Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
Over 6500 Mendelian disorders have been documented, with approximately 4500 genes linked to these conditions. The majority of inherited diseases present in childhood and, currently, lack effective treatments, which imposes significant economic and psychological burdens on families and society. Gene editing, particularly base editing, offers an effective and safe strategy for repairing pathogenic point mutations. It has the potential to become a treatment, even a cure, for rare diseases. Currently, multiple gene editing-related drugs have entered clinical trials. In this chapter, we summarize the various gene editing systems, including CRISPR/Cas, base editing, and prime editing. We then focus on the current research progress of base editing in correcting pathogenic mutations. This includes applications such as building animal models, correcting mutations in various diseases, germline cell editing, delivery methods, and approved clinical trials. Finally, we discuss current challenges related to delivery methods, efficiency, precision, and cost.
Gene Editing Center, School of Life Science and Technology, ShanghaiTech University, Shanghai, China
Yu Zhang
Gene Editing Center, School of Life Science and Technology, ShanghaiTech University, Shanghai, China
Xingxu Huang*
Gene Editing Center, School of Life Science and Technology, ShanghaiTech University, Shanghai, China
The Key Laboratory of Pancreatic Diseases of Zhejiang Province, the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
*Address all correspondence to: huangxx@shanghaitech.edu.cn and ligl@shanghaitech.edu.cn
1. Introduction
On November 16 and December 8 in 2023, the Medicines and Healthcare products Regulatory Agency (MHRA) and the Food and Drug Administration (FDA), respectively, authorized the world’s first gene editing product, Casgevy, for the treatment of sickle cell disease and transfusion-dependent β-thalassemia [1]. It has been 11 years since the initial report on the CRISPR/Cas system’s ability to cut double-stranded DNA [2]. Casgevy is classified as the first generation of genome editing, utilizing CRISPR/Cas9 to create a double-stranded break (DSB) at the erythroid enhancer region of the BCL11A gene [3]. The authorization of Casgevy will set a precedence for the treatment of inherited diseases through gene editing methods. Although Casgevy is the first approved gene editing product, it may not be the most advanced. The CRISPR/Cas system’s ability to induce DSBs, base editing, prime editing, and epigenome editing allows for more precise and versatile DNA editing, paving the way for CRISPR 2.0 [4].
In the post-genome era, it has become increasingly important to clarify the relationship between genotype and phenotype. Additionally, we have confirmed that many inherited diseases are caused by gene mutations. As of December 15, 2023, 7461 types of diseases have been reported, and they are associated with 4871 genes (https://www.omim.org/statistics/geneMap). 6404 of these diseases are single-gene Mendelian disorders and traits. With the widespread availability of sequencing technology, this number will continue to grow. Rare genetic diseases affect millions of people worldwide, and many of them lack effective treatment options. Some genetic diseases can significantly reduce quality of life and lead to premature death [5], especially for developing countries. Gene therapy, including gene addition and gene editing, offers a potential way to correct defective genes [6]. Gene addition using viruses as delivery methods has been proposed for decades, and recently, some products have been approved. However, gene addition is mainly suitable for loss-of-function diseases, while gene editing is more suitable for gain-of-function diseases. The combination of gene addition and gene editing with cell therapy provides an alternative treatment for inherited diseases.
Zinc finger nucleases (ZFN), transcription activator-like effector nucleases (TALEN), and clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein (Cas) systems, particularly CRISPR/Cas9, have significantly advanced the field of gene therapy. All of these methods achieve site-specific gene editing, and several related products using these technologies are currently in clinical trialss [7, 8]. Gene-editing technologies are used not only to correct pathogenic mutations for clinical applications but also to modify immune cells, such as chimeric antigen receptor T (CAR-T) cells [9]. In this chapter, we primarily discuss the application of base editing. In the first section, we present an overview of the development of CRISPR/Cas systems, base editing, and prime editing. We also discuss the potential disadvantage of double-stranded breaks. In the second section, we review the progress made in using base editing to correct inherited pathogenic mutations and present an overview of some preclinical studies. In the third section, we highlight recent clinical trials that have employed base editing.
2. Target gene editing mediated by CRISPR/Cas system
CRISPR/Cas systems are commonly found in bacteria and archaea, where they defend against foreign nucleic acids. CRISPR/Cas systems are typically classified into two classes, with six subtypes: I, III, and IV in class I and II, V, and VI in class II. Recently, Zhang’s group has identified the VII subtype using a deep terascale clustering method [10]. Among the subtypes, CRISPR/Cas9 and CRISPR/Cas12 from type II are commonly utilized. The CRISPR/Cas system performs the function of targeting, and the editing results depend on the repair pathway following double-stranded break or on the functional protein fused to Cas. The CRISPR/Cas system has recently found widespread applications in life science, agriculture, and medicine. Numerous other systems have been developed based on CRISPR/Cas, including CRISPRa, CRISPRi, epigenome editing, base editing (BE), and prime editing (PE). In 2020, Emmanuelle Charpentier and Jennifer A. Doudna were awarded the Nobel Prize in Chemistry for the development of a method for genome editing.
2.1 Gene editing and regulation mediated by CRISPR/Cas
The CRISPR/Cas9 system essentially consists of two components, Cas9 and single guide RNA (sgRNA). The two combine to form a complex that then seeks out and binds to the target site. Initially, the most prevalent use of CRISPR/Cas9 was in disrupting target genes, resulting in insertions or deletions (indels) mediated by nonhomologous end joining (NHEJ). Subsequently, the precise genotypes could be derived with the aid of homology-directed repair. Moving beyond basic DNA editing, developments were made in creating systems like CRISPRa, CRISPRi, and epigenome editing. These do not modify the DNA sequence itself. Fundamentally, all of them are fusion proteins that combine different functionally active proteins with a deactivated Cas protein [11, 12].
2.2 Base editing provides a robust tool for making point mutation
The safety of gene editing products is a critical concern for clinical applications. Double-strand breaks are among the most lethal forms of DNA damage. Prior research has indicated that DSB-based gene editing may lead to the deactivation of p53 expression in responsive cells [13]. Moreover, DSBs can occasionally cause the loss of the targeted chromosome in human primary T cells [14]. The risk escalates when multiple sites are edited within a single cell, potentially leading to genomic rearrangements or fragment deletion [15]. Therefore, the risks associated with DSBs need to be carefully considered, especially in the context of gene therapy products [16].
Base editing, first introduced by Liu’s group [17], is a technique that combines a deaminase enzyme with a catalytically impaired Cas nuclease. This method allows for efficient and precise base conversions without inducing double-strand breaks (DSBs) or necessitating a donor DNA template. In 2017, base editing technology was heralded as the “Breakthrough of the Year”. Among the most utilized base editing systems are cytosine base editors (CBEs), which facilitate C-to-T conversions, and adenine base editors (ABEs), which enable A-to-G conversions. Other base editors, such as C•G-to-G•C base editors (CGBEs) that mediate C-to-G conversions, have also been developed [18]. Base editors incorporate the CRISPR/Cas system and a deaminase enzyme. Various combinations of Cas proteins and deaminases result in distinct editors, each carrying different editing windows or protospacer adjacent motif (PAM) requirements. Continual optimization of base editing systems allows for effective point mutation induction. For instance, Diorio et al. reported using a CBE to introduce point mutations in primary T cells with over 90% efficiency [9]. This manipulation effectively silenced gene expression without inducing DSBs.
Because deaminase binds randomly to ssDNA or ssRNA, base editing systems have been reported to induce sgRNA-independent off-target effects. For CBE, off-target effects occur in both DNA and RNA, while for ABE, off-target effects are mainly found in RNA. Numerous methods have been developed to eliminate these off-target effects, including making mutations in the deaminase, embedding deaminase [19], and using high-fidelity Cas9 and the transformer base editor (tBE) [20]. In addition to engineering base editors, the delivery method can also reduce off-target effects, such as electroporation with CBE protein-replacing plasmids.
Base editing has the potential to introduce point mutations, but its application scope is extensive. One of the most significant applications is correcting pathogenic mutations, as the majority of these are point mutations, making them suitable candidates for base editing. Furthermore, base editing can be used to knock out target genes by introducing stop codons into the coding sequence (CDS), disrupting splicing sites, and destroying start codons [21]. Currently, there are four clinical trials underway using base editing systems, and numerous preclinical studies are in progress [22].
In addition to DNA base editing, RNA base editing and mitochondrial base editing are also emerging technologies that are worth further exploration [23, 24]. By fusing wild-type adenosine deaminase acting on RNA (ADAR) with Cas13, A-to-I base editing in RNA can be achieved, and the evolved ADAR has the ability to convert C-to-U. Even without Cas, native ADAR can be recruited by engineered RNAs to perform A-to-I editing [24]. DddA-derived cytosine base editors (DdCBEs) can convert C-to-T in mitochondrial DNA, while split TALE deaminases (TALEDs) can convert A-to-G in mitochondrial DNA [23].
2.3 Prime editing broadens the editing scope of base editing
Prime editing, first reported by Liu’s group in 2019, involves the fusion of an engineered reverse transcriptase to a catalytically impaired Cas9 endonuclease [25]. Additionally, the guide RNA for prime editing (PE) differs from other editing systems. The PE system utilizes a prime editing guide RNA (pegRNA) that not only specifies the target site but also encodes the desired edit. Prime editing is capable of inducing all 12 possible base-to-base conversions, as well as the insertion or deletion of short sequences. Since its initial publication, various laboratories worldwide have contributed to furthering this groundbreaking technology. With continuous optimization, the PE system has evolved from PE1 to PE6 [26]. Prime editing has also been successfully employed in correcting pathogenic mutations, with numerous preclinical studies reporting its effectiveness [27].
Animal models are crucial for disease research, and CRISPR/Cas9 has become a widely used tool in creating animal disease models [28]. Three main methods exist for producing animal models: embryo microinjection, somatic cell nuclear transfer, and in situ injection. These methods have been commonly employed with CRISPR/Cas9. Base editing, like CRISPR/Cas9, offers a similar approach, and animal models based on base editing have been reported.
Kim et al. delivered the cytidine base editing system into mouse embryos to target the Dmd gene, resulting in Dmd mutant mice that rarely expressed dystrophin protein. For the Tyr gene, newborn pups exhibited an albino phenotype in their eyes [29]. Using a similar method, the same group generated Tyr mutant mice with an albino phenotype using adenine base editing in 2018 [30]. Beyond rodent animals, base editing has also been applied in zebrafish and nonhuman primates. With the CBE4max-SpRY CBE variant, which recognizes nearly all PAM sequences, Rosello et al. created a zebrafish model for melanoma predisposition [31]. In 2023, Zheng et al. simulated a pathogenic site of the tsr2 ribosome maturation factor (tsr2) in zebrafish by injecting a mixture of zSpRY-ABE8e mRNA and sgRNA into embryos. The mutant zebrafish exhibited a phenotype of smaller eyes and a swollen pericardium [32]. Wang et al. generated monkey models with Hutchinson-Gilford progeria syndrome (HGPS). They microinjected RNA of CBE into the embryos, and five out of six newborn monkeys carried the mutation. These HGPS monkeys displayed typical HGPS phenotypes including growth retardation, bone alterations, and vascular abnormalities [33].
The RAG1 and IL2RG genes play crucial roles in the immune system, and mutations in these genes can lead to severe combined immunodeficiency (SCID). Zheng et al. developed an immunodeficient monkey model using the CBE4max system. CBE induced a stop codon at the coding sequence of these two genes, resulting in immunodeficient monkeys that showed the ability to foster tumor growth [34].
Base editing holds significant potential in creating animal models. The primary approach involves microinjecting the mRNA- or RNP-encoding base editing into embryos. In contrast to NHEJ-induced knockout methods, base editing allows for the straightforward generation of desired animals without mosaicism at the F0 generation. However, base editing has limitations in terms of its inability to introduce insertions and deletions. As such, HDR, BE, and PE are complementary techniques for creating various animal disease models.
4. Progress of base editing in correcting inherited pathogenic mutations
Rare genetic diseases affect millions of people worldwide and are often caused by mutations in the genome. Gene therapy offers a fundamental approach to curing these rare diseases. A survey of 1430 researchers agreed that gene therapies will greatly benefit the treatment of patients with rare genetic diseases [5]. While gene addition is useful for loss-of-function mutations, and some approved gene therapy products rely on gene addition, the main reason for its effectiveness is the lack of appropriate techniques to fix defective genes. Correcting these defective genes is a more logical approach for rare genetic diseases. The CRISPR/Cas system provides an effective method to change gene mutations, and the base editing system is the best choice for point mutations. Taking into account efficiency and safety, base editing may be more successful in treating rare diseases. Here, we summarize the research progress of correcting pathogenic mutations using base editing (Table 1).
Selected examples of base editing on correcting pathogenic mutations.
4.1 In vivo and ex vivo gene therapy
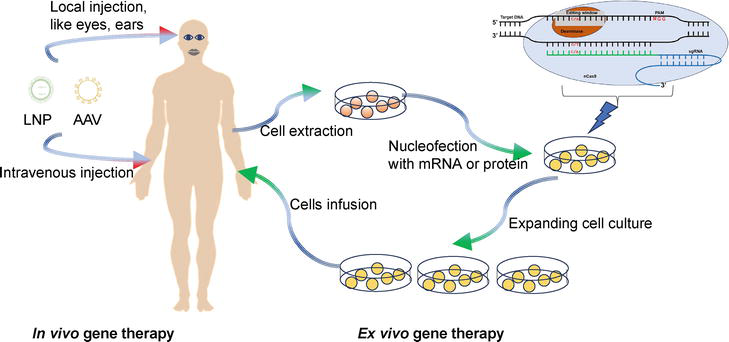
Delivery methods play a crucial role in achieving efficient gene therapy. Gene therapy can be categorized into two types: in vivo and ex vivo [70, 71].Recently, the majority of studies have focused on ex vivo gene therapy. In this approach, cells such as hematopoietic stem and progenitor cells (HSPCs) are extracted from patients and then subjected to the delivery of the editing system. For genetic engineering using base editing, nucleofection with mRNA combined with sgRNA is the primary method [22]. The challenge lies in producing large-scale proteins of base editors, which hinders the application of RNP for base editing purposes. On the other hand, for in vivo gene therapy, adeno-associated virus (AAV) and lipid nanoparticles (LNP) are considered as the main delivery methods for delivering the base editing system to target cells [71] (Figure 1). Enhancing efficiency and safety in delivery methods will significantly advance progress in in vivo gene therapy.
Figure 1.
In vivo and ex vivo gene therapy based on base editing. For ex vivo, the nucleofection is the main method to deliver the editing system into the cells. For in vivo, AAV and LNP are the two main methods to deliver the base editing system into the target cells.
4.2 Hematological disorder
The hematologic system plays a crucial role in multiple vital functions within our bodies, including delivering oxygen and defending against offensive pathogens. Dozens of hematological disorders have been reported [72]. However, in terms of gene therapy, most studies have focused on sickle cell disease (SCD) and β-thalassemia [73, 74]. Both conditions are the result of mutations within the β-globin locus. Antoniou et al. utilized base editing to introduce mutations in the −200 region of the HBG promoter, leading to the activation of fetal hemoglobin [38]. This same strategy has been employed by other research groups [40, 42, 43, 47]. Li et al. implemented this strategy in healthy CD46/β-YAC mice carrying the human β-globin locus. They achieved editing through subcutaneous and intravenous injections, eliminating the need for myeloablation and hematopoietic stem cell transplantation [44]. Badat et al. used the ABE system to correct the HbE mutation to either wild-type (WT) or a normal variant hemoglobin (E26G). Ninety percent of primary human CD34+ cells were edited, and long-term repopulating hematopoietic stem cells (HSCs) were confirmed through xenotransplantation in NSG mice [39]. Editing the mutation in HBB represents a widespread strategy for treating SCD or β-thalassemia. [41, 46].
Fanconi Anemia (FA) is a hematological disorder characterized by a wide range of severe symptoms. The DSB repair pathway is defective in FA, making it difficult to use HDR for mutation correction. Two groups have demonstrated that base editing can provide an effective method to correct mutations in the FANCA gene [35, 36]. Hemophilia is a congenital hemorrhagic disease caused by mutations in coagulation factor VIII (FVIII) or factor IX (FIX). Hiramoto et al. corrected a mutation ((c.947 T > C; I316T) in the F9 gene using CBE in induced pluripotent stem cells [45]. Rong et al. used ABE8e to correct G20519A and then measured the concentration and activity of FIX. [37].
4.3 Neurodegenerative disease
Spinal muscular atrophy (SMA) is a devastating neuromuscular disease that often leads to infant mortality. It is caused by mutations in the SMN1 gene, with the most common mutation being the deletion of exon 7. The function of SMN1 can be partially compensated by SMN2, which differs from SMN1 primarily by a synonymous C·G-to-T·A transition in exon 7. This mutation results in insufficient expression. Arbab et al. used base editing to correct this mutation in Δ7SMA mice. The mice achieved 87% conversions, and their motor function was enhanced [50]. This similar work has been replicated by another group. [48].
Alzheimer’s disease (AD) is a widely recognized condition that affects millions of people worldwide. Mutations in the APP gene are thought to play a role in the development of AD. Guyon et al. used ABE to convert the A673T mutation in SH-SY5Y neuroblastoma cells. Additionally, they identified a novel mutation, E674K, which appeared to further reduce Ab peptide accumulation. This finding may represent a new strategy for the treatment of Alzheimer’s disease [49].
4.4 Inborn errors of metabolism
Inborn errors of metabolism (IEM) encompass over 1500 distinct diseases, primarily resulting from mutations in genes coding for proteins involved in intermediate metabolism. Nearly 25% of IEMs manifest during the neonatal period, emphasizing the importance of early intervention. Numerous gene therapy studies have focused on IEM using CRISPR/Cas9, with the delivery of the editing system as the primary challenge [75]. For base editing, most research in IEM has centered on delivering the editing system to target cells, often using induced pluripotent stem cells (iPSCs), fibroblasts, and mouse models to demonstrate the editing potential [51, 52, 53, 54, 55, 56]. Curiously, most studies have utilized ABE rather than CBE for these corrections (Table 1). Additionally, genes such as ANGPTL3 and PCSK9 have been targeted to prevent atherosclerotic cardiovascular disease (ASCVD) [76, 77].
4.5 Other inherited diseases
Inborn errors of immunity (IEI) are disorders that affect the innate or adaptive immune system. STAT3-hyper IgE syndrome, a representative IEI, is characterized by destructive lung disease and various other symptoms. Eberherr et al. employed ABEs to correct the STAT3 p.R382W mutation in patient fibroblasts and iPSCs. The editing efficiency was approximately 29% in the patient fibroblasts, and the edited cells exhibited functional rescue of STAT3 signaling [57]. CD3δ severe combined immune deficiency (SCID), another life-threatening IEI, is caused by biallelic mutations in the autosomal CD3D gene. McAuley et al. utilized an ABE strategy to restore CD3δ in HSPCs, achieving an editing efficiency of 71.2% [58]. They also highlighted the advantages of base editing compared to standard CRISPR/Cas9 editing, indicating that base editing holds significant promise for future therapeutic applications [78].
Dystrophic epidermolysis bullosa (DEB) is a severe, rare disease characterized by skin blistering and epithelial fragility. It is caused by mutations in the COL7A1 gene. Sheriff et al. employed ABE technology to correct a recurrent COL7A1 nonsense mutation, c.5047 C > T (p.Arg1683Ter), in exon 54 of COL7A1 [67]. Hong et al. further demonstrated the potential of ABE to correct other mutations in the COL7A1 gene [68].
Cystic fibrosis (CF) is a genetic disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. Various mutations of CFTR have been reported, including nonsense and splicing mutations, which have been corrected using base editing [69, 79, 80]. However, the most common mutation, F508del, exceeds the editing scope of base editing. Therefore, prime editing and homology-directed repair (HDR) are alternative choices for this mutation [81].
Duchenne muscular dystrophy (DMD) is a fatal genetic muscle disease that primarily affects males. Numerous research efforts are ongoing to develop effective therapies, including gene therapy based on CRISPR/Cas9 [82]. Xu et al. administered AAV9-iABE-NGA to adult mice, resulting in dystrophin restoration and functional improvement [65]. The off-target effect was low, and no significant toxicity was observed.
Numerous gene therapy studies based on base editing for other inherited diseases are currently underway [61, 64, 83, 84]. The proof-of-concept work has demonstrated the effectiveness and safety of base editing. The development of an effective delivery method in situ will further enhance the application of base editing.
5. Clinical trials using base editing approved by FDA
With the recent approval of the first gene editing product, we can expect to see a surge in the approval of additional gene editing products within the next decade. Currently, there are four products in clinical trials approved by FDA that utilize base editing techniques, produced by Beam therapeutics and Verve therapeutics (Table 2).
Identification code
Intervention / treatment
Phase
Sponsor
Delivery
Conditions
Target
Base editor
NCT05456880
BEAM-101
Phase 1/2
Beam Therapeutics Inc.
Electroporation, Ex vivo HSCs
Sickle Cell Disease and, β-Thalassemia
HBG1/2
ABE
NCT05885464
BEAM-201
Phase 1/2
Beam Therapeutics Inc.
Electroporation, Ex vivo T cells
T-ALL/T-LL, CD7 + AML
CD7, TRAC, PDCD1, CD52
CBE
NCT05398029
VERVE-101
Phase 1
Verve Therapeutics, Inc.
LNP, in vivo
HeFH
PCSK9
ABE
NCT06164730
VERVE-102
Phase 1
Verve Therapeutics, Inc.
GalNAc-LNP, in vivo
HeFH
PCSK9
ABE
Table 2.
Approved clinical trials using base editing.
BEAM-101 and BEAM-201 are two products offered by Beam Therapeutics Inc. BEAM-101 is designed for the treatment of sickle cell disease and β-Thalassemia. It utilizes ABE to target the regulatory element of HBG, with the aim of activating the expression of fetal hemoglobin. The HSCs (hematopoietic stem cells) from patients are electroporated with mRNA encoding the base editor and sgRNA (single guide RNA). The edited cells are then infused back into the patients. BEAM-201, like BEAM-101, is also produced ex vivo. However, BEAM-201 is a CART (chimeric antigen receptor T cell) product designed to treat cancers. It utilizes CBE (cytosine base editor) to knock out CD7, TRAC, PDCD1, and CD52 genes [9].
VERVE-101 and VERVE-102 are developed by Verve Therapeutics, Inc. Both of them utilize an in vivo approach, which involves the use of LNP (lipid nanoparticle) to deliver the ABE (base editor) system targeting PCSK9 to the liver [77]. The difference between them lies in the modification of the LNP for VERVE-102 with galNAc. VERVE-101 is the first in vivo drug to utilize base editing, and it was selected by Nature Medicine as one of the 11 clinical trials that will shape medicine in 2024 [85].
Currently, more than 20 gene editing drugs are in clinical trials, and with the first gene editing drugs approved, we can expect a growing number of gene editing drugs to gain approval in the future. These drugs are being developed not only for inherited diseases but also for cancers, infection diseases, and even HIV by biotech companies and large pharma.
6.1 Gene therapy in utero
Recently, most of these therapies have been directed toward editing somatic cells, through either ex vivo or in vivo methods. Germline editing is currently only permitted for basic research purposes, and it remains forbidden in clinical settings. In utero gene therapy (IUGT) has gained attention, and many researches have been conducted on animals [86]. There are several advantages for IUGT, such as small fetal size, tolerogenic immune system, and prevalent progenitor cells [87]. Transgenic rodent models have demonstrated the long-term safety of IUGT, but several issues remain to be addressed. One concern is the potential risk to the maternal bystander, while another is the risk of germline mutations. Considering the potential risk, disease selection for IUGT is important. The most suitable scene for IUGT is the lethal and morbid perinatal diseases. For different diseases, the best therapy time is before the onset of pathology. Optimization of the delivery methods focused on the fetus will enhance the clinical translation of IUGT.
6.2 Efficiency and precision of base editing
Currently, most base editors have an editing window, during which any C or A can be edited. This characteristic introduces bystander editing, which needs to be considered when using base editing to correct pathogenic mutations. If the aim is to disrupt a gene, bystander editing may be less significant. In the future, narrowing the editing window may help precisely correct pathogenic mutations. CRISPR/Cas requires a PAM sequence to bind the target site. Currently, there are various Cas proteins, including Cas9-SpRY, which do not have the PAM limitation [88]. However, SpRY has the disadvantage of targeting itself when the plasmid is used. Ideally, a Cas protein should have PAM limitations but be able to target different PAM sequences for NA, NT, NG, or NC. For ex vivo therapy, electroporation is commonly used. Electroporation of proteins may help reduce potential off-target effects. In the future, it is important for the engineered editor to easily purify its protein and ensure protein stability.
6.3 Delivery method of the base editing system
Gene therapy can be classified into ex vivo and in vivo methods. In the ex vivo approach, target cells like HSPCs and immune cells are isolated, edited, and then infused into the patient. The procedure for ex vivo therapy is relatively mature; however, it is associated with high costs due to its complexity. In contrast, gene therapy in vivo has the potential to reduce costs. The main obstacle for in vivo therapy is delivery, and viral delivery is the most efficient method currently available. Nonviral delivery systems represent a promising future direction for in vivo gene therapy. Among nonviral delivery systems, LNPs show advantages for liver delivery, but further development of specific delivery methods is needed for other tissues [7].
6.4 Price of gene editing products
Gene therapy drugs offer great hope to patients, but their high cost makes it difficult to apply them to all patients, which may be unfair to those in poverty. Additionally, the high price may result in the delisting of drugs, as seen with Glybera, a gene therapy product approved in 2012 but withdrawn in 2017 due to its exorbitant cost. The complexity of the manufacturing process for these drugs contributes to their high production costs. To address this issue, more reasonable payment methods and the development of off-the-shelf gene therapy drugs may offer potential solutions.
In conclusion, base editing has demonstrated advantages over traditional editing methods based on DSB. Numerous clinical trials using base editing are currently underway. With the obstacles discussed above addressed, we believe that gene therapy has the potential to edit diseases out.
This work was supported by the National Key Research and Development Program of China (2021YFA0804702) and the National Natural Science Foundation of China (82302497). We thank members of Huang’s lab for helpful discussions.
1.Wong C. UK first to approve CRISPR treatment for diseases: What you need to know. Nature. 2023;623:676-677
2.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816-821
3.Frangoul H et al. CRISPR-Cas9 gene editing for sickle cell disease and beta-thalassemia. The New England Journal of Medicine. 2021;384:252-260
4.Ledford H. CRISPR 2.0: A new wave of gene editors heads for clinical trials. Nature. 2023;624:234-235
5.Braga LAM, Conte Filho CG, Mota FB. Future of genetic therapies for rare genetic diseases: What to expect for the next 15 years? Therapeutic Advances in Rare Disease. 2022;3:26330040221100840
6.Roth TL, Marson A. Genetic disease and therapy. Annual Review of Pathology. 2021;16:145-166
7.Huang C, Li Q , Li J. Site-specific genome editing in treatment of inherited diseases: Possibility, progress, and perspectives. Medicine Review (Berl). 2022;2:471-500
8.Liu X et al. Advances in CRISPR/Cas gene therapy for inborn errors of immunity. Frontiers in Immunology. 2023;14:1111777
9.Diorio C et al. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood. 2022;140:619-629
10.Altae-Tran H et al. Uncovering the functional diversity of rare CRISPR-Cas systems with deep terascale clustering. Science. 2023;382:eadi1910
11.Bendixen L, Jensen TI, Bak RO. CRISPR-Cas-mediated transcriptional modulation: The therapeutic promises of CRISPRa and CRISPRi. Molecular Therapy. 2023;31:1920-1937
12.Goell JH, Hilton IB. CRISPR/Cas-based Epigenome editing: Advances, applications, and clinical utility. Trends in Biotechnology. 2021;39:678-691
13.Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nature Medicine. 2018;24:927-930
14.Tsuchida CA et al. Mitigation of chromosome loss in clinical CRISPR-Cas9-engineered T cells. Cell. 2023;186:4567-4582 e4520
15.Dang L et al. Comparison of gene disruption induced by cytosine base editing-mediated iSTOP with CRISPR/Cas9-mediated frameshift. Cell Proliferation. 2020;53:e12820
16.Amendola M, Brusson M, Miccio A. CRISPRthripsis: The risk of CRISPR/Cas9-induced Chromothripsis in gene therapy. Stem Cells Translational Medicine. 2022;11:1003-1009
17.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420-424
18.Koblan LW et al. Efficient C*G-to-G*C base editors developed using CRISPRi screens, target-library analysis, and machine learning. Nature Biotechnology. 2021;39:1414-1425
19.Liu Y et al. A Cas-embedding strategy for minimizing off-target effects of DNA base editors. Nature Communications. 2020;11:6073
20.Wang L et al. Eliminating base-editor-induced genome-wide and transcriptome-wide off-target mutations. Nature Cell Biology. 2021;23:552-563
21.Wang X et al. Efficient gene silencing by Adenine Base editor-mediated start codon mutation. Molecular Therapy. 2020;28:431-440
22.Porto EM, Komor AC. In the business of base editors: Evolution from bench to bedside. PLoS Biology. 2023;21:e3002071
23.Kar B, Castillo SR, Sabharwal A, Clark KJ, Ekker SC. Mitochondrial Base editing: Recent advances towards therapeutic opportunities. International Journal of Molecular Sciences. 2023;24:5798
24.Pfeiffer LS, Stafforst T. Precision RNA base editing with engineered and endogenous effectors. Nature Biotechnology. 2023;41:1526-1542
25.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149-157
26.Doman JL et al. Phage-assisted evolution and protein engineering yield compact, efficient prime editors. Cell. 2023;186:3983-4002 e3926
27.Godbout K, Tremblay JP. Prime editing for human gene therapy: Where are we now? Cell. 2023;12:536
28.Li G et al. Gene editing and its applications in biomedicine. Science China. Life Sciences. 2022;65:660-700
29.Kim K et al. Highly efficient RNA-guided base editing in mouse embryos. Nature Biotechnology. 2017;35:435-437
30.Ryu SM et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nature Biotechnology. 2018;36:536-539
31.Rosello M et al. Disease modeling by efficient genome editing using a near PAM-less base editor in vivo. Nature Communications. 2022;13:3435
32.Zheng S et al. Efficient PAM-Less Base editing for zebrafish modeling of human genetic disease with zSpRY-ABE8e. Journal of Visualized Experiments. 2023;192
33.Wang F et al. Generation of a Hutchinson-Gilford progeria syndrome monkey model by base editing. Protein & Cell. 2020;11:809-824
34.Zheng X et al. Generation of inactivated IL2RG and RAG1 monkeys with severe combined immunodeficiency using base editing. Signal Transduction and Targeted Therapy. 2023;8:327
35.Sipe CJ et al. Correction of Fanconi anemia mutations using digital genome engineering. International Journal of Molecular Sciences. 2022;23:8416
36.Siegner SM et al. Adenine base editing efficiently restores the function of Fanconi anemia hematopoietic stem and progenitor cells. Nature Communications. 2022;13:6900
37.Rong L, Chen D, Huang X, Sun L. Delivery of Cas9-guided ABE8e into stem cells using poly(l-lysine) polypeptides for correction of the hemophilia-associated FIX missense mutation. Biochemical and Biophysical Research Communications. 2022;628:49-56
38.Antoniou P et al. Base-editing-mediated dissection of a gamma-globin cis-regulatory element for the therapeutic reactivation of fetal hemoglobin expression. Nature Communications. 2022;13:6618
39.Badat M et al. Direct correction of haemoglobin E beta-thalassaemia using base editors. Nature Communications. 2023;14:2238
40.Ravi NS et al. Identification of novel HPFH-like mutations by CRISPR base editing that elevate the expression of fetal hemoglobin. eLife. 2022;11:e65421
41.Newby GA et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature. 2021;595:295-302
42.Mayuranathan T et al. Potent and uniform fetal hemoglobin induction via base editing. Nature Genetics. 2023;55:1210-1220
43.Liao J et al. Therapeutic adenine base editing of human hematopoietic stem cells. Nature Communications. 2023;14:207
44.Li C et al. In vivo base editing by a single i.v. vector injection for treatment of hemoglobinopathies. JCI Insight. 2022;7:e162939
45.Hiramoto T et al. PAM-flexible Cas9-mediated base editing of a hemophilia B mutation in induced pluripotent stem cells. Communications Medicine (Lond). 2023;3:56
46.Hardouin G et al. Adenine base editor-mediated correction of the common and severe IVS1-110 (G>a) beta-thalassemia mutation. Blood. 2023;141:1169-1179
47.Han W et al. Base editing of the HBG promoter induces potent fetal hemoglobin expression with no detectable off-target mutations in human HSCs. Cell Stem Cell. 2023;30:1624-1639 e1628
48.Alves CRR et al. Optimization of base editors for the functional correction of SMN2 as a treatment for spinal muscular atrophy. Nature Biomedical Engineering. 2023
49.Guyon A et al. Base editing strategy for insertion of the A673T mutation in the APP gene to prevent the development of AD in vitro. Molecular Therapy - Nucleic Acids. 2021;24:253-263
50.Arbab M et al. Base editing rescue of spinal muscular atrophy in cells and in mice. Science. 2023;380:eadg6518
51.Werder RB et al. Adenine base editing reduces misfolded protein accumulation and toxicity in alpha-1 antitrypsin deficient patient iPSC-hepatocytes. Molecular Therapy. 2021;29:3219-3229
52.Su J et al. In vivo adenine base editing corrects newborn murine model of hurler syndrome. Molecular Biomedicine. 2023;4:6
53.Rovai A et al. In vivo adenine base editing reverts C282Y and improves iron metabolism in hemochromatosis mice. Nature Communications. 2022;13:5215
54.Packer MS et al. Evaluation of cytosine base editing and adenine base editing as a potential treatment for alpha-1 antitrypsin deficiency. Molecular Therapy. 2022;30:1396-1406
55.Leclerc D et al. Gene editing corrects In vitro a G > a GLB1 transition from a GM1 Gangliosidosis patient. CRISPR Journal. 2023;6:17-31
56.Harb JF et al. Base editing corrects the common Salla disease SLC17A5 c.115C>T variant. Molecular Therapy - Nucleic Acids. 2023;34:102022
57.Eberherr AC et al. Rescue of STAT3 function in hyper-IgE syndrome using Adenine Base editing. CRISPR Journal. 2021;4:178-190
58.McAuley GE et al. Human T cell generation is restored in CD3delta severe combined immunodeficiency through adenine base editing. Cell. 2023;186:1398-1416 e1323
59.Nishiyama T et al. Precise genomic editing of pathogenic mutations in RBM20 rescues dilated cardiomyopathy. Science Translational Medicine. 2022;14:eade1633
60.Chai AC et al. Base editing correction of hypertrophic cardiomyopathy in human cardiomyocytes and humanized mice. Nature Medicine. 2023;29:401-411
61.Grosch M et al. Striated muscle-specific base editing enables correction of mutations causing dilated cardiomyopathy. Nature Communications. 2023;14:3714
62.Wu Y et al. AAV-mediated base-editing therapy ameliorates the disease phenotypes in a mouse model of retinitis pigmentosa. Nature Communications. 2023;14:4923
63.Peters CW et al. Rescue of hearing by adenine base editing in a humanized mouse model of usher syndrome type 1F. Molecular Therapy. 2023;31:2439-2453
64.Xiao Q et al. Rescue of autosomal dominant hearing loss by in vivo delivery of mini dCas13X-derived RNA base editor. Science Translational Medicine. 2022;14:eabn0449
65.Xu L et al. Efficient precise in vivo base editing in adult dystrophic mice. Nature Communications. 2021;12:3719
66.Sikrova D et al. Adenine base editing of the DUX4 polyadenylation signal for targeted genetic therapy in facioscapulohumeral muscular dystrophy. Molecular Therapy - Nucleic Acids. 2021;25:342-354
67.Sheriff A et al. ABE8e adenine base editor precisely and efficiently corrects a recurrent COL7A1 nonsense mutation. Scientific Reports. 2022;12:19643
68.Hong SA et al. Therapeutic base editing and prime editing of COL7A1 mutations in recessive dystrophic epidermolysis bullosa. Molecular Therapy. 2022;30:2664-2679
69.Amistadi S et al. Functional restoration of a CFTR splicing mutation through RNA delivery of CRISPR adenine base editor. Molecular Therapy. 2023;31:1647-1660
70.Li Y, Glass Z, Huang M, Chen ZY, Xu Q. Ex vivo cell-based CRISPR/Cas9 genome editing for therapeutic applications. Biomaterials. 2020;234:119711
71.Madigan V, Zhang F, Dahlman JE. Drug delivery systems for CRISPR-based genome editors. Nature Reviews. Drug Discovery. 2023;22:875-894
72.Echeverry G, Dalton A. Hematologic disorders. Anesthesiology Clinics. 2018;36:553-565
73.Park SH, Bao G. CRISPR/Cas9 gene editing for curing sickle cell disease. Transfusion and Apheresis Science. 2021;60:103060
74.Ma L, Yang S, Peng Q , Zhang J, Zhang J. CRISPR/Cas9-based gene-editing technology for sickle cell disease. Gene. 2023;874:147480
75.Leal AF et al. The landscape of CRISPR/Cas9 for inborn errors of metabolism. Molecular Genetics and Metabolism. 2023;138:106968
76.Zuo Y et al. Liver-specific in vivo base editing of Angptl3 via AAV delivery efficiently lowers blood lipid levels in mice. Cell & Bioscience. 2023;13:109
77.Lee RG et al. Efficacy and safety of an investigational single-course CRISPR Base-editing therapy targeting PCSK9 in nonhuman primate and mouse models. Circulation. 2023;147:242-253
78.Malech HL, Notarangelo LD. Gene therapy for inborn errors of immunity: Base editing comes into play. Cell. 2023;186:1302-1304
79.Geurts MH et al. CRISPR-based adenine editors correct nonsense mutations in a cystic fibrosis organoid biobank. Cell Stem Cell. 2020;26:503-510 e507
80.Graham C, Hart S. CRISPR/Cas9 gene editing therapies for cystic fibrosis. Expert Opinion on Biological Therapy. 2021;21:767-780
81.Geurts MH et al. Evaluating CRISPR-based prime editing for cancer modeling and CFTR repair in organoids. Life Science Alliance. 2021;4:e202000940
82.Verhaart IEC, Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nature Reviews. Neurology. 2019;15:373-386
83.Koblan LW et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature. 2021;589:608-614
84.Kaukonen M, McClements ME, MacLaren RE. CRISPR DNA Base editing strategies for treating retinitis Pigmentosa caused by mutations in rhodopsin. Genes (Basel). 2022;13:1327
85.Arnold C, Webster P. 11 clinical trials that will shape medicine in 2024. Nature Medicine. 2023;29:2964-2968
86.Mattar CNZ, Chan JKY, Choolani M. Gene modification therapies for hereditary diseases in the fetus. Prenatal Diagnosis. 2023;43:674-686
87.Bose SK, Menon P, Peranteau WH. InUtero gene therapy: Progress and challenges. Trends in Molecular Medicine. 2021;27:728-730
88.Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nature Biotechnology. 2020;38:824-844
Written By
Guanglei Li, Yu Zhang and Xingxu Huang
Submitted: 21 December 2023Reviewed: 26 December 2023Published: 06 May 2024