Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
Immunotherapy-induced endocrinopathies remain an adverse risk factor for patients undergoing PD-1/PD-L1 and CTLA-4 target immunotherapy treatment. Immunotherapy works by boosting the immune system to target, bind and block tumor receptors that would otherwise allow the cells to camouflage. This paper focuses on thyroid dysfunction, adrenal insufficiency, diabetes mellitus, and hypophysitis, and specifically the clinical presentation, diagnostic approaches, and management of each. All four endocrinopathies often present with generalized symptoms and, therefore, are commonly misdiagnosed. Healthcare professionals must closely monitor symptoms through laboratory testing and, when necessary, diagnostic imaging to appropriately diagnose and treat endocrinopathies. Thyroid dysfunction and adrenal insufficiency are often debated on immunotherapy hold/discontinuation. However, treatment of grade 3–4 diabetes mellitus and hypophysitis results in holding treatment until immune function prognosis decreases or returns to normal. Overall, ongoing research and clinical trials are needed to understand the varying effects of immunotherapy-induced endocrinopathies. Healthcare professionals should always consider patient symptoms, laboratory results and diagnostic imaging to ensure endocrinopathies are not missed and patients receive optimal care.
Thunder Bay Regional Health Science Centre, Thunder Bay, Canada
NOSM University, Thunder Bay, Canada
Alesha Bishop
NOSM University, Thunder Bay, Canada
Hannah Shortreed
NOSM University, Thunder Bay, Canada
Elycia Monaghan
NOSM University, Thunder Bay, Canada
Yue Sun
NOSM University, Thunder Bay, Canada
*Address all correspondence to: olexiy.aseyev@tbh.net
1. Introduction
One cannot read about immunotherapy without encountering its unique history, with one individual coined as the “Father of Immunology”, William B. Coley [1, 2, 3]. Coley is widely regarded as the first individual to endeavor to mobilize the immune system in treating cancer in the l891, producing regression of tumor growth in patients with sarcoma by injecting inactivated Streptococcus pyogenes and Serratia marcescens [2]. These injections were later termed “Coley’s toxins” [1, 3]. Another hallmark of immunotherapy is the initiation of IL-2 (AKA T cell growth factor) as the first cancer immunotherapy in humankind, with FDA approval in 1992 in treating renal cell carcinoma that has metastasized [1, 2].
Immunotherapy is a treatment that boosts the immune system’s power to fight cancer [2]. It focuses on how cancer cells interact with our body’s natural defense system [2, 3, 4]. It considers the complex relationship between tumor cells and immunosurveillance [4]. This interaction involves three key steps. First, our immune system, which has both innate and adaptive components, works to destroy cancer cells [1, 2]. Second, there’s a dynamic balance between the actions of the immune system and cancer cells, where cancer cells adapt to resist the immune attack [2, 3]. Finally, cancer cells may evade detection by the immune system and continue to grow and spread [2, 4]. Immunotherapy is just in its early days, yet it has drastically altered the cancer treatment landscape [1, 3].
Immune checkpoints and their inhibition continue to be an integral part of immunotherapy [1, 2, 3, 4]. Immune checkpoints are receptors found on the surface of T cells, and they serve as the gatekeeper of immune responses in terms of either propagation or inhibition [4, 5, 6, 7]. This gatekeeping is in keeping with the process of immuno-editing, involving the immune system’s ability to keep tumor growth at bay by modifying tumor immunogenicity or modulating tumor escape by decreasing anti-tumor defense responses, or even simultaneously [2]. The way in which immune checkpoints prevent autoimmune dysfunction is in the former option, where negative feedback plays a role in avoiding overactivation of T cells [5]. In the latter option, tumor cells exploit immune checkpoints to weaken the response from the immune system so that they can escape immune modulation and thereby proliferate [4]. Immune checkpoint inhibitors utilize antibodies to inhibit this gatekeeper to allow for antitumor activity [2].
As with other treatments, immunotherapy can cause side effects [8]. Specifically, immune checkpoint inhibitors, a type of immunotherapy, are known to trigger immune-related adverse events (irAEs) [7, 8]. These include conditions like colitis, skin inflammation (dermatitis), liver inflammation (hepatitis), and various disorders related to hormone glands (endocrinopathies) [7, 8]. These inhibitors work by blocking certain immune checkpoint receptors, such as cytotoxic T-lymphocyte antigen 4 CTLA-4 and programmed cell death protein 1 (PD-1) or its ligand (PD-L1), to attack cancer cells [8]. However, this blocking can lead to an excessive immune response against normal cells, resulting in side effects ranging from mild to severe [8]. Detailed explanations of these processes are found in the following sections.
Research shows that about 10% of patients treated with immune checkpoint inhibitors experience endocrinopathies [8]. The most common are thyroid disorders and inflammation of the pituitary gland (hypophysitis) [8]. Less common are problems with the pancreas, adrenal, and parathyroid glands [7, 8]. It is important for both endocrinologists and oncologists to be aware of these potential side effects. Early identification of signs and symptoms, accurate diagnosis, and effective treatment strategies are crucial for successful patient outcomes [8].
2.1 Mechanisms of immunotherapy-induced endocrinopathies
Immunotherapy has become a revolutionary cancer treatment that utilizes the body’s immune system to target and destroy malignant cells [7]. Immune checkpoints are specialized proteins on the surface of immune cells that communicate with T cells to prevent cell destruction [6]. Some tumor cells present this unique mechanism to impersonate normal immune cells [3]. As T cells checkpoint different cells, tumor cells with the required proteins bind to the T cell receptors and bypass destruction [3, 4]. However, immunotherapy inhibits the ability of tumor cells to camouflage by blocking the checkpoint connection [4, 5]. Therefore, the T cell recognizes the tumor cell as a pathogen and initiates cell death [6]. A significant downside of checkpoint inhibitor immunotherapy is its impact on endocrine function [7, 8]. The focus in current research is on two checkpoint inhibitors: PD-1 and its associated protein PD-L1, and CTLA-4 [4, 5, 7]. However, when these checkpoints are blocked by immunotherapy, it leads to increased immune activity [1]. Such an increase can result in autoimmune dysfunction, whereby the immune system mistakenly attacks healthy cells and organs [9].
2.1.1 PD-1/PD-L1
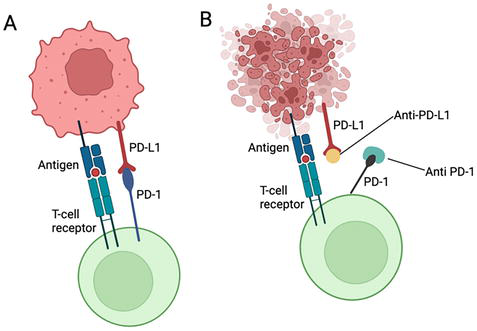
Programmed Cell Death Protein 1 (PD-1) is a receptor commonly associated with immune cells, including T cells used in late immune response [6, 9]. PD-1 co-protein Programmed Cell Death Ligand 1 (PD-L1) receptor is found on tumor cells [6, 9]. The prevalence of PD-L1 receptors is more commonly associated with solid cancers [5]. The mechanism of action results in the binding of PD-1 to PD-L1, therefore labelling the tumor cell as non-pathogenic and therefore, no T cell activation or proliferation of cytotoxic activity occurs [5]. In addition, the PD-1/PD-L1 binding may initiate apoptosis of the T cell, establishing a greater immune tolerance [10]. Overall, the PD-1/PD-L1 receptor binding enables the tumor cell to mimic a healthy cell, escaping an immune response and continuing to proliferate [5, 10]. Immune checkpoint inhibitors overcome these mechanisms by individually binding to PD-1 and PD-L1 receptors, preventing the direct binding of PD-1/PD-L1 depicted in Figure 1 [5, 9] . Without the direct binding of PD-1/PD-L1, the T cell receptor binds to the antigen receptor located on the tumor cell, which is now recognized as a pathogen [5]. The T cell proliferates, surrounds the tumor cell, and releases granzymes or pore-forming proteins to trigger cell death [1, 5].
Figure 1.
(A) PD-1/PD-L1 binding to inhibit T cell activation. (B) PD-1/PD-L1 checkpoint inhibitors preventing binding and therefore T cell activation occurs [11].
2.1.2 CTLA-4
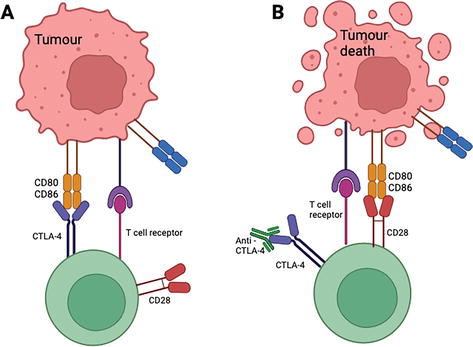
Cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) is an endocytic molecule found on the surface of specific T cells that regulate early T cell proliferation [12]. The binding of CTLA-4 to distinct ligands, CD80 and CD86, located on the tumor cell, can inhibit the activation of T cells [12]. Immunotherapy CTLA-4 checkpoint inhibitors block the binding of CTLA-4 and CD80/CD86, allowing the T cell to proliferate and destroy the tumor cell, pictured in Figure 2 [12].
Figure 2.
(A) CTLA-4 binding to CD80/CD86 inhibiting T cell. (B) Anti CTLA-4 checkpoint inhibitor, binding to CTLA-4, allowing for CD28 to bind to CD80/CD86 resulting in T cell proliferation and tumor death [13].
10% of all patients experience endocrinopathies secondary to checkpoint inhibitors [9]. Thyroid dysfunction is the most prevalent endocrinopathy for patients undergoing PD-1 and CTLA-4 inhibitors, with more significant onset associated with PD-1 [9]. The epidemiology of thyroid dysfunction secondary to immunotherapy is limited [11]. However, thyroiditis presents as a common pathology, potentially arising from PD-1/CTLA-4 disrupting normal thyroid function, resulting in an immune response triggering hypothyroidism or hyperthyroidism [14].
3.1.1 Clinical presentation
Thyroid dysfunction secondary to immune checkpoint inhibitors may present as Hypothyroidism or Hyperthyroidism [11, 14]. Commonly, signs and symptoms of immunotherapy-induced hypothyroidism/hyperthyroidism demonstrate similarities to primary thyroid dysfunction [11]. Hypothyroidism occurs from an underactive thyroid gland, resulting in symptoms including bradycardia, weight gain, constipation, and fatigue [9, 14]. Hyperthyroidism, or thyrotoxicosis, manifests as tachycardia, weight loss, diarrhea, and fatigue and is a consequence of an overactive thyroid gland [9, 14]. It is important to note that these symptoms are representative and often dismissed as other illnesses relating to immunotherapy [9]. Careful analysis should take place when a patient is experiencing subtle or severe symptoms [9]. Hypothyroidism presents more often, with the onset of 71 days with PD-1 alone and 63 days with combined therapy [11, 14]. Hyperthyroidism is less common but still noted, with an onset of 47 days after initial treatment on PD-1 monotherapy and 21 days for combination therapy [11, 14].
3.1.1.1 Case Vignette
An 81-year-old female diagnosed with metastatic lung cancer treated with carboplatin and pemetrexed and four cycles of pembrolizumab immunotherapy presented to the emergency department with symptoms of weakness and fatigue resulting in a near-fall. The patient denied symptoms of nausea, headaches, diarrhea or weight gain. Other than hypertension, the patient’s past medical history is unremarkable. Laboratory results presented an elevated TSH of 178 (normal range = 0.4–4.0 mlU/L) and a free T4 of less than 1 (normal range = 0.9–1.9 ng/dl). Sodium, potassium, chloride, and bicarbonate were all within normal range. Urinalysis was positive for leukocytes and white blood cells. Based on elevated TSH levels, the patient was diagnosed with pembrolizumab-induced hypothyroidism. The thyroid dysfunction presented 3–4 months after initiating pembrolizumab, corresponding with the literature’s onset of 63–71 days. The patient was treated with 50 mcg of oral levothyroxine and prednisone due to significant symptoms and elevated laboratory values. TSH was repeated in 6 weeks. The patient did not receive any diagnostic imaging, most likely due to the severely elevated TSH level used as an indication to begin treatment. Antibodies were also ordered to rule out Hashimoto’s Thyroiditis. The patients’ TSH values decreased after 6 weeks, and they could continue pembrolizumab treatment.
Adapted from our clinical case.
3.1.2 Diagnostic approaches
The detection of thyroid issues, such as hyperthyroidism or hypothyroidism, might not always start with obvious symptoms [6, 14]. It is recommended that patients undergo baseline blood tests before starting immunotherapy [14]. These tests provide a reference point, helping doctors identify any changes in thyroid function that might occur during treatment [6]. Baseline blood tests that should be performed include complete blood count (CBC), comprehensive metabolic panel (CMP), thyroid-stimulating Hormone (TSH) (normal range between 0.4 and 4.0 mlU/L), free thyroxine (T4) (total T4 = 0.4–2.5 mlU/l, FreeT4 = 0.9–1.9 ng/dl), Triiodothyronine (total T3) (0.9–2.8 nmol/L) and urinalysis for baseline kidney function (GFR ≥60) [15]; Liver function test (LFT) may also be considered as part of baseline measurements [15]. Adrenal insufficiency should be ruled out, as symptoms present similar to thyroid dysfunction [15]. Underlying medical conditions, including Hashimoto’s or Graves’s Disease, may produce thyroid antibodies that affect the overall function of the thyroid gland [1]. If a patient’s medical or family history demonstrates a risk for autoimmune disorders, physicians should consider performing a thyroid peroxidase antibody test (TPO) to rule out potential comorbidities that may present as thyroid dysfunction [15].
Hyperthyroidism secondary to immunotherapy diagnostically presents with a TSH below 0.4 mlU/L, whereas hypothyroidism presents with a TSH greater than 4.0 mlU/L [15]. A baseline Electrocardiogram (ECG) would provide important information if the patient has previous or is at risk of cardiovascular complications [15]. If thyroid dysfunction is symptomatic with positive lab results, a thyroid ultrasound or radioactive thyroid uptake scan can be performed (except in the case of pregnancy) to determine if the thyroid gland is underactive or hyperactive [15].
Laboratory results should be interpreted considering potential factors, including steroid use, iodine contrast, and non-thyroid illnesses [16].
3.1.3 Management strategies
Medical interventions for patients experiencing thyroid dysfunction secondary to immunotherapy should be constructed with an individualistic approach [11]. For patients experiencing hyperthyroidism, beta-blockers and antithyroid medications are commonly prescribed [11]. When treating hypothyroidism with an elevated TSH (specifically TSH > 10 mU/L) and symptom presentation, initiating hormone replacement medication is shown to have a high overall survival rate [1, 2]. Levothyroxine (1–1.6 mcg/kg/day) is a first-line treatment and works by replacing the thyroxine hormone that is under-produced by the thyroid gland [14]. Before administering levothyroxine, age and comorbidities should be considered. Other medications used for managing hypothyroidism include propranolol and atenolol [11].
After establishing baseline blood work and diagnostic imaging, TSH and FreeT4 are recommended every 4–8 weeks while receiving immunotherapy and every 3–6 months post-treatment [2, 14, 16]. In patients with diagnosed hyperthyroidism, TSH should be monitored every 2–3 weeks until laboratory results increase or until persistent hyperthyroidism can be established and treated [14]. It is important to note that patients with hyperthyroidism are at increased risk for developing hypothyroidism due to ongoing thyroiditis [15]. For patients experiencing hypothyroidism, TSH should be completed every 6–8 weeks [14]. In addition, cytokines and other inflammatory markers have been associated with tissue damage and inflammatory response, impacting thyroid function [14]. Monitoring inflammatory markers is essential in the early detection of thyroid dysfunction and maintaining regular thyroid activity [11].
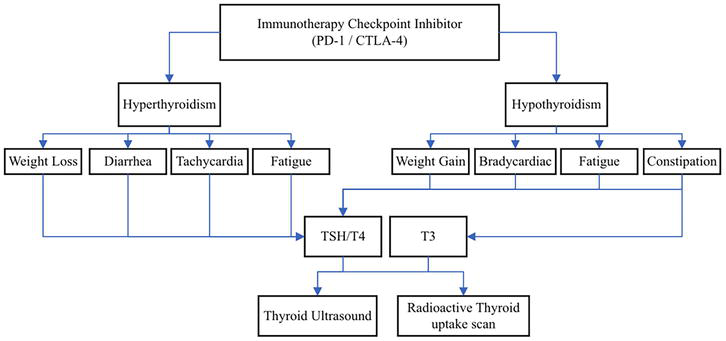
In the case of thyroid dysfunction, discontinuing immunotherapy is deemed unnecessary unless the patient is severely symptomatic [1, 14]. In this case, immunotherapy should be held until symptoms resolve [14]. There are no known immunotherapy dose reduction recommendations regarding thyroid dysfunction [15]. Hyperthyroidism and hypothyroidism with respective symptoms and diagnostic implications are shown in Figure 3.
Figure 3.
Representation of thyroid dysfunction, with presenting symptoms, laboratory testing and diagnostic imaging.
3.2 Adrenal insufficiency
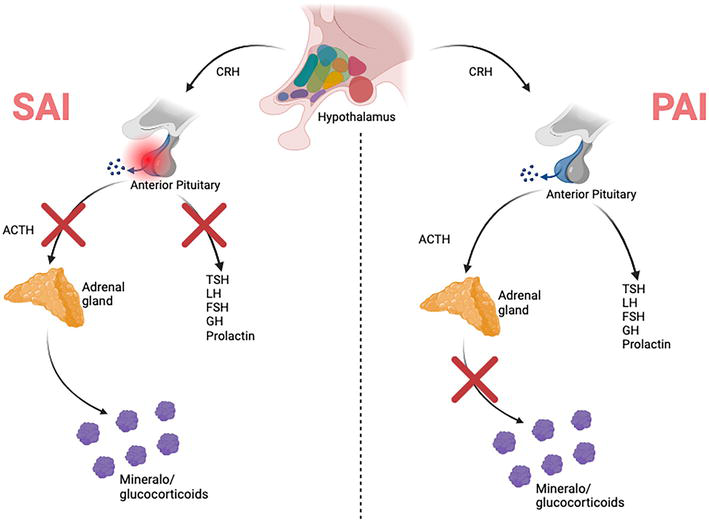
Adrenal insufficiency (AI) is an endocrine adverse event that can occur with the use of immune checkpoint inhibitors (Figure 4) [17, 18, 19, 20]. AI is a particularly salient adverse event as it is often misdiagnosed and therefore not always treated [17, 19]. It is important to differentiate between primary adrenal insufficiency (PAI) and secondary adrenal insufficiency (SAI), with the former being relatively rare and due to lack of glucocorticoid and/or mineralocorticoid production from the adrenal gland cortex [17, 18, 19]. PAI can possibly be lethal, as glucocorticoids and mineralocorticoids have essential roles in homeostasis of sodium, fluids, and energy [18]. SAI is related to inflammation within the pituitary gland, leading to diminished adrenocorticotropic hormone (ACTH) production and therefore loss of cortisol release [17, 18, 19]. Although not immediately life threatening, it may possibly impact other hormones produced by the anterior pituitary, including luteinizing hormone (LH), follicle-stimulating hormone (FSH), growth hormone, prolactin, and thyroid-stimulating hormone (TSH) [17].
Figure 4.
Pathophysiology of PAI versus SAI. PAI is due to the lack of mineralocorticoids and glucocorticoids being released from the adrenal gland. SAI is due to inflammation within the anterior pituitary, thereby causing decreased ACTH release along with possible decrease of other anterior pituitary hormones, including TSH, LH, FSH, GH, and prolactin [17].
3.2.1 Clinical presentation
Patients with both PAI and SAI present with non-specific symptoms such as orthostatic hypotension, nausea and vomiting, weakness, fatigue, and abdominal pain [17, 18, 19, 20]. One sign more so specific to SAI is eosinophilia due to the active inflammatory response within the anterior pituitary [18]. Other laboratory abnormalities include hyponatremia, hypoglycemia [18, 19, 20], hyperkalemia, and decreased morning levels of cortisol [17, 19, 20]. If a patient is experiencing an acute adrenal crisis, they may present with fever, confusion, lethargy, and anorexia [17, 19]. Cases of AI induced by immune checkpoint inhibitors have been observed in 1–2% of patients after receiving either anti-PD-1 or anti-PD-1 combined with CTLA-4 therapy, with this varying depending on types of cancer and immunotherapy being received [19]. AI can also present as syncope along with other non-specific symptoms, which is illustrated by the below case study [17].
3.2.1.1 Case Vignette
Mr. O is a 67-year-old male with metastatic melanoma with various skin lesions and nodules within his axillary lymph nodes and torso. This patient’s past medical history is significant for hypertension, hyperlipidemia, and cerebral vascular accident. Mr. O had received four doses of ipilimumab and nivolumab for treatment of metastatic melanoma. Following his most recent dose of these two immunotherapy agents, PET/CT scan had shown an increase in the amount and size of cutaneous lesions. About 7 days later, this patient started to report gastrointestinal symptoms including nausea and constipation, for which he was prescribed a proton pump inhibitor (PPI). This intervention allowed for improvement of his nausea, but soon after Mr. O had presented to the emergency department following a syncopal episode. After blood work had been ordered, he was noted to have hyponatremia and hypokalemia, for which he received intravenous fluids. This however did little to improve his sodium levels, and Mr. O was later found to have syndrome of inappropriate antidiuretic hormone (SIADH). After this diagnosis had been made, he was given sodium tablets and later discharged home with stable serum sodium levels.
Laboratory tests to assist in the diagnosis of AI include cortisol and ACTH drawn in the morning, along with ACTH stimulation testing to assess for the extent of adrenal insufficiency [17, 18, 20]. In terms of morning cortisol, results consistent with adrenal insufficiency are low morning cortisol, as normal physiology has it so that cortisol levels are normally increased in the morning in accordance with normal circadian rhythms [19]. To distinguish between primary and secondary adrenal insufficiency, morning ACTH levels are usually increased in PAI as a result of the anterior pituitary’s reaction to decreased cortisol prediction from the adrenal gland [20]. Another PAI result would be a diminished aldosterone serum concentration, along with hyponatremia and hyperkalemia [17, 18]; this differs from SAI, where one would expect to see low or normal TSH levels and decreased free thyroxine levels [17, 18].
One imaging modality that would be particularly helpful in ruling out other conditions would be a CT of the head, which would assist in assessing for metastatic spread or hemorrhage [17]. Other imaging that can be ordered includes an MRI of the head/hypophysis, where in SAI the pituitary stalk would be noted as increased in size or the pituitary gland itself would be enhanced [17, 18, 20]. This can be performed with or without contrast and would be indicated in patients with new symptoms of increased intracranial pressure, including new onset severe headaches and visual changes [17]. In terms of imaging the adrenal glands, CT of these structures may assist in the diagnosis of PAI, possibly showing smoothened contours of the glands which would indicate elevated volumes [20].
3.2.3 Management strategies
The main treatment for AI, regardless of it being primary or secondary, is corticosteroids [17, 18, 20]. If symptoms are acute, hydrocortisone is the medication of choice, with the first dose being 100 mg administered intravenously as a form of physiologic stress dosing [17, 20]. Following this, the dose would be tapered down to 50 mg given every 6–8 hours, followed by another taper of 10–20 mg given in the morning and another 5–10 mg given in the early afternoon [17]. Cautious consideration should be given in administering high dose corticosteroids such as methylprednisolone or prednisone 1 mg/kg/day as there are several studies noting an association between high dose corticosteroids and increased mortality [17].
There is debate as to whether one should discontinue taking immune checkpoint inhibitors if AI occurs [17, 20]. One source notes that they should be stopped in all patients with AI [20], while another reference suggests holding immune checkpoint inhibitors until the patient is clinically stable or in patients that have a diagnosis that is ambiguous [17]. Corticosteroids may have to be discontinued as well as they may lead to decreased ACTH, causing amplified AI symptoms [18]. The corticosteroid of choice is usually hydrocortisone [17, 20]. Accompanying this, treatment of additional symptoms slightly differs in cases of PAI versus SAI, with PAI requiring treatment of mineralocorticoid deficiency using fludrocortisone, which will also assist with recovery of hyponatremia and hypotension [17, 20]. SAI, due to its effects on the anterior pituitary, may also result in central hypothyroidism, necessitating use of thyroid replacement therapy [17, 18]. This must be done with consideration of mitigating the risk of adrenal crisis, which involves starting corticosteroids before starting thyroid hormone replacement [17]. Monitoring therapy of SAI should involve evaluating free T4 as opposed to TSH [17]. It should be noted that AI may be a lifelong condition for most patients, however with recovery from AI symptoms and continuation of maintenance therapy, patients may safely continue with ICI therapy [17].
3.3 Diabetes mellitus
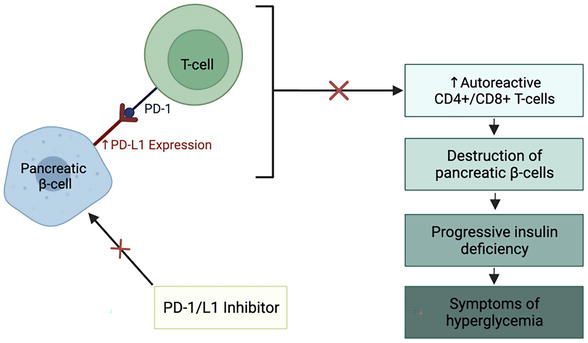
Of the documented endocrinopathies secondary to initiation of immunotherapy, pancreatic endocrine dysfunction is less common but can lead to life-threatening complications such as rapid-onset diabetic ketoacidosis (DKA) [8]. The use of immune checkpoint inhibitors, such as PD-1 and its associated ligand PD-L1, is associated with a 0.2–0.9% incidence of diabetes mellitus (DM) [21]. This includes both new-onset type 1 DM (T1DM) and worsening type-2 DM (T2DM) [8]. The pathophysiology of immunotherapy-induced pancreatic endocrine dysfunction appears to mirror the autoimmune destruction seen with T1DM but with more rapid T-cell-mediated destruction of pancreatic β-cells [8]. The exact mechanisms involved in this process are poorly understood; one pathway, describing PD-1/PD-L1 regulation of T-cell activation, provides some insight into this process and is depicted in Figure 5 [22]. This pathway has been observed and replicated in mouse models and observed indirectly in human studies involving patients with T1DM [22]. Alternative pathways involve the development of autoimmune DM antibodies and CTLA-4 regulation of T-cell activation [22]. However, half of all patients who develop DM secondary to initiation of immunotherapy are found to be autoantibody negative and expression of CTLA-4 in islet cells is unconfirmed, making these pathways more uncertain [22]. Moreover, 70% of patients who develop T1DM secondary to initiation of immunotherapy have a genetic predisposition to DM, suggesting that genetic linkage may play a role in the underlying pathophysiology [8].
Figure 5.
Proposed pathogenesis of diabetes mellitus (DM) secondary to initiation of therapy with immune checkpoint inhibitors. Under normal conditions, pancreatic β-cells express PD-L1, preventing them from being subjected to immune destruction [22]. It has been hypothesized that β-cells can attenuate the immune response in pancreatic islets through upregulation of PD-L1 expression, as seen in patients with T1DM [22]. Immune checkpoint inhibitors that target PD-1 and PD-L1 may, therefore, disrupt this process, leading to activation of autoreactive CD4+ and CD8+ T-cells, destruction of β-cells in the pancreatic islets, progressive insulin deficiency, and subsequent hyperglycemia characteristic of DM [22].
3.3.1 Clinical presentation
New-onset or worsening DM secondary to immunotherapy has been observed in many patients [7]. Clinically, patients can present with symptoms of thirst, polydipsia, polyuria, weight loss, dehydration, and lethargy [8]. Roughly two-thirds of patients can present with DKA, while the remainder suffers from severe hyperglycemia (both insulin-dependent and non-insulin-dependent) [7, 8]. The median time to onset of signs and symptoms following the initiation of immunotherapy typically occurs between 7 and 17 weeks [8]. A few cases have demonstrated that it can take years for evidence of pancreatic endocrine dysfunction to become apparent, while in others, signs and symptoms might appear sooner than 7 weeks, as shown in the Case Vignette [8, 23].
3.3.1.1 Case Vignette
A 73-year-old man with no personal history or family history of diabetes mellitus developed stage IV M1c metastatic melanoma. He was initially treated with vemurafenib, a BRAF inhibitor, and cobimetinib, a MEK inhibitor. After 3 months, his disease had progressed, and a second-line therapy with nivolumab, a PD-1 inhibitor, was initiated at 3 mg/kg every 2 weeks. After 6 weeks of treatment with nivolumab, the patient presented to the emergency department with sudden onset vomiting, abdominal pain, and severe asthenia, which were found to be associated with polyuria-polydipsia syndrome. Investigations revealed the following: glycemia: 27.78 mmol/l; bicarbonate: 18 mmol/l; urinary dipstick test positive for glucose and ketone; creatinine: 177 μmol/l; and glycated hemoglobin (HbA1c): 8.8%. These findings, considering his clinical presentation, were consistent with diabetic ketoacidosis (DKA) resulting from the onset of type 1 diabetes mellitus with acute renal failure. Insulin therapy was initiated, and nivolumab was withheld for 3.5 weeks. This was successful in normalizing his glycemia and alleviating his clinical symptoms. Subsequent laboratory investigations revealed the presence of islet cell autoantibodies (anti-glutamic acid decarboxylase antibody, zinc transporter 8 antibody, and protein phosphatase-like and insulinoma antigen-2 antibody), while C-peptide was undetectable. Retrospective investigations of frozen serum stored at 0 weeks and 3 months before initiating nivolumab treatment showed the presence of these islet cell autoantibodies, while insulin, C-peptide concentrations, and glycemia were found to be normal. The same dose of nivolumab was resumed after 3.5 weeks with no further effects on glycemia. After 3 months of treatment with nivolumab, the tumor showed a complete metabolic response on imaging. In the context of the investigations before and after initiation of treatment with nivolumab and the patient’s clinical presentation, this case report highlights the autoimmune origin of diabetes secondary to treatment with a PD-1 inhibitor in a predisposed patient.
In patients receiving therapy with immune checkpoint inhibitors and presenting with symptoms of new-onset or worsening DM (i.e. polydipsia, polyuria, nausea, and vomiting, etc.,) investigations should include testing for elevated plasma glucose, low or absent C-peptide, and mildly elevated glycated hemoglobin (HbA1c) [8, 25]. The diagnostic criteria for DM in Canada stipulate that a single laboratory test result in the diabetes range (i.e. random plasma glucose ≥11.1 mmol/L or HbA1c ≥ 6.5%) is diagnostic in the presence of symptomatic hyperglycemia [26]. Conversely, in the absence of symptomatic hyperglycemia, a single laboratory test result in the diabetes range requires repeat confirmatory testing (fasting plasma glucose, oral glucose tolerance test, and HbA1c) on a separate day to diagnose DM [26].
Detection of autoantibodies, such as anti-glutamic acid decarboxylase (GAD) and anti-islet cell (ICA), can be used to distinguish between T1DM and T2DM [8, 26]. Of note, autoantibodies are positive in nearly half of patients and may precede or coincide with their development of T1DM [8, 26]. Additionally, in the absence of autoantibodies, T1DM should not be ruled out, as autoantibody levels can vary over time and lack the diagnostic accuracy to necessitate routine use [26]. Alternative autoantibodies for the diagnosis of T1DM include zinc transporter 8, protein phosphatase-like, and insulinoma antigen-2 [24].
Imaging in diagnosing DM secondary to immunotherapy is restricted to non-specific signs, including pancreatic enlargement, pancreatic atrophy, and findings that mirror diffuse pancreatitis [22]. Furthermore, imaging for the diagnosis of DM is not recognized in Canadian diagnostic guidelines, further indicating its limited use as a diagnostic modality [26].
3.3.3 Management strategies
All patients planning to undergo immunotherapy with immune checkpoint inhibitors should be informed of their DM risk and receive education on the warning signs for hyperglycemia and DKA before initiating treatment [22]. Care providers must also be aware of this risk and pursue the necessary diagnostic tests (i.e., plasma glucose, HbA1c, ketones, blood gases, etc.) if DM or DKA is suspected based on the patient’s clinical presentation [22]. Monitoring plasma glucose and HbA1c before initiating treatment and during treatment as a follow-up should also be performed to ensure that the presentation of DM is caught early, and treatment regimens are modified accordingly [22].
In the context of DM secondary to the initiation of immunotherapy, presentation of DKA should be managed according to standard methods (i.e. fluid resuscitation, potassium repletion, etc.) and discontinuation of immunotherapy is recommended until DKA is resolved [7, 22]. Patients whose DKA has been resolved or who have new onset hyperglycemia in the absence of DKA can adhere to treatment recommendations based on their grade of hyperglycemia (as defined by the Common Terminology Criteria for Adverse Events (CTCAE) grading system) [22]. In short, for patients with grade 1 hyperglycemia, immunotherapy may be continued with close laboratory monitoring and without the initiation of insulin therapy [22]. For patients with grade 2 hyperglycemia, immunotherapy can be continued, though insulin therapy should be started to manage clinical symptoms [8, 22]. In patients whose hyperglycemia is a grade 3 or 4, immunotherapy should be withheld, and insulin therapy should be initiated to mitigate the effects of progressive pancreatic endocrine dysfunction [22]. Immunotherapy should be withheld until symptoms and laboratory values are returned to grade 1; at this point, re-administration of immunotherapeutic agents can be considered with close monitoring of blood glucose levels [22]. Moreover, for patients with worsening T2DM after starting immunotherapy, evidence suggests that immunotherapy can continue uninterrupted with additional consideration given to oral antihyperglycemic agents, lifestyle counseling, and vascular complication management [8].
Notably, treatment with glucocorticoid has the potential to worsen pancreatic endocrine dysfunction and exacerbate symptoms of hyperglycemia [7, 8]. This contrasts with the treatment of other immune-related adverse events, including hepatotoxicity and colitis, as glucocorticoids can have adverse effects on blood glucose levels and should, therefore, be avoided in the setting of DM secondary to the initiation of immunotherapy [7, 8, 22].
Finally, management should include referral to a diabetes team and a system to ensure safe and continuous access to insulin for those requiring insulin treatment [7]. Initial patient education through a diabetes specialist is also recommended [7]. DM secondary to the initiation of immunotherapy is generally irreversible, so such measures are necessary to ensure the well-being of patients with this diagnosis [22].
3.4 Hypophysitis
Hypophysitis is induced when the pituitary gland is inflamed due to varied hormone dysfunction [27]. It is a well-established irAE [27]. Variable incidence has been reported in patients treated with immune checkpoint inhibitors [27, 28]. Higher irAE incidence is associated with the male gender, older age, and higher treatment dose [27]. High incidence is also associated with high-dose anti-CTLA-4 inhibitors, ipilimumab, treatment (>3 mg) at 17%, whereas a lower incidence of up to 1% has been reported for patients treated with anti-PD-1/PD-L1 inhibitors [27].
Hypophysitis induced by anti-CTLA-4 is mediated through type II and type IV hypersensitivity and the immune system [27]. More studies are needed about the pathogenesis of anti-PD-1/PD-L1 induced hypophysitis. In contrast to anti-CTLA-4, which affects various pituitary cells equally, anti-PD-1/PD-L1-induced hypophysitis targets ACTH-secreting cells [27]. One proposed mechanism is autoimmunity against corticotrophs and ectopic expression of ACTH in tumors [27]. Preliminary studies suggest hypothalamic autoimmunity as a reason for developing pituitary dysfunction in patients treated with anti-PD-1/PD-L1 [27].
3.4.1 Clinical presentation
There is a wide range of onset times after the initiation of immune checkpoint inhibitors [27, 28]. In general, anti-CLTA-induced hypophysitis presents earlier in treatment than hypophysitis due to anti-PD-1/PD-L1 [27]. For example, in patients treated with ipilimumab, the median time to onset is 9–11 weeks with a range of 1–35 weeks, and the median time to onset for pembrolizumab is 16 weeks with a range of 1–52 weeks [27]. The presenting symptoms for immune checkpoint inhibitor-induced adverse reactions are usually not specific, which include nausea, vomiting, reduced appetite, headache, and dizziness [27, 28]. A large proportion of patients can be asymptomatic at the time of diagnosis [27, 28]. There is an overlap of symptoms with hypopituitarism and that of the underlying malignancy, which adds challenges to diagnosis [28]. Patients on anti-PD-1/PD-L1 usually present with isolated ACTH deficiency without pituitary enlargement [27]. Patients with anti-CTLA-4-induced hypophysitis have more diverse presentations, including symptoms of hypothyroidism, hypogonadism, and visual disturbances [27].
3.4.1.1 Case Vignette
A 33-year-old female diagnosed with metastatic melanoma with involvement of pelvic and inguinal adenopathy and metastasis to the adrenal glands was treated with Ipilimumab/Nivolumab combination therapy. The diagnosis was established with an inguinal lymph node biopsy and PET scan; a repeat PET scan showed the patient’s partial response to treatment. Sixty-four days after treatment initiation, the patient experienced headache, fatigue, palpitations, and joint pains. Laboratory evaluation showed TSH < 0.015mIU/mL (reference range 0.35–3.6), T4 16 mcg/dL (reference range 4.0–11.0), T3 235 ng/dL (58–160) concerning primary hyperthyroidism. Morning cortisol level was 10.8 mcg/dL (3.7–19.4) at the time, which was inconclusive for the diagnosis of adrenal insufficiency. MRI was performed due to worsening headache, which found pituitary gland enlargement with extension into the suprasellar space consistent with hypophysitis. This resulted in cessation of treatment, and the patient was treated with atenolol and prednisone 1 mg/kg/day tapered to 10 mg/day over 10 days. The patient’s symptoms had significantly improved. A repeat MRI showed that the finding associated with hypophysitis had resolved. The patient’s thyroid function returned to normal within 8 weeks. However, she was found to have adrenal insufficiency with undetectable morning cortisol during steroid taper and needed hydrocortisone replacement (15 g daily in divided doses). She was kept on nivolumab maintenance therapy after. Six years later, there was a recurrence of the melanoma, and the patient was rechallenged with Ipilimumab/nivolumab; she started to have symptoms, including headaches, nausea, and tachycardia 33 days after treatment initiation, and MRI again showed findings concerning hypophysitis. The patient was again successfully treated for hypophysitis with high-dose prednisone followed by hydrocortisone maintenance. Symptoms recurred after starting nivolumab maintenance therapy, and immunotherapy was stopped.
Diagnosis or immune checkpoint inhibitor-induced endocrinopathies can be challenging due to variability in symptom onset and overlapping signs and symptoms with adverse reactions from concomitant treatment and malignancy itself [27]. Patients should be monitored for signs of hypophysitis, including headache, visual disturbances, fatigue, gastrointestinal symptoms, and hypotension [27]. Biochemical surveillance of hormone levels is important for diagnosing endocrine adverse reactions [27]. Laboratory investigations for monitoring of pituitary toxicity include TSH, free T4, morning ACTH and cortisol, prolactin, HbA1C, IGF-1, FSH, LH, and testosterone for males and estradiol for premenopausal females [11, 27]. Repeat laboratory investigations can be conducted regularly every 4–6 weeks during immunotherapy and every 6–12 after immunotherapy completion [11, 27]. Electrolytes and fasting glucose are also helpful for diagnostic evaluation [27]. On the rare occasion of suspected diabetes insipidus, serum and urine osmolality should be obtained [23, 27]. A pituitary MRI is recommended for evaluation if there is an abnormality with laboratory investigations to assess for hypophysitis versus metastatic disease [23, 27, 28].
3.4.3 Management strategies
Treatment for hypophysitis depends on the hormone affected and toxicity grade [23]. Treatment mainly involves holding immunotherapy, starting steroids and hormone replacement therapy, close follow up and monitoring [23]. Central adrenal insufficiency is the most common [23]. Patients could also develop central hypothyroidism, diabetes insipidus, hypogonadism and growth hormone deficiency [23]. For Grade 1 (asymptomatic or mild symptoms) and Grade 2 (moderate symptoms, able to perform ADL) toxicity, clinicians may consider holding treatment until the patient is stabilized on hormone replacement therapy [25]. Treatment should be held for Grades 3 and 4 (severe symptoms, medically significant) hypophysitis until stabilization [13, 25].
For patients with primary adrenal insufficiency and hypophysitis, 15–25 mg hydrocortisone in 2–3 divided doses is recommended for hormone replacement therapy [25]. High-dose glucocorticoid (hydrocortisone 100 mg intravenously followed by 50 mg every 6 hours) should be reserved for the treatment of adrenal crisis or compression symptoms from hypophysitis and the pituitary gland [23]. Thyroid hormones should be replaced in symptomatic patients with TSH elevation or asymptomatic patients with TSH levels that persist above 10mIU/L measured 4 weeks apart [23]. TSH should be monitored every 6 weeks while titrating too normal [23]. If thyroid hormone and corticosteroids are both required, the latter should be started several days earlier to prevent precipitation of adrenal crisis [23]. Replacement of gonadal hormones is recommended in men and women of reproductive age and is not advised in the setting of a poor oncologic prognosis [23]. In hypophysitis with active malignancy, growth hormone deficiency should not be treated [23]. Desmopressin is recommended for patients with hypophysitis and confirmed diabetic insipidus [23]. Endocrinopathy may be irreversible and may result in a need for lifelong hormone replacement [23, 28]. Endocrinology consultation is recommended in all cases [23, 28].
3.5 Other (hypogonadism, hypercalcemia)
In addition to central hypogonadism, as seen in hypophysitis, rare cases of immune checkpoint inhibitor-induced primary hypogonadism have been described [29]. WHO database for safety report, VigiBase recorded 13 cases of hypogonadism, and one case was primary hypogonadism [29]. A case study reported the development of bilateral orchitis with ipilimumab/nivolumab treatment, with laboratory investigations showing primary hypogonadism; this adverse reaction resolved without treatment and did not recur with future cycles [13]. This demonstrates that there is the potential for ICI to impact fertility and should be taken into consideration when treating patients of reproductive age.
Hypercalcemia is another adverse reaction of ICI therapy, which occurs via a variety of mechanisms [30]. It could occur due to immune checkpoint inhibitor-induced hyperparathyroidism or in the setting of thyroid dysfunction or adrenal insufficiency [30]. There are reports of hypercalcemia as a result of immune checkpoint inhibitor-induced sarcoid-like granulomas [30]. Furthermore, other reports show hypercalcemia associated with increased production of PTHrP and calcitriol [30]. This is a significant adverse reaction to recognize, as it could be confused with malignancy progression.
The prognosis of various endocrinopathies secondary to initiation of immunotherapy varies depending on which endocrine organ is involved [8]. This is documented with immune checkpoint inhibitors, wherein the thyroid, pituitary, pancreas, and adrenal glands can all be adversely impacted [8]. With thyroid dysfunction, the typical progression of disease occurs within 3–6 weeks, beginning initially with symptoms of hyperthyroidism and culminating in prolonged hypothyroidism [31]. Moreover, hyperthyroidism rarely persists and does not require additional therapy [8, 31]. However, for patients who develop hypothyroidism, long-term thyroid hormone replacement might be necessary for up to two-thirds of patients [8, 31]. Interestingly, hypothyroidism could benefit tumor progression in humans, such as improved response to treatment and increased progression-free survival [31]. However, further evidence is required to help define the role of findings in routine clinical practice [31].
Regarding pituitary dysfunction and hypophysitis, secondary adrenal insufficiency is considered irreversible, and the balance between high-dose glucocorticoid therapy and continued immunotherapy needs to be carefully monitored to ensure best patient outcomes [31]. As with hypothyroidism, several pieces of evidence suggest that patients who develop hypophysitis secondary to initiation of immunotherapy have improved survival outcomes compared to patients who do not [8]. Once again, evidence to support these claims is limited, and the implications on clinical practice guidelines need to be clarified [8, 31].
Less common endocrinopathies, specifically those affecting the pancreatic and adrenal glands, are considered irreversible once they manifest with the initiation of immunotherapy [31]. Long-term management of type 1 diabetes mellitus (T1DM) and primary adrenal insufficiency consists of treatment with insulin therapy and glucocorticoid/mineral corticosteroid therapy, respectively [31]. The prognostic implications of these endocrinopathies on cancer progression and mortality are not well elucidated [8, 31]. However, diabetes mellitus and adrenal insufficiency increase the risk of serious adverse events in patients, such as diabetic ketoacidosis and adrenal crisis, which carry an increased risk of morbidity and mortality [8, 31].
Cancer immunotherapy has revolutionized the field of oncology, offering a new paradigm in cancer treatment [1]. However, endocrine adverse events (EAEs) have emerged as a significant concern [31]. Ongoing research and clinical trials are crucial in understanding the mechanisms behind these EAEs and developing predictive markers or preventive measures [31].
Current research is focused on understanding the pathophysiology of EAEs [31]. It is hypothesized that immune checkpoint inhibitors may trigger an autoimmune response against endocrine organs, leading to EAEs [31]. Clinical trials are underway to validate this hypothesis and identify the specific antigens targeted by the immune system [21]. In parallel, efforts are being made to identify predictive markers for EAEs [21]. Biomarkers such as autoantibodies, cytokines, and genetic polymorphisms are being investigated for their potential to predict the risk of EAEs [21, 31]. Early identification of patients at risk could allow for proactive management strategies, potentially preventing the onset of EAEs [21, 31]. Several potential biomarkers are being investigated for their ability to predict the risk of EAEs [1, 21].
Circulating blood counts, cytokines, autoantibodies, HLA genotypes, microRNA, gene expression profiling, and serum proteins are among the categories of biomarkers being studied [11]. For instance, certain protein markers such as albumin and thyroid-stimulating hormone are readily accessible in current practice and may serve as potential indicators of EAEs [32]. Autoantibodies are another area of interest. The presence of these antibodies may indicate an autoimmune response against endocrine organs, potentially triggered by immune checkpoint inhibitors, a class of drugs used in immunotherapy [31, 32]. This autoimmune response could lead to EAEs, making autoantibodies a potential predictive marker [32]. Cytokines, small proteins involved in cell signaling, are also being explored as potential biomarkers [32]. Changes in cytokine levels could reflect an immune response to immune checkpoint inhibitors and may be associated with the development of EAEs [32].
While the prediction of EAEs remains a major clinical challenge [21], ongoing research into these and other potential biomarkers holds promise for the early identification and proactive management of EAEs in patients undergoing cancer immunotherapy. As our understanding of these markers improves, so too will our ability to minimize the adverse effects of these revolutionary treatments. Moreover, preventive measures are being explored [31, 32]. One approach is the use of immunosuppressive agents to modulate the immune response [32]. However, this must be balanced against the risk of dampening the anti-tumor effect of immunotherapy [32]. Another strategy is the early detection and management of EAEs through regular monitoring of endocrine function [32].
In conclusion, ongoing research and clinical trials are shedding light on the mechanisms behind EAEs in cancer immunotherapy, paving the way for the development of predictive markers and preventive measures. This will undoubtedly enhance our ability to manage these adverse events, improving patient outcomes in the era of cancer immunotherapy. As we continue to unravel the complexities of the immune system and its interplay with cancer, we move closer to the goal of personalized cancer treatment, maximizing efficacy while minimizing adverse events.
Endocrinopathies are common risk factors associated with immune checkpoint inhibitors. Immunotherapy works by stimulating and altering the immune system to detect malignant cells. Most often, PD-L1 and CTLA-4 checkpoint inhibitors may result in the immune system becoming under or overactive, inducing thyroid dysfunction, Adrenal insufficiency, Diabetes Mellitus, and hypophysitis.
Thyroid dysfunction can present as thyroiditis and transition into hypothyroidism (underactive thyroid hormone production) or hyperthyroidism (overactive thyroid hormone production). Monitoring TSH, T4, and T3 (in the case of hyperthyroidism) is essential for thyroid dysfunction management. Treatment of hypothyroidism includes hormone replacement therapy using levothyroxine and beta-blockers for hyperthyroidism. Diagnostic imaging, including thyroid ultrasound and radioactive uptake imaging, can be completed for further thyroid dysfunction detection. Adrenal insufficiency occurs due to either inflammation of the anterior pituitary, as in SAI, or inability of the adrenal gland to produce mineralocorticoids and glucocorticoids, as in PAI. The incidence of the latter is rare and potentially lethal as mineralocorticoids and glucocorticoids are typically essential in the homeostasis of sodium, fluids, and energy, and derangements of these necessary hormones can cause widespread defects. Diagnosis can be made using laboratory tests such as morning draws of cortisol and ACTH, as these are usually high in the morning due to normal circadian rhythms. Morning ACTH, if elevated, is more consistent with PAI, along with decreased serum aldosterone, hyponatremia, and hyperkalemia. Imaging such as head CT and head MRI may elucidate results consistent with enlargement of the pituitary stalk or enhancement of the pituitary gland. Treatment mainly consists of oral corticosteroids, along with fludrocortisone as an additional treatment option in cases of PAI, and thyroid hormone replacement therapy initiated after some time of corticosteroid therapy to mitigate the risk of adrenal crisis. Diabetes Mellitus is less common but life-threatening, with the apparent destruction of T1DM and pancreatic insulin-secreting β-cells.
Diagnostic approaches are centred around diabetes laboratory markers and diagnostic imaging for more severe pancreatic pathologies (pancreatitis). Hypophysitis, induced by CTLA-4 target immunotherapy, results from an inflamed pituitary gland and varies in hormone dysfunction. PD-1/PD-L1-induced hypophysitis targets specific cells known as ACTH-secreting cells found in the pituitary gland. Hypophysitis often presents with generalized symptoms (nausea, vomiting, dizziness, etc.) early in treatment but may become more severe (hypothyroidism, hypogonadism, visual disturbances) when treated with anti-CTLA-4. Laboratory investigations for monitoring of pituitary toxicity include TSH, free T4, morning ACTH and cortisol, prolactin, HbA1C, IGF-1, FSH, LH, and testosterone for males and estradiol for premenopausal females. MRI examination may be completed upon abnormal lab work. The most common treatment for hypophysitis includes holding immunotherapy, beginning steroids or hormone replacement therapy and monitoring.
All management mechanisms should take into consideration patient history and physical examination. Laboratory testing and diagnostic imaging should be used to concrete a diagnosis further. Baseline laboratory should be completed prior to initiating immunotherapy. A summary of endocrinopathies is shown in Table 1.
1.Esfahani K, Roudaia L, Buhlaiga N, et al. A review of cancer immunotherapy: From the past, to the present, to the future. Current Oncology. 2020;27:87-97
2.Khan M, Maker AV, Jain S. The evolution of cancer immunotherapy. Vaccine. 2021;9:1-8. DOI: 10.3390/vaccines9060614 [Epub ahead of print]
3.Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nature Reviews Immunology. 2020;20:651-668
4.Liu C, Yang M, Zhang D, et al. Clinical cancer immunotherapy: Current progress and prospects. Frontiers in Immunology. 2022;13:5-8, 15. DOI: 10.3389/fimmu.2022.961805 [Epub ahead of print]
5.Han Y, Liu D, Li L. PD-1/PD-L1 pathway: Current researches in cancer. 2020. Available from: www.ajcr.us/
6.Nüssing S, Trapani JA, Parish IA. Revisiting T cell tolerance as a checkpoint target for cancer immunotherapy. Frontiers in Immunology. 2020;11:1-7. DOI: 10.3389/fimmu.2020.589641 [Epub ahead of print]
7.Nogueira E, Newsom-Davis T, Morganstein DL. Immunotherapy-induced endocrinopathies: Assessment, management and monitoring. Therapeutic Advances in Endocrinology and Metabolism. 2019;10:1-6. DOI: 10.1177/2042018819896182 [Epub ahead of print]
8.Hattersley R, Nana M, Lansdown AJ. Endocrine complications of immunotherapies: A review. Clinical Medicine, Journal of the Royal College of Physicians of London. 2021;21:E212-E222
9.Smith DA, Germolec DR. Introduction to immunology and autoimmunity. Environmental Health Perspectives. 1999;107(Suppl 5):661-665. DOI: 10.1289/ehp.99107s5661
10.National Cancer Institute. Immunotherapy to Treat Cancer. NCI; 2019. Available from: https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies
11.Baraka B, Abosheaishaa H, Nassar M. Immunotherapy-induced thyroid dysfunction: An updated review. The Egyptian Journal of Internal Medicine. 2023;35:1-3, 6. DOI: 10.1186/s43162-023-00210-7 [Epub ahead of print]
12.Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. American Journal of Clinical Oncology. 2016;39:98-106
13.Brunet-Possenti F, Omgloid V. Immune checkpoint inhibitors-related orchitis. Annals of Oncology. 2017;28:906-907
14.El Sabbagh R, Azar NS, Eid AA, et al. Thyroid dysfunctions due to immune checkpoint inhibitors: A review. International Journal of General Medicine. 2020;13:1003-1009
15.Brahmer JR, Abu-Sbeih H, Ascierto PA, et al. Society for immunotherapy of cancer (sitc) clinical practice guideline on immune checkpoint inhibitor-related adverse events. Journal for ImmunoTherapy of Cancer. 2021;9:1-4, 14-15. DOI: 10.1136/jitc-2021-002435 [Epub ahead of print]
16.Gavrila A. Clinical Thyroidology ® for the Public Development of thyroid problems with immunotherapy drugs for certain cancers is associated with favorable survival. 2020. Available from: https://www.thyroid.org/hypothyroidism/
17.Cherry G. Immune checkpoint inhibitor-related adrenal insufficiency. Seminars in Oncology Nursing. 2021;37(2):1-6. DOI: 10.1016/j.soncn.2021.151131
18.Cui K, Wang Z, Zhang Q , et al. Immune checkpoint inhibitors and adrenal insufficiency: A large-sample case series study. Annals of Translational Medicine. 2022;10:251-251
19.Martella S, Lucas M, Porcu M, et al. Primary adrenal insufficiency induced by immune checkpoint inhibitors: Biological, clinical, and radiological aspects. Seminars in Oncology. 2023;22(52):2-4. DOI: 10.1053/j.seminoncol.2023.11.003 [Epub ahead of print]
20.Salinas C, Renner A, Rojas C, et al. Primary adrenal insufficiency during immune checkpoint inhibitor treatment: Case reports and review of the literature. Case Reports in Oncology. 2020;13:621-626
21.Kennedy R, Awada H, Vura N, et al. Endocrinopathies from checkpoint inhibitors: Incidence, outcomes, and management. Cleveland Clinic Journal of Medicine. 2023;90:307-317
22.Zheng Z, Liu Y, Yang J, et al. Diabetes mellitus induced by immune checkpoint inhibitors. Diabetes Metabolism Research and Reviews. 2021;37(1):2-6. DOI: 10.1002/dmrr.3366
23.Husebye ES, Castinetti F, Criseno S, et al. Endocrine-related adverse conditions in patients receiving immune checkpoint inhibition: An ESE clinical practice guideline. European Journal of Endocrinology. 2022;187:G1-G21
24.Gauci ML, Laly P, Vidal-Trecan T, et al. Autoimmune diabetes induced by PD-1 inhibitor—Retrospective analysis and pathogenesis: A case report and literature review. Cancer Immunology, Immunotherapy. 2017;66:1399-1410
25.Brahmer JR, Lacchetti C, Schneider BJ, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American society of clinical oncology clinical practice guideline. Journal of Clinical Oncology. 2018;36:1714-1768
26.Punthakee Z, Goldenberg R, Katz P. Definition, classification and diagnosis of diabetes, prediabetes and metabolic syndrome. Canadian Journal of Diabetes. 2018;42:S10-S15
27.Prete A, Salvatori R. Hypophysitis. In: Feingold KR, Anawalt B, Blackman MR, et al, editors. Endotext. 2021
28.Park BC, Jung S, Wright JJ, et al. Recurrence of Hypophysitis after immune checkpoint inhibitor Rechallenge. The Oncologist. 2022;27:E967-E969
29.Özdemir BC. Immune checkpoint inhibitor-related hypogonadism and infertility: A neglected issue in immuno-oncology. Journal for ImmunoTherapy of Cancer. 2021;9:1-3. DOI: 10.1136/jitc-2020-002220 [Epub ahead of print]
31.De Filette J, Andreescu CE, Cools F, et al. A systematic review and meta-analysis of endocrine-related adverse events associated with immune checkpoint inhibitors. Hormone and Metabolic Research. 2019;51:145-156
32.Chennamadhavuni A, Abushahin L, Jin N, et al. Risk factors and biomarkers for immune-related adverse events: A practical guide to identifying high-risk patients and rechallenging immune checkpoint inhibitors. Frontiers in Immunology. 2022;13:2-4, 9. DOI: 10.3389/fimmu.2022.779691 [Epub ahead of print]
Written By
Olexiy Aseyev, Alesha Bishop, Hannah Shortreed, Elycia Monaghan and Yue Sun
Submitted: 31 December 2023Reviewed: 08 January 2024Published: 29 March 2024