized and isolated, as a free molecule (Figure 15a) [65].
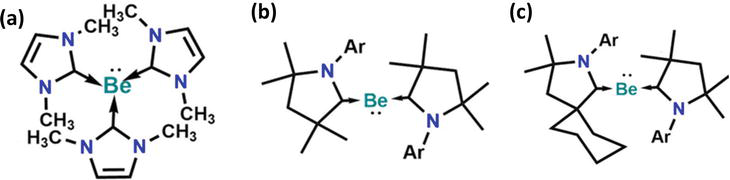
Figure 15.
Rare Be complexes reported a decade ago (a) tri-coordinated beryllium complexes, (b) di-coordinated beryllium complexes, [Be(cAACMe)2], and (c) di-coordinated beryllium complexes with zero oxidation state, [Be(cAACMe)(cAAC)cy]. Ar = 2,6 diisopropylphenyl.
Braunschweig and co-workers pointed out one unexpected result, where they synthesized di-coordinated beryllium complexes, found to be less predicted, as shown in Figure 15b, where two cyclic (alkyl)(amino)carbene (cAAC) ligands stabilize a beryllium atom to form [Be(cAACMe)2] and [Be(cAACMe)(cAAC)Cy], in the formal oxidation state zero (Figure 15b, c) [66].
Lithium, another versatile alkaline metal, can form numerous complexes with NHC that have the ability to deprotonate. Alkaline metal bases are commonly used as deprotonating agents for bis(imidazolium) and related salts. Similarly, bis(trimethylsilyl) amides (HMDS) of sodium, potassium, and lithium are regularly used for deprotonation due to their bulkiness and low nucleophilicity. Fehlhammer’s et al. synthesized the lithium complexes that involve monoanionic BH2-connected bis(NHC)s, having terminal groups of methyl, ethyl, and isopropyl. However, they have solely served as intermediates to produce corresponding gold, palladium, and platinum complexes. Later, Smith et al. synthesized an air and temperature-sensitive lithium complex (Figure 16) having tert-butyl analogue. Despite their high sensitivity, the complex was successfully characterized through NMR and single-crystal X-ray diffraction analysis. The complex forms a dimer consisting of two inequivalent lithium ions with coordination numbers of two and three, respectively [66].
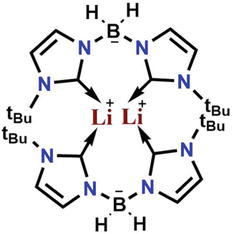
Figure 16.
Lithium complex with imidazolium-based NHC having tert-butyl analog.
Calcium on the other hand, multifaceted alkaline earth metal which shows great potential in organometallic chemistry due to its high lewis acidity and large ionic size. It was investigated that when it comes to stabilizing calcium amides, smaller NHCs are a superior choice in comparison to larger NHCs.
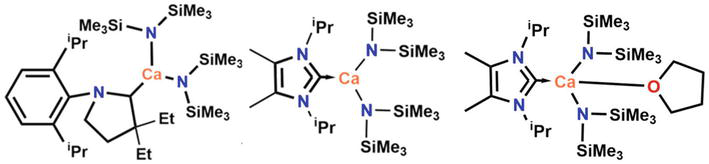
Figure 17.
Typical examples of calcium NHC complexes with imidazolium derivatives based NHC.
Recent years have seen significant improvements in the production of molecular alkaline earth metal catalysts and reagents. Beryllium and magnesium molecular aryloxide and alkoxide complexes as prospective starting points for the synthesis of light, alkaline earth metals. Bis-NHC beryllium/magnesium dichloride complex, (sIPr)2BeCl2/(sIPr)2BeCl2 is created when beryllium chloride or magnesium chloride reacts with two equivalents of 1,3-diisopropyl-4,5-dimethylimidizol-2-ylidine (presented in Figure 18). The terminal aryloxide (sIPr)Be(ODipp)2 and alkoxide dimer [(sIPr)Mg(OEt)Me]2, [(sIPr)Mg(OEt)Brl]2 and [(sIPr)Be(OEt)Cl]2 are produced with lithium diisopropylphenoxide (LiODipp) or sodium ethoxide (NaOEt), respectively depicted in Figure 18 [68].
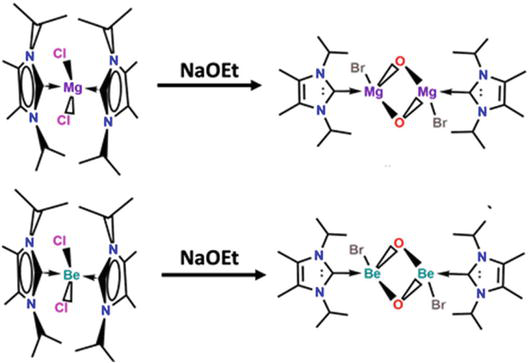
Figure 18.
The synthesis of Be and Mg NHCs [(sIPr)Mg(OEt)me]2 and [(sIPr)Be(OEt)Cl]2.
2.2 Coordination with p-block element
Synthetic targets for stable heavy main group element radicals are difficult. Despite several methods for stabilizing such odd-electron entities, there are not many heavier pnictogen-centered radicals. Radical cations with two coordinated pnictogen centers [(MeCAAC)EGa(Cl)L][B(C6F5)4] (MeCAAC = [H2C(CMe2)2NDipp]C; Dipp = 2,6-I-Pr2C6H3; E = Sb, Bi, As; L = HC[C(Me)NDipp]2) synthesized by one-electron oxidation of L(Cl)Ga-substituted pnictinidenes (MecAAC)EGa(Cl)L (E = BiIII, SbII, AsI) presented in Figure 19 [69].
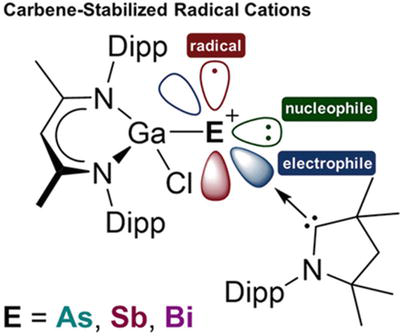
Figure 19.
A rare examples of main group NHCs complex [(MeCAAC)EGa(Cl)L][B(C6F5)4] (MecAAC = [H2C(CMe2)2NDipp]C; Dipp = 2,6-I-Pr2C6H3; E = Sb, Bi, As; L = HC[C(me)NDipp]2). The figure is reprinted with permission from ref. [
Synthetic chemists have used NHCs, cyclic alkyl amino carbenes, and aberrant NHCs as neutral ligands for stabilizing odd compounds containing main group elements as a result of the isolation of stable NHC. Below are a few of the instances showing the silicon NHC complex presented in Figure 20 [70].
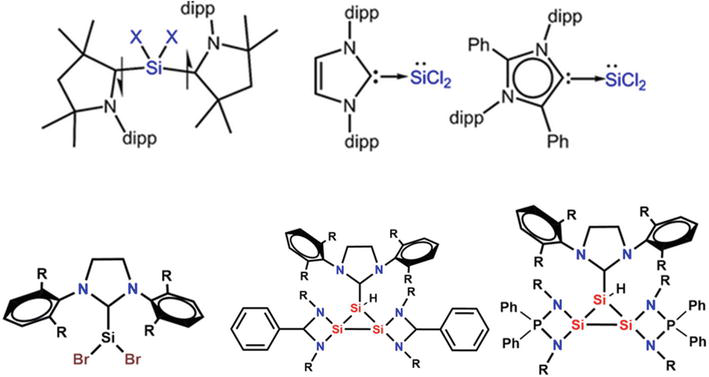
Figure 20.
Few examples of SI NHC complexes.
2.3 Coordination with transition metal
The two most common techniques for producing transition metal-NHC complexes are carbene transfer procedures or the utilization of free carbenes (isolated or synthesized
The potential of carbenes with an inorganic backbone as ligands in transition metal chemistry is illustrated, given examples where it acts as a better-donor than classical carbenes. This also highlights the significant impact of the inorganic backbone on the carbon ligand’s coordination capacity. Currently, being researched is the synthesis of analogues with less bulky substituents on the nitrogen atoms next to the carbene carbon. An examples of W NHCs is illustrated where imidazolium based OTf- is complexes with W by the exposer with radiation presented in Figure 21 [71].
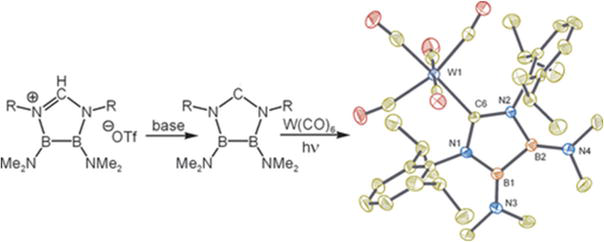
Figure 21.
A W NHCs complex with imidazolium based OTf-. Reprinted with the permission from ref
The chemotherapy medication cisplatin PtCl2(NH3)2 has gained significant importance. The development of new platinum-based medications with lower toxicity is a vibrant and highly active area of research because despite having a number of serious side effects that significantly restrict the drug’s efficacy. The usage of NHCs ligands has shown to be a potential method for creating cisplatin alternatives a such example is depicted in Figure 22. For the creation of prospective metallodrugs, not only platinum NHC complexes but also Au, Ag, Pd, Cu, Ru, or Ir NHC complexes have been studied. The antiproliferative effect of platinum (II) NHCs complexes carrying dithiocarbamate ligands was tested against several cancer cell lines. These complexes exhibit remarkable stability in solution [72].
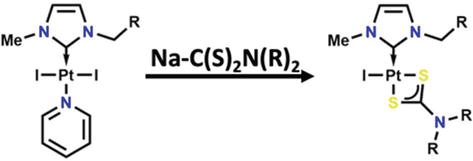
Figure 22.
Dithiocarbamate ligand is used with platinum (II) NHCs complexes as an alternative for cisplatin PtCl2(NH3)2.
Because of their powerful propensity to donate electrons, NHCs make excellent alternatives for phosphine ligands because they display more permanent metal–ligand interaction, which increases the robustness of the complex. In addition to complexes made of noble metals, Fe-NHC complexes have lately drawn a lot of interest because they might be used as affordable, ecologically friendly, and potent catalyst precursors. In homogeneous catalysis, well-defined and
Nonheme iron(II) catalysts with tetradentate nitrogen ligands have cis-coordination geometry, resulting in two cis-oriented binding sites at the iron center. Instances of this include BPMEN and BQEN (Figure 23), which each use two pyridine/quinoline and amine donors. It has been demonstrated that these instances provide iron complexes useful for difficult C▬H oxidation processes. Initial results on the hydrosilylation of acetophenone catalyzed by the novel Fe-NHC complex [73] using the bis(imidazo[1,5-a]pyridine-3-ylidene) ligand, which has two amine moieties in addition to two NHC groups.
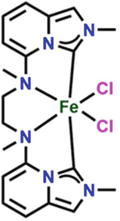
Figure 23.
Bis(imidazo[1,5-a]pyridine-3-ylidene is used with the iron to form Fe-NHC complex.
Using the transmetalation reaction between the equivalent silver(I) complex [Ag2L2](PF6)2 and KAuBr4 in the presence of AgPF6, one may create gold(III) complexes ([AuL2](PF6)3 (L = diNHC)). t-butyl and other bulky wingtip substituents hinder the production of the bis(diNHC) gold(III) complex, which results in the synthesis of the dinuclear gold(I) complex [Au2L2](PF6)2 following a redox rearrangement presented in Figure 24 [74].
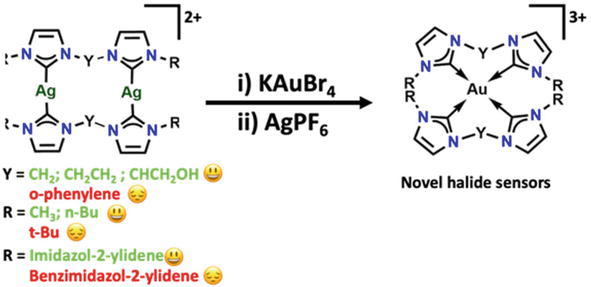
Figure 24.
Preparation of Au-NHCs using reinstallation reaction where [Ag2L2](PF6)2 and KAuBr4 reacts with AgPF6, yields gold(III) complexes ([AuL2](PF6)3 (L = diNHC)).
The sodium salt Na[Ph2P(O)eC(H)eSO2Ph] has been synthesized and isolated as a methanide ligand system for transition metal complexes. The structural elements of this ligand have been shown to facilitate the separation of nucleophilic late transition metal NHCs complexes. By using a straightforward salt metathesis procedure, the appropriate Ru(cymene) chlorido complex was quickly made accessible. However, dehydrohalogenation of the chlorido complex resulted in the development of a cyclometallated Ru complex as opposed to the NHCs complex, as was the case with previously reported thio- and iminophosphoryl-tethered ligand systems (Figure 25) [75].
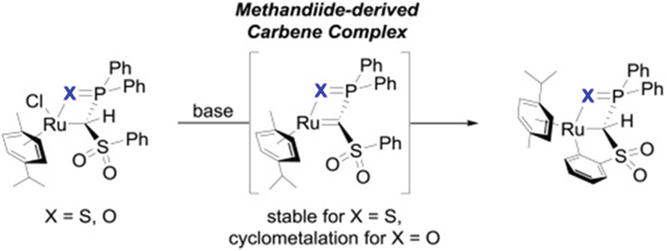
Figure 25.
Dehydrohalogenation of the chlorido complex resulted in the development of a cyclometallated Ru complex. Reprinted with permission from ref [
Iron(0) species with coordination unsaturation have drawn a lot of attention in recent years due to the resurgence of iron catalysis [76]. These species are thought to be reactive intermediates in processes like alkyne cyclotrimerization, the Diels-Alder reaction between dienes and alkynes, hydrogenation of alkenes and alkynes, and others. Coordinatively unsaturated iron(0) complexes are known to be responsive to C▬H bond activation processes. For instance, it is known that in an intramolecular C(sp3)H bond oxidative addition reaction, four coordinate iron(0) complex [Fe(PMe3)4] in solution phase to reversibly transform into the iron(II) complex [Fe-(PMe3)3(CH2PMe2)H] (Figure 26) [76].
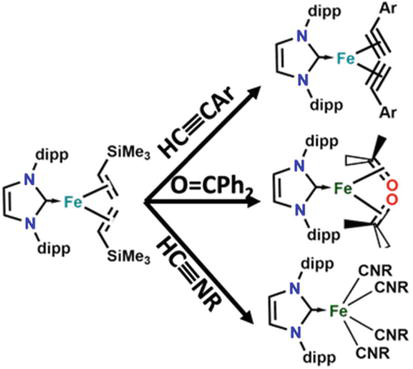
Figure 26.
Addition reaction to yield different derivatives on the Fe-NHCs complexes.
2.4 Complexes with inner transition metal
Due to their distinct electrical characteristics, lanthanide organometallic reagents and catalysts have gained increasing interest over the past two decades in the activation of chemical bonds and polymerization. Due to their excellent stability, lanthanide amides are one of the most useful groups among them. In order to investigate their reactivity, numerous lanthanide amides have been synthesized using various kinds of anionic spectator ligands. Despite the fact that certain of the homoleptic Ln[N(SiMe3)2]n complexes have already been employed as organometallic reagents and catalysts, there are currently no practical methods to control their reactivity and selectivity.
NHC ligands can stabilize the heteroleptic amido-ytterbium(II) alkynyl complex and anilido complex. This is most likely due to the NHC’s relativistically strong interaction with the divalent ytterbium ion and appropriate steric effect. Furthermore, rac-LA’s ROP has strong activity for NHC-stabilized samarium-(II) bis(amido) complexes even in the absence of an alcohol initiator (Figure 27). Ongoing research examines the reactivity of heteroleptic ytterbium(II) alkynyl and anilido complexes stabilized by NHC as well as the catalysis of samarium(II) bis(amido) complexes [78].
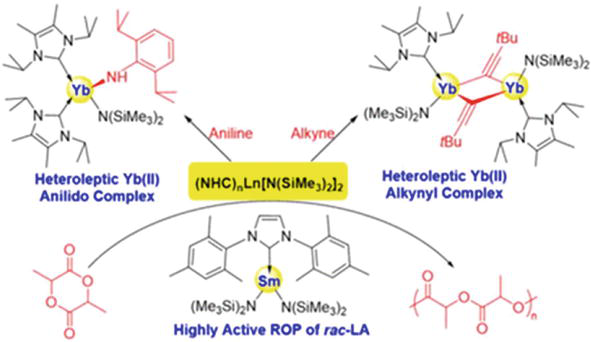
Figure 27.
NHC ligands can stabilize the heteroleptic amido-ytterbium(II) alkynyl complex and anilido complex. Rac-LA’s ROP has strong activity for NHC-stabilized samarium-(II) bis(amido) complexes even in the absence of an alcohol initiator reprinted with permission from ref. [
3. Use of NHCs in organic synthesis
Divalent carbon compounds called carbenes are a common building component in chemical synthesis. They are specifically employed in the synthesis of small rings. Diazo compounds and ketene are traditional sources of carbenes and easily undergo thermal, photochemical, or metal-induced cleavage. Additionally, a-haloalkyl lithium compounds [79] and trihalomethylmercury compounds have been shown to be promising organometallic precursors. The choice of precursor and the manner of formation both affect the reactivity of the carbene species.
NHCs complexes contain metal-stabilized carbenes. These compounds can be divided into two classes: the “Fischer-type” and the “Schrock type”, named after their discoverers. Subdivision into “carbene complexes” and “alkylidene complexes”, which is encountered now and then, makes little sense didactically and has at most only historical significance [80].
4. Photophysics of NHCs
The NHC-based metal complexes exhibit very interesting photophysical properties and many of them showed applications in OLEDs. Therefore, the origin of the electronic transitions in the NHC metal complexes and fluorescence emission bands has been discussed. A brief note on the design principles and the structure of NHC metal complex and their photophysical properties are highlighted in the book chapter.
Fluorescent/luminescent complexes that consist of N-heterocyclic carbine (NHC) ligands have been studied quite extensively over the last two decades [81, 82, 83]. Unlike many of the other ligands, the NHC ligands are strong σ-donors and stabilize most of the coordination complexes which in turn have shown very interesting applications in material chemistry [83, 84, 85, 86, 87, 88, 89]. The NHC ligands tend to push the d−d transitions to a shorter wavelength (high-energy) side thereby the designed metal−NHC complexes becomes fluorescent/luminescent (Figure 28). Photophysical properties such as UV-absorption, fluorescence, phosphorescence, and singlet-state lifetimes of NHC-metal complexes are important indicators to assess the utility of the compounds in optoelectronic applications. Hence, the photophysical properties of different NHC-metal complexes were studied and documented in the literature [81, 82, 83].
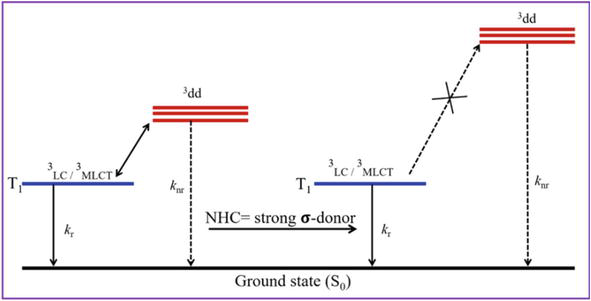
Figure 28.
Schematic representation of excited states of metal-NHC complexes emphasizing the NHCs as strong-donors ligands and destabilization of 3dd metal cantered states [
For example, photophysical properties of the coordination polymers supported by a pyridyl-substituted NHCs ligand were reported along with the monomers [90]. Electronic absorption spectra of monomers NHC1 and NHC2 exhibit absorption bands between 260 and 270 nm which are attributed to the π–π* transition and these are similar to the precursor of [HCH3impy]PF6 molecule. Because of the dissociation, the polymeric NHC-complexes show similar absorption respective to their monomers in solution which is evident from the electronic absorption spectra of NHC3, NHC4, NHC5 & NHC6 and its similarity with that of the monomer, NHC2. It was found that that all the coordination polymers as well as NHC monomers are photo-luminescent upon UV excitation in both solution and solid-state phases. While carbine precursors exhibit an emission band at 435 nm, the monometallic Au-containing monomer species, NHC2, shows an emission band at 541 nm in solid state. It was observed that upon cooling the solid samples to 77 K, the emission band shifts toward blue side (506 nm). Similarly, the nitrile-containing coordination polymers show intense bands at 480, 474, 522, and 469 nm at room temperature for the acetonitrile, benzonitrile, benzylnitrile, and nitrite containing polymers NHC3, NHC4, NHC5 & NHC6, respectively. Upon cooling these solid samples to 77 K, the emission bands sharpen and blue-shifted (Figures 29 and 30).
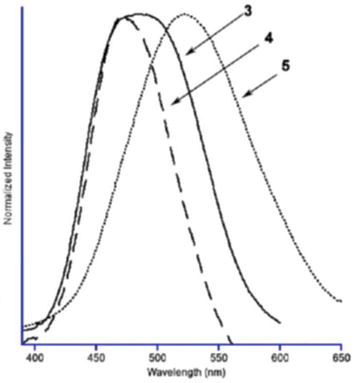
Figure 29.
Room temperature emission spectra of solid samples of the nitrile-containing polymers NHC3 (3), NHC4 (4), and NHC5 (5). The nitrate-containing polymer has a spectrum nearly identical to that of NHC4.
Figure 30.
NHC-based metal complexes.
The same research group has also reported four NHC coordination polymers by modifying pyridyl-substituted N-methylimidazole backbones, {[AuAg(CH3impy)2(L)](BF4)2}n and {[AuAg(CH3impy)2(NO3)]NO3}n where L is CH3CN, C6H5CN, or C6H5CH2CN [91]. The electronic absorption spectra and fluorescence emission properties are found to be similar to the other polymers NHC3, NHC4, NHC5 & NHC6. Further, the data also suggest that the optical properties of the polymers can be tailored by making appropriate changes to the ligands and composition of the polymers.
In another study, the photophysical properties of picolyl-substituted NHC-bridged triangular complexes of Ag(I) and Cu(I) were reported in different solvents [92]. In acetonitrile, the electronic absorption spectra of Cu-containing complexes NHC10a, NHC10b, and NHC10c exhibit two distinct absorption bands: a single absorption band between 253 and 265 nm and additional peaks at 374, 366, and 373 nm, respectively. Based on the ligand precursor absorption study, these absorption bands are attributed to the pyridyl π-π* transition and MLCT, respectively. Upon UV-excitation, the Cu-complexes NHC10a, NHC10b, and NHC10c emit at 459, 427, and 429 nm in acetonitrile, whereas in the solid state, these bands shift to 433, 429, and 432 nm, respectively. The emission maxima of the complexes are roughly following their substitution pattern on the pyridine ring. The excitation spectra suggest that NHC10a, NHC10b, and NHC10c show that these emission bands are effectively populated by two states. The emission maxima hardly change with a change in the solvent. On the other hand, the silver-containing NHC-bridged triangular complexes are found to be less emissive partly because of the dissociation of the complexes in acetonitrile (Figure 31).
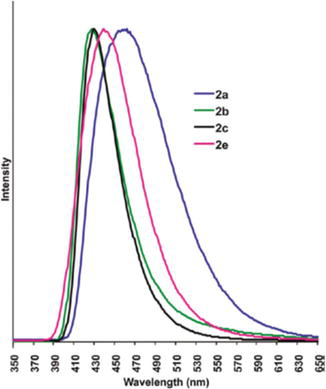
Figure 31.
Normalized room-temperature emission spectra of NHC10a, NHC10b, and NHC10c in acetonitrile.
Photophysical properties of pyrazole-linked bis(imidazolium) based multidentate mixed N,C-ligands, and corresponding tetranuclear and trinuclear silver–NHC complexes were reported [93]. UV-visible absorption spectra of precursor H2L2PF6, silver–NHC complex NHC8, and nickel–NHC complex NHC9 in acetonitrile show maximum absorption of 248, 245, and 251 nm, respectively. The small spectral difference is attributed to the absorption of the quinoxaline moiety in these three compounds and the dominance of the HOMO electrons localized on quinoxaline π-orbitals instead of metal-ligand bonding in the silver(I) and nickel(II) complexes. The solid-state luminescent properties reveal that the emission spectrum of precursor ligand shows a single emission band at 416 nm and nickel–NHC complex NHC9 exhibits a single broad emission at 471 nm. However, the silver–NHC complex NHC8 was found to be non-fluorescent due to the decomposition of the complex upon excitation. It was also found that by varying the N-alkyl groups of the silver cluster, photoluminescent properties can be tuned, considering the fact that luminescence originates from intraligand or metal-based excitation states. Luminescence properties of imidazolium salts [H3L1](PF6)2 and [HL4](PF6), and multinuclear silver complexes that consist of pyrazole-functionalized NHC ligands were reported [94] and it was observed that the ligand [H3L1](PF6)2 shows a single broad band at 401 nm, whereas its tetranuclear complex NHC12 shows two broad emission bands centered at 362 and 463 nm. Similarly, the ligand [HL4](PF6) exhibits emission bands at 371 and 461 nm, while its silver complex NHC11 emits a broad band at 442 nm. The intense emission behavior of the Ag(I)–NHC complexes is attributed to the intraligand excited states or metal-centered excited states (Figures 32 and 33).
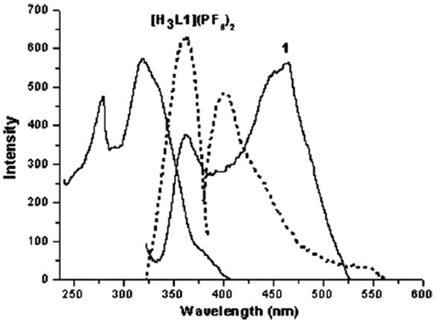
Figure 32.
The solid-state emission (right) and excitation (left) spectra of [H3L1](PF6)2 (dashed) and NHC8 (solid).
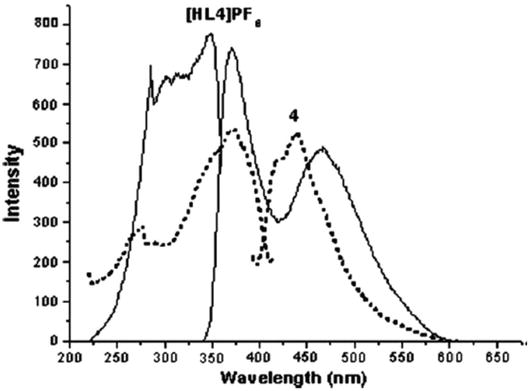
Figure 33.
The solid-state emission (right) and excitation (left) spectra of [HL4](PF6) (dashed) and NHC9 (solid).
Photophysical properties of cyclometalated alkynyl platinum(II) complexes (NHC13-18) baring tridentate pyridine-based NHC ligands were studied in detail [95]. The absorption spectrum of [PtII(CNC)Cl][PF6] consists of a strong absorption band at 272 nm and a moderately intense absorption band at 364 nm. Molar absorption coefficients (ε) are estimated to be of the order of 103–104 dm3mol−1 cm−1 in acetonitrile. The shorter wavelength absorption band cantered 272 nm is assigned π-π* transitions of the C-N-C pincer ligands, whereas the longer wavelength absorption band (364 nm) is assigned to the metal-to-ligand charge-transfer (MLCT) [dπ(Pt) → π*(C^N^C)] transitions which are partly mixed with the IL π-π* transitions of the C^N^C pincer ligands. However, upon substitution of alkynyl ligands in place of chloro ligand, the low-energy absorption bands are red-shifted and situated between 383 and 471 nm with ε values of the order of 103 dm3mol−1 cm−1 whereas the shorter wavelength band remained unchanged. The high-energy absorption band (277–291 nm) is assigned to the IL π-π* transitions of the C☰CR and C^N^C pincer ligands and whereas the lower-energy band (383–471 nm) is attributed to as an admixture of the MLCT [dπ(Pt) → π*(C^N^C)] transition and alkynyl-to-C^N^C ligand-to-ligand charge-transfer (LLCT) [π(C☰CR)π* → (C^N^C)] transition. As compared to the alkylalkynyl complex NHC13, the low-energy absorption band of the arylakynyl complexes NHC14-18 is red-shifted which is attributed to the π-donating nature of the arylakynyl ligands. The longer wavelength absorption maxima of the NHC14-18 complexes are as follows: NHC14 (376) > NHC15 (405) > NHC16 (422) > NHC17 (439) > NHC18 (471 nm), which is consistent with the π-electron-donating ability of the arylalkynyl ligand. The red shift of the longer wavelength absorption band is assigned to the negative solvatochromism in which the dipole moments of the complexes decrease upon electronic excitation (Figure 34).
Figure 34.
NHC based metal complexes.
Fluorescence study reveal that the chloroplatium(II) precursor complex, [PtII(C^N^C)Cl][PF6], is non-fluorescent in acetonitrile solvent due to the presence of low-energy d–d orbitals that promote the quenching of the 3MLCT/3 IL state through nonradiative processes. On the other hand, the alkynylplatinum(II) complexes NHC13-17 exhibit strong structureless fluorescence emission bands in acetonitrile whose emission maxima are found between 497 and 631 nm. (Figure 35). The fluorescence emission maxima of complexes NHC13-17 vary based on the substituent on the phenyl ring of the alkynyl ligands. For example, the complex NHC17 which contains the most electron-rich methoxy group show emission maxima at 631 nm whereas the complex NHC14, which consists of the most electron-poor trifluoromethyl substituent exhibits emission maxima at 500 nm. This shift is consistent with the assignment of 3MLCT/3LLCT in which the electron-donating substituents on the phenyl ring raises the energy level of the dπ(Pt) and π(C☰CR) orbitals which in turn the lowers CT energy. In other words, the fluorescence emission maxima of the complexes NHC13-17 can be modulated from green to yellow with the substitution of alkynyl/arylalkynyl ligands. More interestingly, unlike many of the NHC-metal complexes, the complexes NHC13-15 are found to be highly fluorescent, which is evident from the estimated fluorescent quantum yields. The observed large Stokes shifts and long fluorescence emission lifetimes (microseconds) further confirm the emitting state has a triplet character and is assigned to the 3MLCT [dπ(Pt)→π*(C^N^C)] state with the mixing of a 3LLCT [π(C☰CR)π*→(C^N^C)] state. In solution, the complex NHC18 is found to be non-fluorescent at room temperature, which is attributed to photoinduced electron transfer (PET) quenching of the emissive 3MLCT state by amine functional group. The non-fluorescent nature can also be interpreted in terms of the low-lying non-emissive 3LLCT excited state that originated from the higher-lying π (C☰CR) orbital of the aminophenyl alkynyl unit. Further, the complexes NHC13-18 are fluorescent in the solid state as well as in glass matrices at 77 K, in which vibronically structured emission bands were observed between 461 and 512 nm.
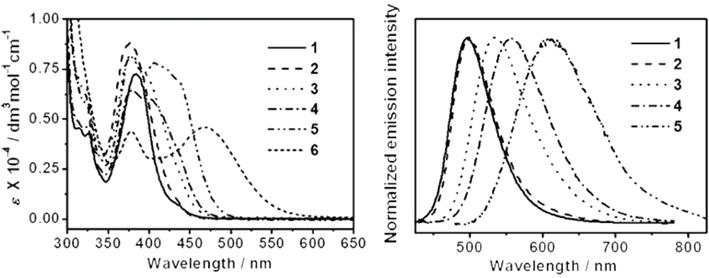
Figure 35.
Electronic absorption spectra of NHC13–18 in acetonitrile (left) and normalized emission spectra of NHC13–17 in degassed CH3CN at 298 K.
Lee and co-workers have reported photophysical properties of a square-planar Pt(II) complex (NHC19) baring a pyridine-based pincer-type NHC ligand, [PtII(C^N^C)(Cl)]Cl where C^N^C is 2,6-bis(1-butylimidazol-2-ylidenyl)pyridine [96]. The hydrated form of the complex shows exhibits strong fluorescence emission at 614 nm (orange) with a lifetime of 34 μs (quantum yield, φ = 0.30), whereas the anhydrate form of the complex shows intense fluorescence emission at 550 nm with a lifetime of 22 μs (quantum yield, φ = 0.35). In other words, the photoluminescence of the complex in the solid state switches from orange to green upon hydration and dehydration, respectively. Based on TDDFT calculations, the lowest electronic transition is attributed to the metal-metal to ligand charge transfer (MMLCT) and the observed fluorescence emission band to the 3MMLCT (Figure 36).
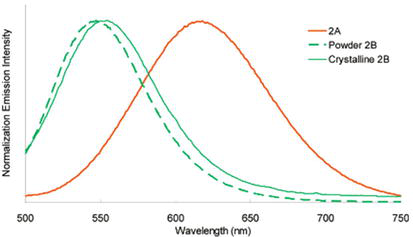
Figure 36.
Fluorescence emission spectra of hydrated and dehydrated NHC19 at 300 K in the solid state. 2A.
In another study, the photophysical properties of a new class of cyclometalated pyridine NHC (NHC) Pt(II) complexes with electronically different alkyne derivatives (NHC20-23) have been investigated [97]. The electronic absorption spectra of NHC20-23 in dichloromethane solvent display a strong band below 300 nm and a moderately intense absorption band between 320 and 420 nm. The spectroscopic data combined with the DFT and TDDFT calculations demonstrate that shorter wavelength absorption bands mainly originate from the 1LLCT[πalk→πN^C*] and 1MLCT[d→πN^C*] transitions. The UV/vis absorption spectra of the four complexes were also studied in different solvents and a negative solvatochromic behavior was observed, which demonstrates the different dipole moments for both ground and excited states. It is interesting to note that this observation is similar to the bipyridine Pt(II) dialkyne complexes behavior which is reported in the literature [98, 99, 100].
The complexes NHC20–23 are non-fluorescent in solution at room temperature which is quite different than the previously reported bipyridine Pt(II) and bis-NHC Pt(II) complexes [98, 99, 100, 101]. The solvent molecules promote the thermal quenching of the fluorescence emission in solution at room temperature [102]. However, the 1 wt % of complexes NHC20-23 in poly(methyl methacrylate) (PMMA) matrix gives a broad and structureless single fluorescence emission band whose maxima is found between 459 and 480 nm. Surprisingly fluorescence quantum yields are found to be very high, 51–59%. The fluorescence emitting state has been attributed to a mixed excited state of 1LLCT, 3LLCT, 1MLCT, and 3MLCT based on related complexes fluorescence behavior [103, 104, 105]. It was observed that with an increase in the weight percentage of the complex from 10 to 80 wt % in the PMMA films, a new fluorescence emission band emerged in the longer wavelength region (575–595 nm) at the expense of shorter wavelength band w-energy emission was observed. However, the overall fluorescence quantum yield decreased to 20 wt % due to an increase in the concentration and its self-quenching [106]. Further, based on the excitation spectral analysis, the longer fluorescence emission band has been attributed to the excimer emission rather than a ground-state dimer which generally arises due to the strong metal-metal interactions. The ground-state dimer formation was ruled out from the UV-visible spectral measurements in which the MMLCT band is absent with an increase in the concentration of the molecules. The PMMA films show different fluorescence emission bands at 460 and 570 nm due to the presence of monomeric and excimeric species and cover the entire visible region which essentially produces the white light. The CIE coordinates of complex NHC23 at a concentration of 20 wt % in PMMA were found to be x = 0.31 and y = 0.33 and these are very close to the white light CIE coordinates (x = 0.33, y = 0.33) (Figures 37 and 38).
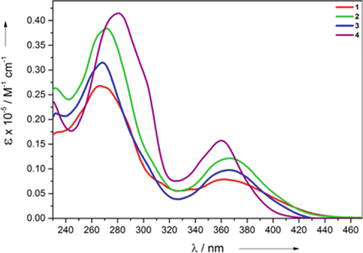
Figure 37.
UV/Vis spectra of NHC20 (c = 1.62 × 10–5 M), NHC21 (c = 4.58 × 10–5 M), NHC3 (c = 1.87 × 10–5 M), and NHC4 (c = 3.39 × 10–5 M) in CH2Cl2.
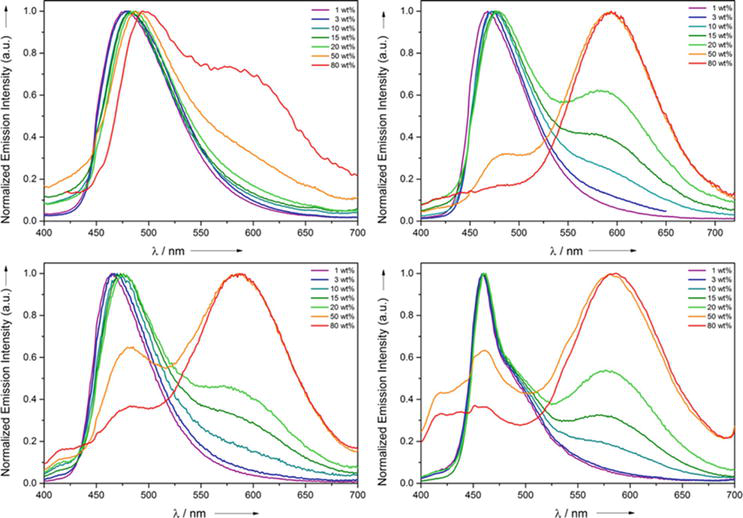
Figure 38.
Concentration-dependent emission spectra of complexes NHC20 (top left), NHC21 (top right), NHC22 (bottom left), NHC23 (bottom right) in PMMA films.
Bis-NHC silver complex baring acridine chromophoric derivative was synthesized and photophysical properties were studied [107]. The complex NHC24 is shown to be fluorescent and emits blue light in solution state with CIE coordinate x = 0.154 and y = 0.061 (Figure 39).
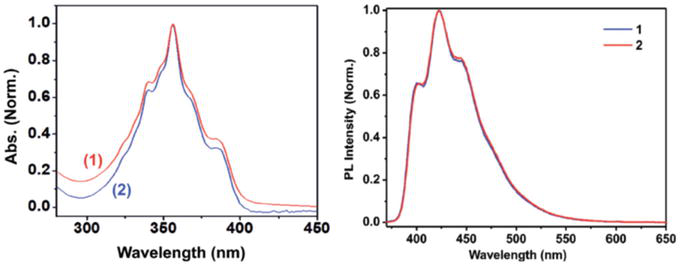
Figure 39.
Electronic absorption and photoluminescence of NHC24 complex (red line) and its precursor ligand (blue line).
Photophysical and electrochemical properties of five iridium bis(carbene) complexes NHC25, NHC26, NHC27, NHC28, and NHC29 were reported by Cheng and co-workers [108]. Electroluminescent (EL) devices of NHC26, NHC27, and NHC29 were fabricated as dopant emitters. The EL devices emit deep-blue light with external quantum efficiencies of 15.2, 14.1, and 7.6%, and the CIE coordinates were estimated to be of (0.14, 0.27), (0.14, 0.18) and (0.14, 0.10), respectively.
Phosphorescent three-coordinated NHC copper complexes that consist of anionic nonconjugated di(2-pyridyl)-dimethyl borate ligand have been reported by Thompson and co-workers [109]. The complexes NHC30, NHC31, and NHC32 emit sky-blue, yellow, and orange light in solid-state with quantum yields of 0.8 to 0.7, and to 0.16, respectively. Further, the emission lifetimes are observed to be 11, 15, and 7.5 μs, respectively which shows a modest increase in the emission lifetimes: 36, 17, and 21 μs, respectively at 77 K. Therefore, the emission of the NHC30, NHC31, and NHC32 complexes at room temperature is attributed to the phosphorescent in nature.
Inspired by the promising photochemical properties of ruthenium polypyridine complexes, Chung and co-workers have synthesized Ruthenium complexes NHC33 and NHC34 of NHC [110]. These NHC complexes are designed to be structural analogs of [Ru(bpy)3]2+ and [Ru(terpy)2]2+, respectively. The electronic absorption spectrum of the Complex 14 is blue-shifted (368 nm) as compared to the [Ru(bpy)3]2+ complex. Similarly, complex 15(PF6−) shows two absorption bands with a maximum of 343 and 382 nm, and as compared to the [Ru(terpy)2]2+, these two band are highly blue-shifted. The blue-shifts of the MLCT absorption bands are assigned to the relative electron-richness of the NHC ligands over the bipyridine and terpyridine ligands. Complex NHC33 is nonemissive in acetonitrile at room temperature. In sharp contrast to [Ru(terpy)2]2+, the complex 15 (PF6−) exhibits a strong fluorescence emission with a maximum at 532 nm. Further, the terpy-like complex NHC34 displays a long fluorescence lifetime of 820 ns in MeCN and 3100 ns in H2O (Figure 40).
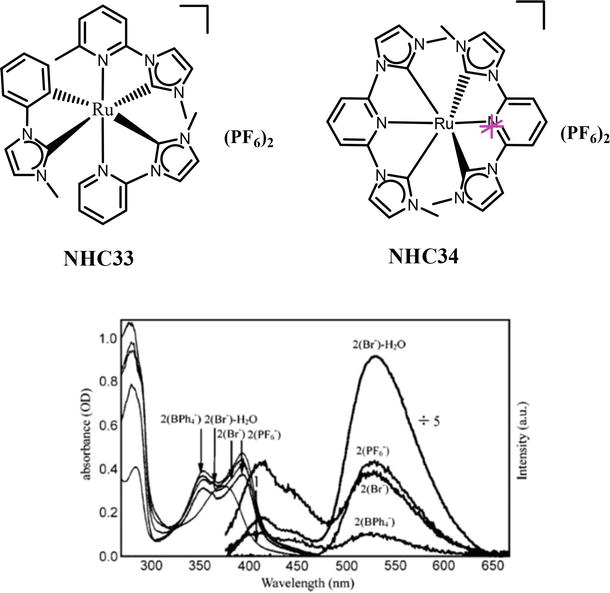
Figure 40.
Absorption and emission spectra, with excitation at 354 nm, of Ru2+ complexes in acetonitrile (if not specified) and water under argon.
5. Conclusion
The strong σ-donating nature of the NHC is the key factor in manipulating the optical properties of NHC complexes. The NHC ligands shift the d−d transitions to a shorter wavelength region and the complexes become luminescent. The studies on NHC-based metal complexes of copper, silver, platinum, iridium, and coinage metals reveal that several metal complexes are luminescent and some of them are even found in applications in organic light-emitting diodes (OLEDs). However, serval studies also reveal that the quantum yields of the metal complexes are not sufficient to be used in applications. This perhaps can be overcome by modifying the NHC ligand with electron-rich substituents which is expected to reduce the HOMO-LUMO gap and push the d-d transitions toward the UV-region. Apart from the NHC, the cyclic (alkyl)(amino)carbene (CAACs) were found to the promising in tuning the photophysical properties. The CAACs reduce the HOMO−LUMO gap as compared to NHCs and the CAAC is found to be more electrophilic in nature. It has been found that luminescent CAAC metal complexes exhibit sub-microsecond lifetimes which make them suitable for OLED applications. The challenge remains open in this research to master the structure, bonding, and property in order to tailor very stable and highly luminescent materials based on abundant and cheap metals such as iron, copper, zinc, and silver complexes.
Acknowledgments
SS thanks the Department of Chemistry, BHU, for infrastructure and IoE – BHU (faculty incentive grant) for financial assistance.
References
- 1.
Davies HML, Manning JR. Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion. Nature. 2008; 451 :417-424 - 2.
Hopkinson MN, Richter C, Schedler M, Glorius F. An overview of N-heterocyclic carbenes. Nature. 2014; 510 :485-496 - 3.
de Frémont P, Marion N, Nolan SP. Carbenes: Synthesis, properties, and organometallic chemistry. Coordination Chemistry Reviews. 2009; 253 (7–8):862-892 - 4.
Chinoporos E. Carbenes. Reactive Intermediates Containing Divalent Carbon. Chemical Reviews. 1963; 63 :235 - 5.
Lappert MF, R.S. Rowe the role of group 14 element carbene analogues in transition metal chemistry. Coordination Chemistry Reviews. 1990; 100 :267 - 6.
Arduengo AJ, Harlow RL, Kline M. A stable crystalline carbene. Journal of the American Chemical Society. 1991; 113 :361-363 - 7.
Sau SC, Pradip KH, Swadhin KM, Michele S, Guy B. Stable abnormal N-heterocyclic carbenes and their applications. Chemical Society Reviews. 2020; 49 (4):1233-1252 - 8.
Lin IJB, Vasam CS. Preparation and application of N-heterocyclic carbene complexes of Ag(I). Coordination Chemistry Reviews. 2007; 251 (5–6):642-670 - 9.
Wanzlick H-W, Schikora R. Aspects of Nucleophilic Carbene Chemistry. Angewandte Chemie. 1960; 72 :494 - 10.
Igau A et al. ‘Analogous.alpha.,.alpha.’-bis-carbenoid, triply bonded species: Synthesis of a stable. lambda. 3-phosphino carbene-.lambda.5-phosphaacetylene’. Journal of the American Chemical Society. 1988; 110 (19):6463-6466 - 11.
Schubert U. Structural consequences of bonding in transition metal carbene complexes. Coordination Chemistry Reviews. 1984; 55 :261 - 12.
Bertrand G, Reed R. λ3-Phosphinocarbenes λ5-phosphaacetylene. Coordination Chemistry Reviews. 1994; 137 :323 - 13.
Runbo P, Liancheng H, Yue Z, Xinping W. The dynamic Lewis acid–carbene hybrid: Pushing the electrophilicity of carbenes to the limit. Journal of the American Chemical Society. 2023; 145 (40):21733-21737 - 14.
Benhamou L, Chardon E, Lavigne G, Bellemin-Laponnaz S, César V. Synthetic routes to N-heterocyclic carbene precursors. Chemical Reviews. 2011; 111 :2705-2733 - 15.
Emmanuel BP-R, Karen G-S, Daniel M-E. Mercaptopyridine-functionalized MIC-Pd(II)-PEPPSI complexes: Synthesis and hemilability effect in catalytic applications. Organometallics. 2023; 42 (19):2893-2901 - 16.
Jazzar R, Soleilhavoup M, Bertrand G. Cyclic (alkyl)- and (aryl)-(amino)carbene coinage metal complexes and their applications. Chemical Reviews. 2020; 120 :4141-4168 - 17.
Melaimi M, Jazzar R, Soleilhavoup M, Bertrand G. Cyclic (alkyl)(amino)carbenes (CAACs): Recent developments. Angewandte Chemie, International Edition. 2017; 56 :10046-10068 - 18.
Soleilhavoup M, Bertrand G. Cyclic (alkyl)(amino) carbenes (CAACs): Stable carbenes on the rise. Accounts of Chemical Research. 2015; 48 :256-266 - 19.
Lavallo V, Canac Y, Präsang C, Donnadieu B, Bertrand G. Stable cyclic (alkyl)(amino)carbenes as rigid or flexible, bulky, electron-rich ligands for transition-metal catalysts: A quaternary carbon atom makes the difference. Angewandte Chemie, International Edition. 2005; 44 :5705-5709 - 20.
Frey GD, Lavallo V, Donnadieu B, Schoeller WW, Bertrand G. Facile splitting of hydrogen and ammonia by nucleophilic activation at a single carbon center. Science. 2007; 316 :439-441 - 21.
Zhao L, Zeng X. Transition-metal-catalyzed reactions promoted by cyclic (alkyl or aryl)(amino)carbene ligands. Chemistry. 2022; 8 :2082-2113 - 22.
Wang Y, Tran PM, Lahm ME, Xie Y, Wei P, Adams ER, et al. Activation of ammonia by a carbene-stabilized dithiolene zwitterion. Journal of the American Chemical Society. 2022; 144 :16325-16331 - 23.
Raup DEA, Cardinal-David B, Holte D, Scheidt KA. Cooperative catalysis by carbenes and Lewis acids in a highly stereoselective route to γ-lactams. Nature Chemistry. 2010; 2 :766-771 - 24.
Crudden CM et al. Ultra stable self-assembled monolayers of N-heterocyclic carbenes on gold. Nature Chemistry. 2014; 6 :409-414 - 25.
Mahoney JK et al. Air-persistent monomeric (amino) (carboxy) radicals derived from cyclic (alkyl)(amino) carbenes. Journal of the American Chemical Society. 2015; 137 :7519-7525 - 26.
Pony Yu R, Hesk D, Rivera N, Pelczer I, Chirik PJ. Iron-catalysed tritiation of pharmaceuticals. Nature. 2016; 529 :195-199 - 27.
Ernst JB et al. Molecular adsorbates switch on heterogeneous catalysis: Induction of reactivity by N-heterocyclic carbenes. Journal of the American Chemical Society. 2017; 139 :9144-9147 - 28.
Wang G et al. Ballbot-type motion of N-heterocyclic carbenes on gold surfaces. Nature Chemistry. 2017; 9 :152-156 - 29.
Légaré MA et al. Nitrogen fixation and reduction at boron. Science. 2018; 359 :896-900 - 30.
Ishii T, Kakeno Y, Nagao K, Ohmiya H. N-heterocyclic carbene-catalyzed decarboxylative alkylation of aldehydes. Journal of the American Chemical Society. 2019; 141 :3854-3858 - 31.
Han Y-F, Huang Y, Liu H, Gao Z-H, Zhang C-L, Ye S. Photoredox cooperative N-heterocyclic carbene/palladium-catalysed alkylacylation of alkenes. Nature Communications. 2022; 13 :5754 - 32.
Narouz MR et al. N-Heterocyclic carbenefunctionalized magic-number gold nanoclusters. Nature Chemistry. 2019; 11 :419-425 - 33.
Hamze R et al. Eliminating nonradiative decay in Cu(I) emitters: >99% quantum efficiency and microsecond lifetime. Science. 2019; 363 :601-606 - 34.
Davies AV, Fitzpatrick KP, Betori RC, Scheidt KA. Combined photoredox and carbene catalysis for the synthesis of ketones from carboxylic acids. Angewandte Chemie, International Edition. 2020; 59 :9143-9148 - 35.
Franz M et al. Controlled growth of ordered monolayers of N-heterocyclic carbenes on silicon. Nature Chemistry. 2021; 13 (9):828-835 - 36.
Huynh HV. Electronic properties of N-heterocyclic carbenes and their experimental determination. Chemical Reviews. 2018; 118 :9457-9492 - 37.
Runyon JW et al. Carbene-based Lewis pairs for hydrogen activation. Australian Journal of Chemistry. 2011; 64 :1165-1172 - 38.
Dröge T, Glorius F. The measure of all rings — N-heterocyclic carbenes. Angewandte Chemie, International Edition. 2010; 49 :6940-6952 - 39.
Heinemann C, Müller T, Apeloig Y, Schwarz H. On the question of stability, conjugation, and “aromaticity” in imidazol-2-ylidenes and their silicon analogs. Journal of the American Chemical Society. 1996; 118 :2023-2038 - 40.
Vivancos Á, Segarra C, Albrecht M. Mesoionic and related less heteroatom-stabilized N-heterocyclic carbene complexes: Synthesis, catalysis, and other applications. Chemical Reviews. 2018; 118 :9493-9586 - 41.
Mąkosza M, Fedoryński M. Dichlorocarbene and analogs: Discovery, properties and reactions. Russian Chemical Bulletin. 2021; 70 (11):2045-2050 - 42.
Hine J, Dowell AM. Carbon dihalides as intermediates in the basic hydrolysis of haloforms. III. Combination of carbon dichloride with halide ions. Journal of the American Chemical Society. 1954; 76 :2688-2692 - 43.
Hine J, Langford PB. Methylene derivatives as intermediates in polar reactions. IX. The concerted mechanism for α-eliminations of haloforms. Journal of the American Chemical Society. 1957; 79 :5497-5500 - 44.
Hine J. Divalent Carbon. New York: Ronald Press Co.; 1964 - 45.
Green SP et al. Thermal stability and explosive hazard assessment of diazo compounds and diazo transfer reagents. Organic Process Research & Development. 2019; 24 (1):67-84 - 46.
Liu W et al. Copper-catalyzed oxidation of hydrazones to diazo compounds using oxygen as the terminal oxidant. ACS Catalysis. 2021; 11 (5):2676-2683 - 47.
Wei F et al. Gold carbene chemistry from diazo compounds. Science Bulletin. 2015; 60 (17):1479-1492 - 48.
Hutchins RO, Maryanoff B, Milewski C. Selective reduction of aliphatic ketones and aldehydes to hydrocarbons with sodium cyanoborohydride and P-toluenesulfonyl hydrazide in dimethylformamide-sulfolane. Journal of the American Chemical Society. 1971; 93 (7):1793-1794 - 49.
Visser P et al. Reactivity of carbenes and ketenes in low-temperature matrices. Carbene co trapping, Wolff rearrangement, and ketene−pyridine ylide (zwitterion) observation. Journal of the American Chemical Society. 1996; 118 (50):12598-12602 - 50.
Simmons HE, Smith RD. A new synthesis of cyclopropanes from olefins. Journal of the American Chemical Society. 1958; 80 (19):5323-5324 - 51.
LeGoff E. Cyclopropanes from an easily prepared, highly active zinc-copper couple, dibromomethane, and olefins. The Journal of Organic Chemistry. 1964; 29 (7):2048-2050 - 52.
Simmons HE, Smith RD. A new synthesis of cyclopropanes. Journal of the American Chemical Society. 1959; 81 (16):4256-4264 - 53.
Petrellis PC, Griffin GW. Photofragmentation of oxirans. Precursors for phenylcyanocarbene and phenylmethoxycarbonylcarbene. Chemical Communications. 1967;(14):691-692 - 54.
Saba S, Brescia A-M, Kaloustian MK. One-pot synthesis of cyclic amidinium tetrafluoroborates and hexafluorophosphates; the simplest models of N5,N100 methenyltetrahydrofolate coenzyme. Tetrahedron Letters. 1991; 32 :5031 - 55.
Delaude L, Szypa M, Demonceau A, Noels AF. Synthesis and application of new N-heterocyclic carbene ruthenium. Advanced Synthesis and Catalysis. 2002; 344 :749 - 56.
Leuthausser S, Schwarz D, Plenio H. Tuning the electronic properties of N-heterocyclic carbenes. Chemistry - A European Journal. 2007; 13 :7195 - 57.
Kantchev EAB, Ying JY. Practical One-Pot, Three-Component Synthesis of N-Heterocyclic Carbene (NHC) Ligated Palladacycles Derived from N,N -Dimethylbenzylamine. Organometallics. 2009;28 :289 - 58.
Leuthausser S, Schmidts V, Thiele CM, Plenio H. π-Face Donor Properties of N-Heterocyclic Carbenes in Grubbs II Complexes. Chemistry: A European Journal. 2008; 14 :5465 - 59.
Fleckenstein C, Roy S, Leuthausser S, Plenio H. Sulfonated N-heterocyclic carbenes for Suzuki coupling in water. Chemical Communications. 2007;(27):2870 - 60.
Arduengo AJ III, Krafczyk R, Schmutzler R, Craig HA, Goerlich JR, Marshall WJ, et al. Imidazolylidenes, imidazolinylidenes and imidazolidines. Tetrahedron. 1999; 55 :14523 - 61.
Van Veldhuizen JJ, Garber SB, Kingsbury JS, Hoveyda AH. A Recyclable Chiral Ru Catalyst for Enantioselective Olefin Metathesis. Efficient Catalytic Asymmetric Ring-Opening/Cross Metathesis in Air. Journal of the American Chemical Society. 2002; 124 :4954 - 62.
Alcarazo M, Roseblade SJ, Alonso E, Fernandez R, Alvarez E, Lahoz FJ, et al. 1,3-Bis( N,N -dialkylamino)imidazolin-2-ylidenes: Synthesis and Reactivity of a New Family of Stable N-Heterocyclic Carbenes. Journal of the American Chemical Society. 2004;126 :13242 - 63.
Enders D, Meiers M. Diastereo- and Enantioselective Synthesis of Differently N,O -Protected 1,3-Amino Alcohols with Three Neighbouring Stereogenic Centers. Synthesis. 2002:2542 - 64.
Dinger MB, Nieczypor P, Mol JC. Adamantyl-Substituted N -Heterocyclic Carbene Ligands in Second-Generation Grubbs-Type Metathesis Catalysts. Organometallics. 2003;22 :5291 - 65.
Dutton JL, Frenking G. New avenues in S-block chemistry: Beryllium(0) complexes. Angewandte Chemie International Edition. 2016; 55 (43):13380-13382 - 66.
Arrowsmith M et al. Neutral zero-valent S-block complexes with strong multiple bonding. Nature Chemistry. 2016; 8 (9):890-894 - 67.
Obi AD et al. Carbene–calcium silylamides and amidoboranes. Organometallics. 2022; 41 (21):3064-3072. DOI: 10.1021/acs.organomet.2c00464 - 68.
Walley JE et al. N-heterocyclic carbene-supported aryl- and alk- oxides of beryllium and magnesium. Catalysts. 2019; 9 (11):934. DOI: 10.3390/catal9110934 - 69.
Krüger J et al. Single-electron oxidation of carbene-coordinated pnictinidenes–entry into heteroleptic radical cations and metalloid clusters. Inorganic Chemistry. 2022; 61 (15):5878-5884. DOI: 10.1021/acs.inorgchem.2c00249 - 70.
Ghosh, M.l Khan, S. N-heterocyclic silylenes in coinage metal chemistry: An account of recent advances. Dalton Transactions. 2021; 50 (31):10674-10688 - 71.
Pei R, He L, Zhao Y, Wang X. The dynamic Lewis acid–carbene hybrid: Pushing the electrophilicity of carbenes to the limit. Journal of the American Chemical Society. 2023; 145 :21733-21737 - 72.
Díez-González S, N-Heterocyclic Carbenes. From Laboratory Curiosities to Efficient Synthetic Tools. 2nd ed. The Royal Society of Chemistry; 2017 - 73.
Weskamp T et al. A novel class of ruthenium catalysts for olefin metathesis. Angewandte Chemie, International Edition. 1998; 37 (2490–2493):20 - 74.
Huang J et al. Olefin metathesis-active ruthenium complexes bearing a nucleophilic carbene ligand. Journal of the American Chemical Society. 1999; 121 (2674–2678):21 - 75.
Scholl M et al. Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene ligands. Organic Letters. 1999; 1 :953-956 - 76.
Viciano M et al. C−H oxidative addition of bisimidazolium salts to iridium and rhodium complexes, and N-heterocyclic carbene generation. A combined experimental and theoretical study. Organometallics. 2006; 25 :1120-1134 - 77.
Pan Z, Gao D, Zhang C, Guo L, Li J, Cui C. Synthesis and reactivity of N-heterocyclic carbene stabilized lanthanide(II) bis(amido) complexes. Organometallics. 2021; 40 (11):1728-1734 - 78.
Clement ND et al. Oxidative addition of imidazolium salts to Ni (0)and Pd (0): Synthesis and structural characterization of unusually stable metal–hydride complexes. Angewandte Chemie, International Edition. 2004; 43 :1277-1279 - 79.
Lappert MF, Pye PL. Electron-rich olefin-derived neutral mono- and bis-(carbene) complexes of low-oxidationstate manganese, iron, cobalt, nickel, and ruthenium. Journal of the Chemical Society Dalton Transactions. 1977;(21):2172-2180 - 80.
Wang HMJ, Lin IJB. Facile synthesis of silver (I)−carbene complexes. Useful carbene transfer agents. Organometallics. 1998; 17 (972–975):23 - 81.
Visbal R, Gimeno MC. N-heterocyclic carbene metal complexes: Photoluminescence and applications. Chemical Society Reviews. 2014; 43 :3551-3574 - 82.
Strassner T. Phosphorescent Platinum(II) Complexes with C∧C* Cyclometalated NHC Ligands. Accounts of Chemical Research. 2016; 49 :2680-2689 - 83.
Amouri H. Luminescent Complexes of Platinum, Iridium, and Coinage Metals Containing N -Heterocyclic Carbene Ligands: Design, Structural Diversity, and Photophysical Properties. Chemical Reviews. 2023;123 :230-270 - 84.
Crabtree RH. Recent Developments in the Organometallic Chemistry of N-Heterocyclic Carbenes. Coordination Chemistry Reviews. 2007; 251 :595 - 85.
Herrmann WA, Kocher C. N-Heterocyclic Carbenes. Angewandte Chemie, International Edition in English. 1997; 36 :2162-2187 - 86.
Dušková T et al. N-heterocyclic carbenes with three and six fluorous ponytails and their highly fluorophilic Rh and Ir complexes. Journal of Organometallic Chemistry. 2023; 986 :122605 - 87.
Bourissou D, Guerret O, Gabbai FP, Bertrand G. Stable Carbenes. Chemical Reviews. 2000; 100 :39-91 - 88.
Diez-Gonzalez S, Nolan SP. Stereoelectronic parameters associated with N-heterocyclic carbene (NHC) ligands: A quest for understanding. Coordination Chemistry Reviews. 2007; 251 :874-883 - 89.
Lin JCY, Huang RTW, Lee CS, Bhattacharyya A, Hwang WS, Lin IJB. Coinage metal-N-heterocyclic carbene complexes. Chemical Reviews. 2009; 109 :3561-3598 - 90.
Catalano VJ, Etogo AOJ. Luminescent coordination polymers with extended Au(I)-Ag(I) interactions supported by a pyridyl-substituted NHC ligand. Organometallic Chemistry. 2005; 690 :6041-6050 - 91.
Catalano VJ, Etogo AO. Preparation of Au(I), Ag(I), and Pd(II) N-Heterocyclic Carbene Complexes Utilizing a Methylpyridyl-Substituted NHC Ligand. Formation of a Luminescent Coordination Polymer. Inorganic Chemistry. 2007; 46 :5608-5615 - 92.
Catalano VJ, Munro LB, Strasser CE, Samin AF. Modulation of metal-metal separations in a series of Ag(I) and intensely blue photoluminescent Cu(I) NHC-bridged triangular clusters. Inorganic Chemistry. 2011; 50 (17):8465-8476 - 93.
Xia C, Yang K, Sun W, Lu X, Xie JJ. Silver(I), nickel(II) N-heterocyclic carbene complexes based on bidentate bis-imidazolium salt with a quinoxaline linker: Syntheses, structures, and characterization. Coordination Chemistry. 2017; 70 (4):615-625 - 94.
Zhou Y, Zhang X, Chen W, Huayu Qiu H. Synthesis, structural characterization, and luminescence properties of multinuclear silver complexes of pyrazole-functionalized NHC ligands containing Ag-Ag and Ag-π interaction. Journal of Organometallic Chemistry. 2008; 693 :205-215 - 95.
Leung SY-L, Lam ES-H, Lam WH, Wong KM-C, Wing-Tak Wong W-T, Yam VW-W. Luminescent Cyclometalated Alkynylplatinum(II) Complexes with a Tridentate Pyridine-Based N-Heterocyclic Carbene Ligand: Synthesis, Characterization, Electrochemistry, Photophysics, and Computational Studies. Chemistry - A European Journal. 2013; 19 :10360-10369 - 96.
Lee C-S, Sabiah S, Wang J-C, Hwang W-S, Lin IJB. Water-Induced Changes of Photoluminescence of a Pincer-Type N-Heterocyclic Carbene Platinum(II) Complex. Organometallics. 2010; 29 :286-289 - 97.
Bachmann M, Suter D, Blacque O, Venkatesan K. Tunable and Efficient White Light Phosphorescent Emission Based on Single Component N-Heterocyclic Carbene Platinum(II) Complexes. Inorganic Chemistry. 2016; 55 (10):4733-4745 - 98.
Connick WB, Geiger D, Eisenberg R. Excited-State Self-Quenching Reactions of Square Planar Platinum(II) Diimine Complexes in Room-Temperature Fluid Solution. Inorganic Chemistry. 1999; 38 :3264-3265 - 99.
Hissler M, Connick WB, Geiger DK, McGarrah JE, Lipa D, Lachicotte RJ, et al. Platinum Diimine Bis(acetylide) Complexes: Synthesis, Characterization, and Luminescence Properties. Inorganic Chemistry. 2000; 39 :447-457 - 100.
Hissler M, McGarrah JE, Connick WB, Geiger DK, Cummings SD, Eisenberg R. Platinum diimine complexes: Towards a molecular photochemical device. Coordination Chemistry Reviews. 2000; 208 :115-137 - 101.
Zhang Y, Garg JA, Michelin C, Fox T, Blacque O, Venkatesan K. Synthesis and Luminescent Properties of cis Bis-N -Heterocyclic Carbene Platinum(II) Bis-Arylacetylide Complexes. Inorganic Chemistry. 2011;50 :1220-1228 - 102.
Wang X, Chang YL, Lu JS, Zhang T, Lu ZH, Wang SN. Bright Blue and White Electrophosphorescent Triarylboryl-Functionalized C^N-Chelate Pt(II) Compounds: Impact of Intramolecular Hydrogen Bonds and Ancillary Ligands. Advanced Functional Materials. 2014; 24 :1911-1927 - 103.
Hua F, Kinayyigit S, Cable JR, Castellano FN. Platinum(II) Diimine Diacetylides: Metallacyclization Enhances Photophysical Properties. Inorganic Chemistry. 2006; 45 :4304-4306 - 104.
Pomestchenko IE, Castellano FN. Solvent switching between charge transfer and intraligand excited states in a multichromophoric platinum (II) complex. The Journal of Physical Chemistry. A. 2004; 108 :3485-3492 - 105.
Damrauer NH, Cerullo G, Yeh A, Boussie TR, Shank CV, McCusker JK. Femtosecond Dynamics of Excited-State Evolution in [Ru(bpy)3]2+. Science. 1997; 275 :54-57 - 106.
Penzkofer A, Lu Y. Fluorescence quenching of rhodamine 6G in methanol at high concentration. Chemical Physics. 1986; 103 :399-405 - 107.
Prabusankar G, Muthukumaran N, Vaddamanu M, Raju G, Velappan K, Sathyanarayana A, et al. Blue-emitting acridine-tagged silver(I)-bis-N-heterocyclic carbine. RSC Advances. 2019; 9 :7543 - 108.
Hsieh C-H, Wu F-I, Fan C-H, Huang M-J, Lu K-Y, Chou P-Y, et al. Design and Synthesis of Iridium Bis(carbene) Complexes for Efficient Blue Electrophosphorescence. Chemistry - A European Journal. 2011; 17 :9180-9187 - 109.
Krylova VA, Djurovich PI, Conley BL, Haiges R, Whited MT, Williams TJ, et al. Control of emission colour with N-heterocyclic carbene (NHC) ligands in phosphorescent three-coordinate Cu(i) complexes. Chemical Communications. 2014; 50 :7176-7179 - 110.
Son SU, Park KH, Lee Y-S, Kim BY, Choi CH, Lah MS, et al. Synthesis of Ru(II) Complexes of N-Heterocyclic Carbenes and Their Promising Photoluminescence Properties in Water. Inorganic Chemistry. 2004; 43 :6896-6898