
Characterization of cryptic shock [91].
Abstract
Patients with progressing hemorrhagic shock (HS) die rapidly of cardiac arrest by insufficient venous return or within days by second-hit multiple organ dysfunction/failure (MOD/MOF). Once earliest source control has been effectuated and macro-hemodynamics is normalized, only three variables affect mortality, namely microcirculation, temperature, and oxygen. Late, non-immediate, mortality is usually preceded by a period of cryptic shock, essentially a disease of microcirculation in the midst of a seemingly functional macrocirculation. The persistence of the effects of ischemia-reperfusion toxemia (IRT) underlying a subclinical cryptic shock is the fundamental pathogenetic factors for clinical observed second hit deterioration. Rewarming a hypothermic hemorrhagic patient and administration of supplementary high dosages of oxygen are standard practices for the management of hemorrhagic shock in acute phase. A complete shift of paradigm prospects an answer to the above tactics’ limitations, drawbacks, and contra-indications. Mild-to-moderate hypothermia, titrated supplementary oxygen, and timely-given vasodilators should instead be given during damage control surgery (DCS) for advanced shock with the aim of reducing ischemia repercussion injury (IRI) e microcirculation dysfunction. A new strategy is suggested: preoperative hypothermia and titrated oxygen before hemorrhage source control, and intra-operative vasodilation and anti-inflammatory tactics after source control.
Keywords
- hypothermia
- oxygen
- vasodilation
- cryptic shock
- acidosis
- ischemia-reperfusion
- MODS/MOF
1. Introduction
1.1 Changing the paradigms
Once earliest source control has been effectuated and macro-hemodynamics is normalized, only three variables affect mortality, namely microcirculation, temperature, and oxygen.
Microcirculation optimization is crucial for prognosis of HS; hypothermia is still seen as an enemy and high flow oxygen advised in advanced HS till recently.
Can the interaction between the three variables be manipulated for improving prognosis of advanced unstable progressing HS?
Advertisement
2. Hypothermia in hemorrhagic shock
In hemorrhagic shock, re-warming the patient with normothermic/warm fluids and devices like the Bair Hugger is standard practice, assumedly and principally to prevent accidental hypothermia from blood loss, general anesthesia, and environmental exposure of open cavities and cold fluids [2].
Spontaneous hypothermia in hemorrhagic shock is not an independent risk factor for complications or mortality, which is correlated rather with the severity of the injury [3, 4].
Surely warming makes the patient more comfortable and prevents or stops the shivering accompanying hypothermia [5, 6], but is this practice really worthy, not to say safe [7]?
Cold increases α-adrenergic vasoconstrictive response; likewise heat increases beta-agonist response, with changes that are independent from the activity of metabolizing enzymes affected by temperature [8].
Hypothermia decreases blood flow to all organs of the body in different manner, the skeletal muscle and extremities being the most sensitive to the reduction, followed by the other internal organs, the lungs as the last. The metabolism of all drugs, in particular opioids and muscle relaxants, is slowed, and the lower is the temperature the lower and more distanced dosages are required [9].
The increase or normalization of temperature neutralizes the initial compensatory vasoconstrictive phase conveniently contributed by hypothermia, predisposing to a faster passage to a decompensated phase; moreover, hypothermia reduces the amount of fluids necessary to maintain pressure following vasoconstriction [10]. Since Hippocrates time and in World War I, it was noted that shocked patients had a higher survival when exposed to cold rather than warm environment, due to the vasoconstriction effect of cold [11].
The rationale for preventing or delaying the onset of hypothermia-related coagulopathy [12] is rather weak argument when compared to the beneficial effects on hemodynamics and metabolism in the overall computation. Hypothermia-induced coagulopathy is clinically significant at around 32°C, indicating a terminal stage of shock [5], while coagulopathy in early stage is determined by hypoxia and trauma itself, compounded by iatrogenic intervention and dilution of coagulation factors [13, 14]. Even at lower deep levels, coagulopathy remains a reversible phenomenon. In experiments on animals, spontaneous mild hypothermia does not affect coagulation in a model of controlled hemorrhagic shock [15] as predictably instead does hypothermia induced with intravenous cold solutions in uncontrolled hemorrhage models, where dilution of coagulations factors and the increased volume-driven pressures of the extra fluids become a contributing factor [16].
There is overwhelming convincing experimental evidence in animals of different size, proving the increased survival with induced intravenous and surface hypothermia compared to normothermia. This is not achieved without problems with both methods. Surface hypothermia is rather uncomfortable and intravenous fluids induce dilution coagulopathy, decreased DO2 due to Hb curve shift to left, and must be accompanied by pharmacological control of shivering. Another drawback of surface cooling is that may accelerate death during severe hemorrhage shock because it causes reflex vasoconstriction that increases arterial pressure and bleeding speed before cooling down metabolism, which preserve cell functions [17]. For all these reasons, the systemic intravenous not-hematic fluids and surface hypothermia have been abandoned. The basic benefits of hypothermia in respect to normothermia were otherwise evident.
The experimental models of controlled hemorrhagic shock in small and big animals have confirmed the historical observations in war surgery and, ominously, the indisputable fact that hypothermia supports arterial pressure, decreases heart rate and oxygen consumption, and prevents lactate levels’ rising [18, 19, 20, 21]. It appears also to be in prospective more advantageous in advanced, uncontrolled hemorrhagic shock stages [21].
Very few studies have looked into the relation between hypothermia and hemorrhage in humans, on the claim coagulation increases bleeding by interfering with coagulation.
There is no evidence that mild hypothermia-induced coagulopathy increases mortality by increasing the rate of bleeding in elective surgery with patients kept at 36–34°C [22, 23] or in trauma patients [24], nor is evidence it increases mortality as early coagulopathy or as late coagulopathy [25, 26].
It can be postulated that the increased bleeding in the initial stages of hypothermia is consequence of vasoconstriction increasing pressure, hence bleeding rate speed, whereas in the late stages, both the vasoconstricting effect prevailing on the fading hemodynamic pump pressures and the positive effect on metabolism actually decrease mortality by preventing, slowing, or blocking further blood loss and maintaining cellular metabolism to a minimum.
A limit of the experimental studies lies in extrapolating conclusions from results obtained in laboratory on rats, dogs, and pigs, who have, respectively, much less and more fat mass than average humans, a different response to catecholamines, and a different surface-to-volume ratio. Finally, the cognitive functions tested after extracorporeal circulation are incomparable with the ones in humans [21, 27].
The main reason for the bad reputation of hypothermia in hemorrhagic shock has been the confusion between the early dynamics of spontaneous accidental hypothermia and the late metabolic hypothermia of advanced shock, between the various causes of hypothermia, and between early and late hypothermia.
Accidental, environmental, hypothermia respond to external rewarming; metabolic and cellular hypothermia does not, and only oxygenation with restoration of the capacity to produce adequate adenosine triphosphate (ATP) can reverse the latter.
2.1 Cellular effects of hypothermia
2.1.1 Protection from ischemia
ATP preservation is the essential life-preserving mechanism. ATP content is decreased in hemorrhagic shock but is regenerated once blood flow is restored in time. Cellular function remains viable up to a limit (warm ischemia period) depending on mitochondria function and up to a certain point can be re-established with restoration of perfusion [28, 29, 30].
Hypothermia diminishes the oxygen demand of the body (6–7% per 1°C cooling), protecting the most oxygen-dependent organs of the body, brain, and heart, against hypoxic damage [31]. The drop in body temperature in response to hypoxia, due to energy depletion, and a drop in heat production, unlike the ventilatory response to hypoxia, do not depend on the activation of peripheral chemoreceptors. This is the cause of metabolic hypothermia occurring in advanced HS [32].
Preservation of adenosine triphosphate (ATP) and glycogen stores in the ischemic myocardium areas, and of creatine phosphate in the non-ischemic areas appear to be as the determinant mechanisms for the benefit of hypothermia in ischemic myocardium [33, 34].
In brain cells, hypothermia reduces basal cellular metabolism by reducing oxygen uptake and consumption, with consequent shift of Hb-O2 dissociation curve to the left and preservation of ATP content in all cells by stopping its breakdown [35, 36] and at some extent lactate and acidosis increase [37].
Ischemic neurons release neuroexcitory amines, especially glutamate, which in turn activates N-methyl-D-aspartate (NMDA) channels; once the channels are activated, calcium enters the cells and accumulates activating multiple destructive enzymes, with result of neuronal death. Hypothermia reduces the release of glutamate and another neuro-transmitter glycine needed for the NMDA receptor activation and prevents calcium entry restricting membrane permeability [38, 39, 40], so preventing cell death [41]. The reduction of calcium and lactate pathways accounts for the cell preserving effect of hypothermia in the brain.
Advertisement
3. Protection against ischemia-reperfusion
Hypothermia creates ischemic tolerance by several mechanisms: metabolic depression, reduced need for oxygen and energy, blundered immune and inflammatory response, and increased cell membranes tightening. Even more important is its protection from ischemia-reperfusion injury [42, 43].
The protective of hypothermia, whether induced or spontaneous, on I/R injury in the heart bases on the protection mitochondrial permeability transition pores, reduction of calcium overload, regulation of cellular signaling (Akt pathways, heat-shock proteins, extracellular-regulated kinase, etc.), and an overall reduction of the inflammatory response.
These events were observed when hypothermia was induced during the ischemic episode and disappeared after reperfusion [44, 45], hence the crucial importance of installing any form of therapeutic hypothermia before reperfusion.
This effect of hypothermia is timing/speed of onset-dependent. If hypothermia is present before ischemia installs can be life saving, otherwise it is not beneficial and may actually accelerate
Systemically induced therapeutical hypothermia (TH) at a core body temperature of 33–35°C has been shown to be beneficial not only for neonates with ischemic encephalopathy but also for comatose adults post out-of-hospital cardiac arrest with both shockable and non-shockable rhythms [54, 55, 56, 57]. Acute brain damage from ischemia-reperfusion may further lead to other distal organ IRI [58].
Current clinical practice to prevent IRD with systemic therapeutic hypothermia involves whole-body cooling at core temperatures not below 34°C, threshold that can lead to severe complications [59, 60, 61].
Therapeutic mild hypothermia (≤34°C) reduces neurologic disability and cerebral palsy in neonatal hypoxic encephalopathy without statistically significant reductions in mortality during the neonatal period, infancy or later childhood, or in seizures at any age. Mortality at 18–24 months was though more reduced in high-income countries and not in low-income ones. The treatment should be instituted in term and late preterm infants with moderate-to-severe hypoxic ischemic encephalopathy, initiated early (within 180–360 min), and be protracted ≥48 hours [62].
This drawback stimulated the development of specialized storage solutions, integrating the hypothermic effect [64, 65].
Cellular swelling from intracellular edema at deep hypothermic levels can be explained by the temperature-dependent decrease in the activity of the Na/K-ATPase pump causing an increase in cytosolic Na + concentration and subsequent water retention. The increase of iron, ROS, and cytosol Ca++ in the first few hours of hypothermia accounts for the swelling [66, 67].
Survival in elective surgery on non-cardiovascular patients has been reported with induced temperatures down to around 5°C, around 10°C for cardiac surgery patients [68].
3.1 Clinical effects
The beneficial effects of hypothermia on heart oxygen consumption are more, the sooner the wanted temperature is reached [69].
Following the experience with deep hypothermia in thoracic aorta, a temperature target of 20°C can be seen as a safe limit for both heart and brain safety in models of extracorporeal circulation for cardiac arrest by exsanguination [70, 71]: It is safer and faster to reach than the 10°C reached with experimental big animals at a speed of 2°C/min [72].
Two-thirds of patients with trauma present with mild hypothermia, and a 10% with moderate one [73]; deep hypothermia <32°C occurs in a quarter of hypotensive shocks.
The 32°C temperature is the threshold below which significant coagulopathy occurs [5, 12, 15].
Below 26°C a rise of systemic and pulmonary vascular resistance can be noted [74].
Below 29°C dysrhythmias appear; below 24°C CA is a rule; no survival is normally the rule below 9–10°C of core temperature of spontaneous hypothermia [75].
Hypothermia decreases blood flow to all organs of the body in different manner, the skeletal muscle and extremities being the most sensitive to the reduction, followed by the other internal organs, the lungs as the last. The metabolism of all drugs, in particular opioids and muscle relaxants, is slowed so lower and more distanced doses are required, the more the lower is the temperature [9].
In ischemic hearts during mild hypothermia, the heart rate decreases while cardiac contractility is preserved, thus reducing myocardial work and oxygen consumption; the cardiac output is decreased, but the stroke volume and blood pressure are maintained.
In normal hearts, the increase in vascular resistance is caused by the increase in catecholamines release yielding to an increase of cardiac output, oxygen demand, tachycardia, and tachypnea.
With advancing hypothermia and concomitant slowing of metabolism cardiac, the cardiac output and oxygen demand decrease. Despite the protective role of hypothermia on tissue survival, profound and prolonged cooling eventually leads to circulatory failure by a direct effect on coronaries and microcirculation.
Hypothermia at 32°C in healthy coronary arteries slows coronary blood flow and increases microvascular resistance; furthermore, by increasing NO release, it exacerbates endothelium-dependent vasodilatory response. The alteration of the NO endothelium-dependent vasodilatory response makes patients with chronic ischemic heart disease and chronic heart failure prone to ischemic damage [76].
With the deepening of hypothermia, the heart slows down pumping, dysrhythmias with high risk of cardiac arrest (CA) are common with hypothermia at ≤29°C, and dysrhythmic CA with very few anecdotal exceptions is a rule <24°C. Dysrhythmias and coronary vasoconstriction is what causes CA in accidental hypothermia.
The microcirculation stagnation (no flow phenomenon) and the hemoconcentration (decreased plasma volume), due to a leakage of plasma in capillaries, cause an increase of blood viscosity and concentration and aggregation of RBC with rouleaux formation in the microcirculation, in the end contributing with the effect on the coronaries and/or myocardium to the patient’s demise.
3.2 Drawbacks vs. benefits of therapeutic hypothermia
The collapse is fatal if left untreated, but it can be buffered temporarily by interventions aimed at increasing peripheral resistance [79, 80]. It occurs only during rewarming in accidental hypothermia and does not occur during rewarming and weaning of the CPB appears.
Timing of temperature manipulation is crucial for its benefits, as diriment is the synchronous reading of shock progression and dynamics and the distinction of the cause of hypothermia.
Moreover, systemic hypothermia with CPB is the major contributing factor to prevent or control systemic IRI [83].
The only study on the effects of hypothermia on microcirculation confirms that mild hypothermia at 34°C either protects or does not affect microcirculation. In an experiment on sheep, one group was kept in normothermia and another sent in hypothermia at 34°C with gastric circulation of cold water, both were made to bleed in a controlled volume hemorrhage. Microcirculation parameters were measured at hemodynamic stability and shock: Cortical renal, intestinal villi, and sublingual microcirculation were assessed with incident dark field illumination (IDF) video-microscopy; intestinal and renal blood flows were measured by an ultrasonic flowmeter, and mucosal PCO2 was measured by air tonometry. Mild hypothermia does not worsen the microcirculatory derangements induced by hemorrhagic shock in the three most hit beds, with peritubular capillaries most sensitive to changes of regional and tissue perfusion than intestinal and sublingual beds [84].
Advertisement
4. Oxygen and hemorrhagic shock
The administration of oxygen at normobaric levels
It is commonly assumed that 100% oxygen,
There is no evidence of increased survival or of benefits with concentrations at a FiO2 > 0.4–0.5 in an uncontrolled hemorrhagic shock before source control, contrarily to moderate or mild hypothermia [85, 86, 87] or in controlled shock without hypoxemia [88].
As a matter of fact, hyperoxia has the opposite effect: The ischemic CNS response and the chemoreceptor response get in fact neutralized by hyperoxia in uncontrolled hemorrhage before or without source control, especially if a blood transfusion “giving a lift to that oxygen” is also running.
This might be as important reason for the better results of the “scoop and run” policy in humans and the scarce or no interference with hemodynamics before source control [89].
Oxygen in those scenarios is often given empirically without endotracheal intubation with masks with an average FiO2 of ≤0.6, therefore not affecting significantly PaO2 at the extent of neutralizing the two lifesaving reflexes.
In decompensated hypotensive shock and in absence of post-traumatic respiratory failure not requiring intercostal drain, for example, severe pulmonary contusion, patients should initially be kept at concentrations not higher than 40% FiO2. Subsequently, 10% increases titrated to a SaO2 of 90-94% can be effected according to arterial blood gases.
Even in a fainted patient—fainting
Total intravenous anesthesia with solely ketamine has in fact been safely done on not hemorrhagic or shocked patients and on patients with severe hemorrhages on spontaneous ventilation, breathing only oxygen-air, with no intubation or sophisticated monitoring other than clinical observations (pers. obs.).
Preoxygenation with 100% O2 is only advised by several anesthetic societies to provide enough time during endotracheal intubation and prevent periods of hypoxia potentially occurring like difficult airway scenarios at expenses of temporary absorption atelectasis. It is also advised in CA resuscitation until ROSC.
Advertisement
5. Cryptic hemorrhagic shock
5.1 Ischemia-reperfusion and systemic inflammatory response
Ischemia-reperfusion phenomenon represents the continuation, local reverberation, and systemic amplification of the effects of ischemia to tissues.
Acute ischemia yields to nitrosative-oxidative stress with accumulation of by-products such as reactive oxygen and nitrogen species (ROS/RNS) that disrupts the mitochondria’s enzymatic pathways and membranes, provoking an unduly intracellular accumulation of calcium, eventually leading to cell death
The stressed or dying cell itself, when reperfused, triggers an accentuation of local toxicity by calling inflammatory cells, which reverberate further damage and other cells’ death. With the restoration of the circulation, toxic and inflammatory mediators are spread to remote organs [91, 92]. Preferred targets of the toxemia are the lung, the brain, the heart, the liver, and the kidney, due to their microcirculation peculiar anatomical structure.
The inflammatory mediators released as a consequence of reperfusion appear also to activate endothelial cells in remote organs that are not exposed to the initial ischemic insult. This distant response to I/R can result in leukocyte-dependent microvascular injury that is characteristic of the multiple organ dysfunction syndrome [93].
The impaired endothelium-dependent dilation in arterioles enhanced fluid filtration and leukocyte plugging in capillaries, and the trafficking of leukocytes and plasma protein extravasation in postcapillary venules.
The level of hypotension is a major determinant of the systemic inflammatory response arising during hemorrhagic shock [94]. The experimental works of Douzinas have proven the nexus between the level of ischemia and the IR-triggered inflammatory response, after previous indirect studies had correlated the level and duration of ischemia with the level of the inflammatory response [95, 96].
The phenomenon is very similar to a crush injury evolving in crush syndrome or to the toxemia and inflammatory shock following an ANP [91, 97, 98].
The gut is normally resistant to ischemia [27] but when is hit, as for its permeability, it acts as a rebound platform for a systemic spread of toxins, necrotic or inflammatory factors, and bacteria translocation even in the absence of sepsis. In a systemic IR toxemia (IRT), like the one induced by a HS, the gut is a formidable multiplier of toxemia augmented with the local bacteria translocation in addition to the inflammatory and toxic cascade. Furthermore, intestinal I/R increases luminal epithelial permeability yielding to ingress of bacteria and exit of bacteria and enterotoxins in the circulation, which can result in sepsis and multiple organ failure [99, 100].
An analogue situation occurs when the primary ischemic site is the gut itself such as in acute mesenteric ischemia or a gangrenous colon volvulus [101].
Another organ with highly permeable microcirculation, hit in any inflammatory, septic to toxic shock is the lung.
Endotoxemia in the absence of infection predisposes to infections in distant organs in the first postoperative week.
The IR local injury and systemic toxemia is not the only side effect of late inadequately treated ischemia.
A direct blunt trauma if significant or massive can give a post-traumatic inflammatory response (PTIR) of a SIR sepsis-driven, with the same endotheliopathy as underlying main pathophysiological mechanism [93, 98].
A master review describes the molecular intracellular damages induced by PTIR or post-trauma SIR after blunt trauma [102].
A reverberation and persistence of inflammatory response and endothelial dysfunction of arterioles is the underlying pathophysiological mechanism triggering a cascade of events leading to death in the first week or so.
No
The resulting in increased microcirculation permeability with localized and distant organ fluid loss into the interstitial space, cytokine systemic storm and an inflammatory cascade reactions that can lead to reversible or irreversible end-organ dysfunction. Impaired endothelium-dependent dilation in arterioles, enhanced fluid and protein extravasation, platelets and leukocyte plugging in capillaries account for the clinical effects. At microcirculation capillary level, extravascular fluid hampers the transport of oxygen and decreases substrates availability, affecting all energetic processes including would dealing [102]. The reverberation between intracellular, local, regional, and systemic damage by specific intracellular and external molecules, activated in a trauma, especially a blunt one, and acting as intermediary and messengers for the SIR fuse, nonetheless the involvement
5.2 What is cryptic shock?
The persistence of the above inflammatory, immunosuppressive, toxic, and catabolic dynamics is the reason for late, non-immediate, mortality.
Mortality after the first 6–24 hours is related to the speed and efficacy of source control, and in the first week to first month, mortality is MOD/F driven with or without sepsis [103].
The commonly used term “second hit mortality” is valid and acceptable if referred only to the timing peak of mortality. The underlying pathophysiological mechanisms are in fact a continuum [91, 98, 104, 105].
Cryptic (subclinical, persisting, silent, latent, unresolved, insidious, and refractory) hemorrhagic shock (CHS) ensues is an untreated or inadequately or late treated shock [91], carrying a 50–60% mortality [106, 107].
CHS is essentially a disease of microcirculation, where a normal macrocirculation, CaO2, and DO2 do not guarantee adequate VO2 after source control or source elimination.
A situation of subclinical shock that will abut in MOD/F is then present and should be prevented, identified, and managed early before evolves
Monitoring of the solely macro-hemodynamics variables may lack sufficient predictive value on the evolution of a critical patient. Often in ICU macro-hemodynamics variables are seen within normal range; nevertheless, patients still deteriorate and die all of a sudden, hit by a rapid onset multiple organ dysfunction/failure (MODS/MOF) despite reassuring values [91, 106]. It is what kills in ICU patients with normal macro-hemodynamic variables.
When macro- and microcirculation are in evident dys-synchrony [108, 109], it is essential to address the crucial role of the microcirculation/tissues interaction and hence restore physiological levels of CaO2, DO2, and VO2.
Despite restoration of the macrocirculation, the sublingual microcirculation is seen impaired for at least 72 hours in hemorrhagic shock [107].
5.3 Characterization of cryptic hemorrhagic shock
Whereas in a septic or toxic shock the persisting anomaly is the persistence of sepsis factors, mediators or by-products and in a toxic shock is the underlying systemic presence of toxic and necrosis by-products [93, 98]; cryptic hemorrhagic shock is characterized by the presence of an underlying persisting ischemia-reperfusion toxemia (IRT), persisting acidosis and a dysfunctional microcirculation, and endotheliopathy in advanced stages, when the arterial gate system suffers itself of hypoxia (Table 1) [27, 91, 93].
| • Trend towards tachycardia, not explained otherwise |
| • MODS/MOF with normal macro-haemodynamics |
| • Presence of inflammatory or toxic mediators in the blood |
| • ↓SvO2/ScvO2 < 65–70% |
| • NBE > 4 mmol/L |
| • Lactic acid >4 mmol/L |
| • Microcirculation imaging (spread hetereogenity of capillaries, reduced capillary density,microcirculatory flow reduction, tissue edema). |
Table 1.
Acidosis, a direct function of ischemia/hypoxia/hypoperfusion, is monitored with the rising levels, in temporal order, of NBE, LA, and pH [91, 110, 111]. Whereas ischemia-reperfusion phenomenon depends on the entity and duration of tissues relative ischemia before the index operation, acidosis is a direct function of hypoxia or infection or indicates the presence of a ischemia-reperfusion toxemia.
Different abnormalities can be observed with direct visualization of the sublingual microcirculation with hand-held vital microscopes or HVMs [orthogonal polarization spectral (OPS), sidestream dark field (SDF), and incident dark field (IDF)] in a situation of cryptic shock: spread heterogeneity of capillaries, reduced capillary density due to hemodilution and anemia, microcirculatory flow reduction due to vasoconstriction or micro-tamponade, and tissue edema [112, 113].
Other methods, such as near-infrared spectroscopy (NIRS) sublingual capnometry and cerebral oxygen saturation and skeletal muscles oxygenation, have also been used in ICU to assess microcirculation in hemorrhage, cardiac surgery, and trauma [114, 115, 116, 117, 118, 119, 120].
NADPH fluorescence levels are the nearest method to measure visually tissue oxygen content [121].
Advertisement
6. Prevention of second hit mortality
To prevent or limit the ischemia-reperfusion toxemia, to prevent and buffer the effects of endotheliopathy in the arteriolar system, and to adjust amount of oxygen entering the life units [98] is essential management for preventing the second hit MOD/F morbidity and mortality.
Management, therefore, must be multidirectional.
While acidosis is managed by reversing hypoxemia and rapidly controlling or eliminating the sources (stop hemorrhage in a hemorrhagic shock and remove all infected necrotic ischemic gangrenous contaminated tissues/fluids in septic shock/inflammatory shock), or optimize cardiac output (cardiogenic shock), and only at extreme levels require bicarbonate buffering, IR toxemia dynamics allow space for therapeutical strategies and manipulations in pre- and post-source control and in postoperative period.
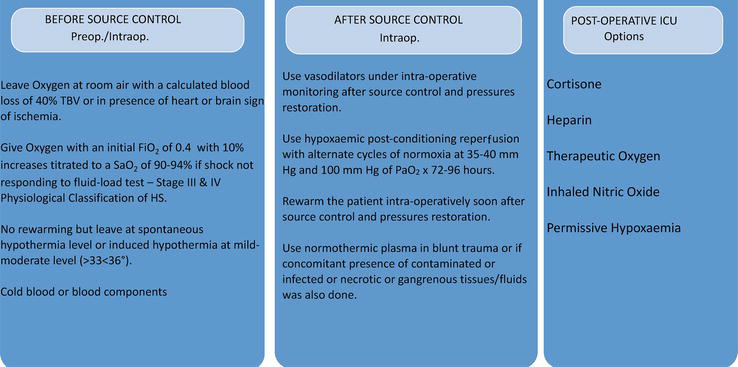
The strategy proposed to prevent second hit IRT MODS/F can be summarized in the following: (i) the earliest or an early timed surgery preventing early mortality and the incidence and level of a second hit MOD/F; (ii) appropriate application of damage control surgery (DCS) (Table 2); (iii) awareness in postoperative phase of the signs characterizing the status of a cryptic subclinical shock heralding the second hit; (iv) exploiting the benefits of hypothermia preoperatively (cold blood and blood components (plasma, RBC, platelets, and no reheating)), and shivering control, (v) hypoxemic or normoxemic reperfusion post-DCS; (vi) plasma in the presence of SIR syndrome or post-traumatic inflammatory response,
| Time from from injury to source control > 90-120 min in patients with haemodynamic instability |
| i Fluid-resistant hypotension or temporary (20-30 min) response to 500-1000 ml rapid infusion elicited after 2 hours from onset of a haemorrhage |
| ii in presence of signs of cardiac or brain ischemia |
| i ScvO2 < 65–70%; (ii) NBE > 6–8 mmol/L; (iii) pH < 7.2 - (values in trends). |
| i Convenience for the surgeon before transfer, ii) Unreachable bleeding, e.g. terminal ICA, high IJV, sacral vessels, retro-hepatic veins iii) Intra-Operative Hypotension, iv) Operation lasting more than 60 min on patients hemodynamically unstable before operation, v) co-presence of fecal peritonitis or of gangrenous tissues/fluids anywhere in the body, vi) tension or gap between gut and abdominal wall margins, vii) high risk of anastomosis/abdominal wall healing failure (edematous, ischemic, congested, cyanotic, not oozing margins). |
| THAI (timing, hypotension, acidosis, intra-operative) criteria |
Once earliest source control has been effectuated and macro-hemodynamics is normalized, only three variables affect mortality, namely temperature, oxygen, and microcirculation.
6.1 Managing microcirculation
While upstream circulation derangement’ correction is effected with an early or the earliest intervention of source control and venous return optimization, it is the effect of the hypoxemia distally to the tissues that determines the prognosis [123, 124].
In a progressive shock, microcirculation is put under distress and decompensates at three different moments.
First, when Crit-HbO2 level is reached, bleeding source/s control and blood replenishment or judicious blood component restoration have not yet been accomplished.
Second, being HS, likewise CS, a vasoconstricting shock with the arteriolar reflexes system aiming to keep blood in the macrocirculation at expenses of microcirculation and tissue perfusion, when exogenous vasoconstrictors are used in the situation of a progressing hypotensive shock not responding to fluids during THR Stage III, in a phase of arterioles unresponsiveness Stage IV, or postoperatively in ICU, more distal hypoxia occurs [27, 110, 125].
Third, when circulation all of a sudden is restored and the nitrosative/oxidative stress products are enhanced and reverberated locally, and amplified systemically with damage to other organs, provided with specific special microcirculation, namely lung, liver, and kidney—the ischemia–-reperfusion injury (IRI), which can become systemic ischemia-reperfusion toxemia (IRT), leading to the second hit of MODS/F.
6.2 Vasodilatation
In late phase septic shock, when vasoconstriction takes over the initial vasodilation [93], NO donors protect also against hypothermia and hypoxia/acidosis [126].
Nitroglycerin decreases systemic vascular resistance (SVR), heart work, and oxygen consumption in severe heart failure and cardiogenic shock, a vasoconstricting situation [127].
In experimental studies on small animals, low dose nitrites and other extrinsic NO donors in low doses (extrinsic acetylcholine, sodium nitroprusside, and nitroglycerin) have also shown to enhance microcirculation flow and perfusion, reverse arteriolar vasoconstriction, and increase capillary perfusion and venous return, improving central cardiac function and prevented further tissue ischemia, when administered in early, reversible, and vasoconstrictive phase of hemorrhagic shock and detrimental in late stages [128, 129, 130, 131].
This is interpreted with the major chances of temporary timed benefits, when the arteriolar system is not in advanced hypoxemic derangement. Experimental studies were done also with animals filled with fluid after control hemorrhage and did not study a situation of pre- and post-source control. In the author’s view, source control is the cut off for a rational use in humans, after restoration of pressures.
The timing of administration is therefore the key factor for a beneficial usage of vasodilators in HS.
Understanding the hemodynamics of shock (vasodilator type the septic shock in the initial phases, vasoconstrictor types the hemorrhagic, the cardiogenic and the advanced septic shock) [93] is diriment for a safe use of vasodilators.
NO donors can be administered intra-operatively, after source control and with an optimal venous return and SAP of ≥90 and MAP of ≥60 mmHg, under continuous real-time monitoring.
No hypotension should be allowed to occur intra-operatively or postoperatively.
Dosages should be titrated to maintain mean an arterial pressure (MAP) > 65–70 mm Hg under real-time continuous monitoring and a detectable response in the microcirculation. The safety of VD in SS and CS points toward a benefit in HS.
CS and HS are microcirculation vasoconstriction shocks, due to the occurring reflex response, whereas SS is vasoconstricting only un advanced stages, being a vasodilating in the initial reversible stages as for its direct effect on endothelium [93].
Intravenous infusion of vasodilators in physiological conditions has proved safe and generally beneficial in all vasoconstricting shocks, namely advanced septic shock and cardiogenic shock. In HS, intra-operative iatrogenic vasodilation soon after source control is a therefore potentially beneficial, despite a not clear mechanism.
A potential benefit of systemic infusion of nitrites or NO donors is the protective effect on patients with coronary artery disease by counteracting the vasoconstriction effect of drugs (
The benefits of a systemic vasodilatation with nitrites and NO donors have also been confirmed by NO inhalation, with its anti-inflammatory effect in preventing and counteracting the IRI to the lung, manifesting in postoperative period with secondary acute lung injury by vasoconstriction or by I-R from splanchnic organs such as gut and liver by NO [132, 133, 134].
A reduction of I-R damage in the liver from any etiology, whether primarily inflammatory or secondary hemorrhagic with NO eNOS-produced has also been noted [135].
The timing of administration with beneficial effects of iNOS inhibitors in the vasodilative phase and of NO inhalation during the vasoconstrictive phase emphasizes again the crucial importance of timing and synchronous monitoring of its effects. Areas that are lacking iNOS have less NO-induced vasodilation and become underperfused.
Vasodilation creates also an optimal micro-hemodynamics for oxygenation and removal of CO2 buffering of acidosis and milieu normalization and IR inflammatory and toxic product removal.
Intra-operative internal and external warming under full oxygenation and after source control helps or can substitute vasodilators action. The same strategy can be applied in HS.
External warming can be added soon after source control and in postoperative period.
To bring up internal temperature up to 36°C can already be done intra-operatively. In experimental conditions on animals with different sizes than humans, the recommended warming speed from extreme temperatures of 10–15°C in large size animals is 0.5°C per minute [81].
6.2.1 Addressing ischemia-reperfusion
Systemic hypothermia and the hypoxic/ischemic post-conditioning strategy, despite the lack of clinical data pointing to a decrease in HS mortality, remain to date the best therapeutical assets available to prevent or attenuate the damage of IRI.
The benefits of hypothermia at mild moderate levels have been delineated experimentally [21].
Besides the prevention and attenuation of the direct ischemic insult, hypothermia gives protection against free radicals in I-R injury and protects against inflammation by tightening cell membranes, decreasing capillary permeability, leukotriene production, and edema [42, 43].
This effect is exploited in preventing IRI on the brain during ROSC [50, 51] in decreasing the size and performances of myocardium post-infarction [44, 45].
6.3 Systemic hypothermia pre-source control
Despite local hypothermia in AMI reduces infarct size and IRI without improving the overall outcome, its findings are ominously translatable to HS with global ischemia situation and low DO2.
If local or regional hypothermia prevents the escalation of IR damage to heart and brain even before ischemic infarction installs, even more so systemic hypothermia may decrease the level IRD/I in HS systemic hypoperfusion and hypoxemia.
Hypothermia can be conveniently exploited preoperatively either not counteracting spontaneous accidental hypothermia or by inducing a mild-moderate level.
It should be controlled or brought and kept at the least to >30°C for drugs be effective, > 32°C to avoid the nuisance of coagulopathy requiring packing and consuming time for completion of the index operation, and below 36°C core temperature to avoid the negative effects of unnecessary high temperature in tissues that have already suffered a condition of relative hypoxia and exploit its beneficial effect on buffering IRI.
Practically, if present
Clinical experience with cold stored low-titer whole blood (LTWB) has been seen not to confer disadvantages on hemodynamic and safety aspects compared to normothermic blood, with the advantage of healthier platelets up to 2 weeks and higher retention of Hb at 24 hours [136, 137, 138, 139, 140, 141].
If hypothermia is used as part of the treatment in HS, the entire strategy should change starting from preoperative management and anesthesia.
There is overwhelming convincing evidence in animals of different size, proving the increased survival with intravenous and surface-induced hypothermia compared with normothermia. This is not achieved without problems. Surface hypothermia would be more uncomfortable, and intravenous fluids induce dilution coagulopathy and decreased DO2 due to Hb curve shift to left and must be accompanied by pharmacological control of shivering and in its accidental form not to be counteracted, resorting to small doses of pethidine for shivering.
Therapeutic hypothermia, after reperfusion following resuscitation with fluids, predictably cannot prolong survival in volume-controlled HS [142].
Normothermia is predictably beneficial after source control and resuscitation [143]. During surgery after source control, it is rewarming, in fact, that increases survival, and not hypothermia; likewise, it is hypothermia, which is beneficial and increases survival before source control.
In a study under volume-controlled induced HS on medium-sized animals spontaneously breathing under a GA with a vasodilating agent such as halothane, hypothermia induced after exsanguination with extracorporeal shunt cooling at 35+/−0.5°C resulted in an improved survival compared to normothermia [144]. The study was biased by the beneficial effect on the vasodilating halothane. In any case, postoperative hypothermia is not an acceptable situation or a possible iatrogenic tactics in HS. It remains beneficial in normovolemic conditions such as post-CA ROSC period or neonatal encephalopathy.
For an indirect proof of the beneficial effect of cold blood and blood components global ischemia scenario is CPB. Systemic hypothermia with CPB is the major contributing factor to prevent or control systemic IRI [49]. The different outcomes between pre- and post-source control hypothermia can be explained only by the global and more uniform distribution of hypothermia with ECLS, method not practical or convenient in postoperative period when the comfort of the patient is a main therapeutic target.
The key universal word to HS management, together with a rapid or earliest source/s control, is timing.
The protective effect of hypothermia from ischemic damage, whether induced or spontaneous, is timing/speed of onset-dependent. If hypothermia is present before ischemia installs is or maybe lifesaving, otherwise it is not beneficial and may actually accelerate
6.4 Oxygen-titrated reperfusion
Besides hypothermia, ischemia and hypoxemia have been studied to test the effects of their variations on the IR dynamics.
Excess of O2 therapy during significant hemorrhage intensifies the physiological compensatory responses of vasoconstriction and blood flow redistribution [145]. Therefore, compared with room-air breathing, high flow or high concentration O2 therapy deteriorates both hemodynamics and tissue/cellular hypoxia, in spite of the significantly higher arterial blood oxygenation.
Moreover, the abundance of oxygen supply in the initial phase of reperfusion produces a burst of ROS generation. This opens to the question of how possibly attenuating IRI by manipulating the oxygen content and titrate it to the cells needs.
“
The opposite tactics, “
A closely related technique involves initiation of transient episodes of ischemia in a remote tissue or organ at the time of reperfusion (remote
A variation from the alternating cycles of ischemia-reperfusion is “
“
The advantage of ischemic position is in the possibility to modulate PaO2 using different levels of FiO2 without compromising perfusion. In this way, ROS and the intracellular pH are kept at a minimum, at the same time oxygen is available to mitochondria for ATP generation/storing, and metabolic waste is not slowed.
Another potential advantage of hypoxemic reperfusion compared to the use of antioxidants is that it aims to prevent ROS production rather than eliminate their deleterious effects.
Moreover, hypoxemic reperfusion may be advantageous compared to post-conditioning strategies since blood flow is restored offering better replenishment from metabolic wastes.
Two experimental studies highlight the relevance of oxygenation on the perfusion in determining IRI and overall prognosis in resuscitation of a progressive hemorrhagic shock.
In a study, tissue oxygenation (PO2 e tPO2) improved at a FiO2 of 1 in normovolaemia and at blood volume losses of less than 20%. Instead, at significant, for more than 50% blood volume losses, high inspired oxygen admixtures lead to precipitous reduction of tissue oxygenation, similar to that of animals breathing in-room air. An even worse outcome was observed by inducing hypoxemia (breathing at FiO2 = 0.15) without resuscitation; all parameters deteriorated, and the animals had an earlier death. Hypoxemia combined with hypoperfusion accelerated the tPO2 fall considerably [155].
An experiment
In the last 7 years there has not been any study or
Only one study has showed that the more advanced is shock, the lesser oxygen is required. In it, where rats were made to bleed 70% of their TBV in a controlled HS experiment, a 3-week-old hypoxically stored RBCs, made hypoxic using an O2 depletion system, scored like fresh RBC and better than conventional 3-week-old stored RBC in terms of hemodynamics and organ injury, during resuscitation, and in terms of oxidative stress, RNA/DNA injury, and lipid per oxidation, following reperfusion [157].
6.5 Normothermic plasma
Trauma causes a systemic inflammatory response, which, contrarily to sepsis and inflammatory shock [91, 98] where is regularly and primarily hit, in trauma with or without hemorrhage is more present in severe blunt trauma, due to the relative increased response for the relative increase of soft tissue injuries in respect to a penetrating one where the inflammatory reaction is present only mainly along the track of the offense weapons trajectories. Moreover, SIR messengers and factors are lost in a relative bigger amount in a penetrating trauma than a blunt one where tissue contusions are universally present compared to a penetrating trauma. Endothelial cell damage and glycocalyx shedding of capillaries and arterioles are the main target, yielding to coagulopathy, further inflammation, increased vascular permeability, and dyslexia that may lead to death. The microcirculation derangement is mirrored by worsening of flow, density, and heterogeneity of capillaries within microvessels, as seen with sublingual incident dark field video-microscopy [158].
The vascular endothelium plays a central role in maintaining organ homeostasis through its regulation of vascular tone, coagulation, inflammation, and barrier function and the vital interaction with the distal tissues on gas exchange. Dysregulation of normal endothelial function occurs during major injuries with severe tissues trauma, hemorrhagic shock, and burns, where it gives rise to systemic microvascular thrombosis, inflammation, loss of barrier integrity, coagulopathy, and respiration dysregulation. The endotheliopathy of trauma involves a complex interplay between the glycocalyx, von Willebrand factor (VWF), expressed on its surface, and platelets. Upon exposure to subendothelial proteins such as collagen, VWF binds platelets to the injured vasculature. The endothelium, which provides an anticoagulant and platelet-repellent surface in the resting state, becomes highly procoagulant and attracts platelets and leukocytes when its protective glycocalyx is depleted. Hyperadhesive VWF also binds leukocytes to normal ECs remote from the injury site [159].
Normothermic FFP is the most effective fluid in restoring the endothelial glycocalyx and junctions, in reducing endothelial permeability, and in attenuating the early inflammation/coagulation response. The mechanism is not clear but is likely due to the effects of the several components of FFP as the same beneficial effects have been seen also with plasma-derived products such as prothrombin complex concentrate (PCC) and lyophilized and spray-dried plasma. The protective effects of FFP are diminished by post-thaw storage at 4°C for 5 days and are time sensitive with most efficacy with the first 3 hours post trauma [160].
A decrease of 30 days mortality was found with plasma when compared to no plasma, but only in blunt trauma [161]. In some blunt trauma, such as a predominantly orthopedic blunt poly-trauma a variable post-traumatic inflammatory response ensues. In these scenarios, the inflammatory cascade has more chance of being retained inside circulation, damaging the endothelium and microcirculation, than a penetrating trauma where the inflammatory avalanche gets lost with the blood loss. Presumably, following the loss of inflammatory or toxic mediators and factors with the blood loss, there is a notable absence or a comparably lesser amount of SIR and IRT in penetrating injury, while the post-traumatic inflame response is very much present and longer lasting in a blunt injury where viable but damaged tissue contusions or hematoma continue being a source of SIR or IRT. This explains why plasma has benefits in blunt injury when added to blood.
Another advantage of plasma in blunt trauma is that it helps the release of VWF from ECs. This action is prevented by the anti-fibrinolytic agent tranexamic acid, providing a direct link between endotheliopathy and fibrinolysis during acute trauma [162].
These findings make normothermic plasma an important fluid in postoperative management of a HS from a blunt poly-trauma. An advantage can further come in decreasing plasma amenable coagulopathies, if already present and missed.
6.6 Postoperative therapeutic adjuncts
Cortisone has two major potentially negative side effects in the immediate postoperative period: healing interference and impairment, and curbing the inflammatory response to IRI/T. While healing is not impaired if given in the first 3 days, not anyway enough to interfere with healing processes of an anastomosis or abdominal/chest wall union, and can anyhow be counteracted by high doses of vitamin A, its anti-inflammatory property is a major problem. Its use with intent to prevent or curb the IRI to lungs kidneys and liver needs to be well pondered.
The damages to lung alveoli microcirculation and structure due to vasoconstriction and the excess of ROS at superior normobaric doses are the drawback to avoid.
Hyperbaric oxygen is not an option too.
6.7 Permissive hypoxemia
Mild permissive hypoxemia (PaO2 55–80 mmHg; SpO2 88–92%) results in improved outcomes also in patients at risk or with actual early acute lung injury, included those with ARDS or with ARF from other causes. Preliminary results with low FiO2 at 0.5 in a mix population have confirmed the safety and therapeutic efficacy of a “permissive hypoxemia” tactics, with improvement of in-hospital mortality, a longer ventilator-free day period, and an improvement of the 28-day mortality in an ARDS subset and a worse outcome in high or hypoxemic concentrations [168].
Advertisement
7. Perspective
On these bases, a strategy can be drawn for timely application before or soon after source control in an advanced shock requiring DCS [111], when IR toxemia and cryptic shock are very likely to install and determine prognosis (Table 3).
Table 3.
Strategies.
The framework conceptualized by Convertino of a similarity between extreme physical exercise requiring increasing VO2 and the need to ensure sufficient VO2 after normalization of CaO2 and DO2 [169] as well as the conclusions of a study by Gutierrez et al. showing clearly high FiO2 cannot compensate for the low cardiac output and absence of tissue perfusion and O2 transport does not necessarily correlate with sufficient or optimal VO2 that is in fact limited only by the diffusion of oxygen from capillaries to cells [167] and address the crucial questions and research directions to follow, if we want to diminish late HS mortality by preventing, buffering, and managing the pathophysiological mechanisms leading to
The real-time
References
- 1.
Singer M, Matthay MA. Clinical review: Thinking outside the box – An iconoclastic view of current practice. Critical Care. 2011; 15 :225 - 2.
Rauch S, Miller C, Bräuer A, Wallner B, Bock M, Paal P. Perioperative hypothermia-a narrative review. International Journal of Environmental Research and Public Health. 2021; 18 :8749 - 3.
Mommsen P, Andruszkow H, Frömke C, Zeckey C, Wagner U, van Griensven M, et al. Effects of accidental hypothermia on posttraumatic complications and outcome in multiple trauma patients. Injury. 2013; 44 :86-90 - 4.
Nishi K, Takasu A, Shinozaki D, Sakamoto K, Yamamoto Y, Sakamoto T. Hypothermia does not hasten death during uncontrolled hemorrhagic shock presenting as the “triad of death” in rats. Acute Medicine & Surgery. 2015; 2 :29-34 - 5.
Jurkovich GJ, Greiser WB, Luterman A, Curreri PW. Hypothermia in trauma victims: An ominous predictor of survival. The Journal of Trauma. 1987; 27 :1019-1024 - 6.
Gentilello LM, Jurcovich GJ, Stark MS, Hassantash SA, O'Keefe GE. Is hypothermia in the victim of major trauma protective or harmful? Annals of Surgery. 1997; 226 :439-447 - 7.
Convertino VA, Cap AP. Should patients with haemorrhage be kept warm? The Journal of Physiology. 2010; 588 (Pt. 17):3135 - 8.
Matheny JL, Ahlquist RP. Role of neuronal and extraneuronal factors in temperature mediated responsiveness of adrenoceptors. Archives Internationales de Pharmacodynamie et de Thérapie. 1976; 224 :180-189 - 9.
Jung KT, Bapat A, Kim Y-K, Hucker WJ, Lee K. Therapeutic hypothermia for acute myocardial infarction: A narrative review of evidence from animal and clinical studies. Korean Journal of Anesthesiology. 2022; 75 :216-230 - 10.
Nishi K, Takasu A, Shibata M, Uchino S, Yamamoto Y, Sakamoto T. Hypothermia reduces resuscitation fluid volumes required to maintain blood pressure in a rat Hemorrhagic shock model. The Journal of Trauma. 2012; 72 :130-135 - 11.
Swan H. Clinical hypothermia: A lady with a past and some promise for the future. Surgery. 1973; 73 :736-758 - 12.
Gubler KD, Gentilello LM, Hassantash SA, Maier RV. The impact of hypothermia on diluitional coagulopathy. The Journal of Trauma. 1994; 36 :847-851 - 13.
Brohi K, Cohen MJ, Ganter MT, Schultz MJ, Levi M, Mackersie RC, et al. Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. The Journal of Trauma. 2008; 64 :1211-1217 - 14.
Floccard B, Rugeri L, Faure A, Denis MS, Boyle EM, Peguet O, et al. Early coagulopathy in trauma patients: An on-scene and hospital admission study. Injury. 2010; 43 :26-32 - 15.
Iwamoto S, Takasu A, Sakamoto T. Therapeutic mild hypothermia: Effects on coagulopathy and survival in a rat hemorrhagic shock model. The Journal of Trauma. 2010; 68 :669-675 - 16.
Wu X, Kochanek PM, Cochran K, Nozari A, Henchir J, Stezoski SW, et al. Mild hypothermia improves survival after prolonged, traumatic hemorrhagic shock in pigs. The Journal of Trauma. 2005; 59 :291-301 - 17.
Takasu A, Ishihara S, Anada H, Sakamoto T, Okada Y. Surface cooling, which fails to reduce the core temperature rapidly, hastens death during severe hemorrhagic shock in pigs. The Journal of Trauma. 2000; 48 :942-947 - 18.
Takasu A, Norio H, Sakamoto T, Okada Y. Mild hypothermia prolongs the survival time during uncontrolled hemorrhagic shock in rats. Resuscitation. 2002; 54 :303-309 - 19.
Wu X, Stezoski J, Safar P, Nozari A, Tisherman SA. After spontaneous hypothermia during hemorrhagic shock, continuing mild hypothermia (34 degrees C) improves early but not late survival in rats. The Journal of Trauma. 2003; 55 :308-316 - 20.
George ME, Mulier KE, Bellman GJ. Hypothermia is associated with improved outcomes in a porcine model of hemorrhagic shock. The Journal of Trauma. 2010; 68 :662-668 - 21.
Hildebrand F, Radermacher P, Ruchholtz S, Huber-Lang M, Seekamp A, Flohè S, et al. Relevance of induced and accidental hypothermia after trauma-haemorrhage. Intensive Care Medicine Experimental. 2014; 2 :16 - 22.
Schmied H, Kurz A, Sessler DI, Kozek S, Reiter A. Mild hypothermia increases blood loss and transfusion requirements during total hip arthroplasty. Lancet. 1996; 3 :289-292 - 23.
Rajagopalan S, Mascha E, Na J, Sessler DI. The effects of mild perioperative hypothermia on blood loss and transfusion requirement. Anesthesiology. 2008; 108 :71-77 - 24.
Lester ELW, Fox E, Holcomb JB, Brasel K, Bulger E, Cohen MJ, et al. The impact of hypothermia on outcomes in massively transfused patients. Journal of Trauma and Acute Care Surgery. 2019; 86 :458-463 - 25.
Bonanno FG. Early coagulopathy in trauma and major bleeding: Is it time to challenge the dogma? Trauma. 2021; 1 :1-4 - 26.
Bonanno F. Facts and myths of coagulopathies in trauma and major bleeding - to clear the fog. In: Advancement and New Understanding in Medical Sciences. Vol. 3. Ch 6. Bengal, India. 2023. DOI: 10.9734/bpi/anums/v3/7338C - 27.
Bonanno FG. Hemorrhagic shock: The physiology approach. Journal of Emergencies, Trauma, and Shock. 2012; 5 :285-295 - 28.
Marubayashi S, Takenaka M, Dohi K, Ezaki H, Kawasaki T. Adenine nucleotide metabolism during hepatic ischemia and subsequent blood reflow periods and its relation to organ viability. Transplantation. 1980; 30 :294-296 - 29.
Chaudry IH. Cellular mechanisms in shock and ischemia and their correction. The American Journal of Physiology. 1983; 14 :R117-R134 - 30.
Storey KB, Storey JM. Tribute to P. L. Lutz: Putting life on ‘pause’ – Molecular regulation of hypometabolism. The Journal of Experimental Biology. 2007; 210 :1700-1714 - 31.
Wood S. Interactions between hypoxia and hypothermia. Annual Review of Physiology. 1991; 53 :71-85 - 32.
Steiner AA, Branco LGS. Hypoxia-induced anapyrexia: Implications and putative mediators. Annual Review of Physiology. 2002; 64 :263-288. DOI: 10.1146/annurev.physiol.64.081501.155856 - 33.
Hale SL, Kloner RA. Myocardial temperature in acute myocardial infarction: Protection with mild regional hypothermia. The American Journal of Physiology. 1997; 273 :H220-H227 - 34.
Simkhovich BZ, Hale S, Kloner RA. Metabolic mechanism by which mild regional hypothermia preserves ischemic tissue. Journal of Cardiovascular Pharmacology and Therapeutics. 2004; 9 :83-90 - 35.
Stone GH, Donnelly C, Frobese AS. The effect of lowered body temperature on the cerebral hemodynamics and metabolism of man. Surgery, Gynecology & Obstetrics. 1956; 103 :313-317 - 36.
Kramer AS, Sanders AP, Lesage AM, Woodhall B, Sealy WC. The effect of profound hypothermia on preservation of cerebral ATP content during circulatory arrest. The Journal of Thoracic and Cardiovascular. 1968; 56 :699-709 - 37.
Erecinska M, Thoresen M, Silver IA. Effects of hypothermia on energy metabolism in mammalian central nervous system. Journal of Cerebral Blood Flow and Metabolism. 2003; 23 :513-530 - 38.
Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic-ischemic brain damage. Annals of Neurology. 1986; 19 :105-111 - 39.
Busto R, Globus MY-T, Dietrich WD, Martinez E, Valdes I, Ginsberg MD. Effect of mild hypothermia on ischemia-induced release of neurotransmitters and free fatty acids in rat brain. Stroke. 1989; 20 :904-910 - 40.
Gonzalez-Ibarra FP, Varon J, Lopez-Meza EG. Therapeutic hypothermia: A critical review the molecular mechanisms of action. Frontiers in Neurology. 2011; 2 :4 - 41.
Ding D, Moskowitz SI, Li R, Lee SB, Esteban M, Tomaselli K, et al. Acidosis induces necrosis and apoptosis of cultured hippocampal neurons. Experimental Neurology. 2000; 162 :1-12 - 42.
Jurkovich GJ, Pitt RM, Curreri PW, Granger DN. Hypothermia prevents increased capillary permeability following ischemia-reperfusion injury. The Journal of Surgical Research. 1988; 44 :514-521 - 43.
Childs EW, Udobi KF, Hunter FA. Hypothermia reduces microvascular permeability and reactive oxygen species expression after hemorrhagic shock. The Journal of Trauma. 2005; 58 :2 - 44.
Kohlhauer M, Berdeaux A, Ghaleh B, Tissier R. Therapeutic hypothermia to protect the heart against acute myocardial infarction. Archives of Cardiovascular Diseases. 2016; 109 :716-722 - 45.
Yamada KP, Kariya T, Aikawa T, Ishikawa K. Effects of therapeutic hypothermia on Normal and ischemic heart. Frontiers in Cardiovascular Medicine. Feb 2021; 15 (8):642843. DOI: 10.3389/fcvm.2021.642843 - 46.
Gilbert M, Busund R, Skagseth A, Nilsen PA, Solbø JP. Resuscitation from accidental hypothermia of 13.7 degrees C with circulatory arrest. Lancet. 2000; 355 (9201):375-376 - 47.
Kanemoto S, Matsubara M, Noma M, Leshnower BG, Parish LM, Jackson BM, et al. Mild hypothermia to limit myocardial ischemia-reperfusion injury: Importance of timing. The Annals of Thoracic Surgery. 2009; 87 :157-163 - 48.
Erlinge D, Götberg M, Noc M, Lang I, Holzer M, Clemmensen P, et al. Therapeutic hypothermia for the treatment of acute myocardial infarction-combined analysis of the RAPID MI-ICE and the CHILL-MI trials. Therapeutic Hypothermia and Temperature Management. 2015; 5 :77-84 - 49.
Tahsili-Fahadan P, Farrokh S, Geocadin RG. Hypothermia and brain inflammation after cardiac arrest. Brain Circulation. Jan-March 2018; 4 (1):1-13. Available from:http://www.braincirculation.org . DOI: 10.4103/bc.bc_4_18 - 50.
Sandroni C, Cronberg T, Sekhon M. Brain injury after cardiac arrest: Pathophysiology, treatment, and prognosis. Intensive Care Medicine. 2021; 47 (12):1393-1414 - 51.
Doherty DR, Parshuram CS, Gaboury I, Hoskote A, Lacroix J, Tucci M, et al. Hypothermia therapy after pediatric cardiac arrest. Circulation. 2009; 119 :1492-1500 - 52.
He S, Weng Y, Sun S, Chen W, Wu X, Li Z, et al. Comparison of the durations of mild therapeutic hypothermia on outcome after cardiopulmonary resuscitation in the rat. Circulation. 2012; 125 :123-129 - 53.
Lindsay PJ, Buell D, Scales DC. The efficacy and safety of pre-hospital cooling after out-of-hospital cardiac arrest: A systematic review and meta-analysis. Critical Care. 2018; 22 :66 - 54.
Cotten CM, Shankaran S. Hypothermia for hypoxic-ischemic encephalopathy. Expert Review of Obstetrics and Gynecology. 2010; 5 :227-239 - 55.
Ma H, Sinha B, Pandya RS, Lin N, Popp AJ, Li J, et al. Therapeutic hypothermia as a neuroprotective strategy in neonatal hypoxic-ischemic brain injury and traumatic brain injury. Current Molecular Medicine. 2012; 12 :1282-1296 - 56.
Chang M. Therapeutic hypothermia for newborns with hypoxic ischemic encephalopathy. Neonatal Medicine. 2013; 20 :2-11 - 57.
Zhang XW, Xie JF, Chen JX, Huang YZ, Guo FM, Yang Y, et al. The effect of mild induced hypothermia on outcomes of patients after cardiac arrest: A systematic review and meta-analysis of randomised controlled trials. Critical Care. 2015; 19 :417 - 58.
Robba C, Battaglini D, Samary CS, Silva PL, Ball L, Rocco PRM, et al. Ischaemic stroke-induced distal organ damage: Pathophysiology and new therapeutic strategies. Intensive Care Medicine Experimental. 2020; 8 :23 - 59.
Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Critical Care Medicine. 2009; 37 :S186-S202 - 60.
Vaity C, Al-Subaie N, Cecconi M. Cooling techniques for targeted temperature management post-cardiac arrest. Critical Care. 2015; 19 :103 - 61.
Nolan JP, Sandroni C, Bottiger BW, Cariou A, Cronberg T, Friberg H, et al. European resuscitation council and European society of intensive care medicine guidelines 2021: Post-resuscitation care. Resuscitation. 2021; 161 :220-269 - 62.
Mathew JL, Kaur N, Dsouza JM. Therapeutic hypothermia in neonatal hypoxic encephalopathy: A systematic review and meta-analysis. Journal of Global Health. 2022; 12 :04030 - 63.
Taylor MJ, Bailes JE, Elrifai AM, Shih SR, Teeple E, Leavitt ML, et al. A new solution for life without blood. Asanguineous low-flow perfusion of a whole-body perfusate during 3 hours of cardiac arrest and profound hypothermia. Circulation. 1995; 91 :431-444 - 64.
Roskott AMC, Nieuwenhuijs VB, Dijkstra G, Koudstaal LG, Leuvenink HGD, Ploeg RJ. Small bowel preservation for intestinal transplantation: A review. Transplant International. 2011; 24 :107-131 - 65.
Michel SG, Madsen JC. Current perspectives in transplant medicine: Hypothermic oxygenated perfusion. Transplant Research and Risk Management. 2016; 8 :25-30 - 66.
Petrat F, de Groot H, Sustmann R, Rauen U. The chelatable iron pool in living cells: A methodically defined quantity. Biological Chemistry. 2002; 383 :489-502 - 67.
Rauen U, de Groot H. Mammalian cell injury induced by hypothermia - the emerging role for reactive oxygen species. Biological Chemistry. 2002; 383 :477-488 - 68.
Stephen CR, Dent SJ, Hall KD, Smith WW. Physiologic reactions during profound hypothermia with cardioplegia. Anesthesiology. 1961; 22 :873-881 - 69.
Ziganshin BA, Elefteriades JA. Deep hypothermic circulatory arrest. Annals of Cardiothoracic Surgery. 2013; 2 :303-315 - 70.
Mohiyaddin S, Nanjaiah P, Saad AO, Acharya MN, Khan TA, Davies RH, et al. Suspended animation: The past, present and future of major cardiothoracic trauma. ANZ Journal of Surgery. 2018; 88 :678-682 - 71.
Moffatt SE, Mitchell SJB, Walke JL. Deep and profound hypothermia in haemorrhagic shock, friend or foe? A systematic review. Journal of the Royal Army Medical Corps. 2018; 164 :191-196 - 72.
Alam HB, Chen Z, Honma K, Koustova E, Querol RI, Jaskille A, et al. The rate of induction of hypothermic arrest determines the outcome in a swine model of lethal hemorrhage. The Journal of Trauma. 2004; 57 :961-969 - 73.
Tsuei BJ, Kearney PA. Hypothermia in the trauma patient. Injury. 2004; 35 :7-15 - 74.
Cooper KE, Hunter AR, Keatinge WR. Accidental hypothermia. International Anesthesiology Clinics. 1964; 24 (2):999-1014 - 75.
Lott C, Truhla A, Alfonzo A, Barelli A, Gonzalez-Salvado V, Hinkelbein J, et al. European resuscitation council guidelines 2021: Cardiac arrest in special circumstances. Resuscitation. 2021; 161 :152-219 - 76.
Bobi J, Solanes N, Dantas AP, Ishida K, Regueiro A, Castillo N, et al. Moderate hypothermia modifies coronary Hemodynamics and endothelium-dependent vasodilation in a porcine model of temperature management. Journal of the American Heart Association. 2020; 9 (3):e014035. DOI: 10.1161/JAHA.119.014035 - 77.
Taniguchi Y, Lenhardt R, Sessler DI, Kurz A. The effect of altering skin-surface cooling speeds on vasoconstriction and shivering thresholds. Anesthesia and Analgesia. 2011; 113 :540-544 - 78.
Polderman KH, Herold I. Therapeutic hypothermia and controlled normothermia in the intensive care unit: Practical considerations, side effects, and cooling methods. Critical Care Medicine. 2009; 37 :1101-1120 - 79.
Tveita T, Ytrehus K, Myhre ESP, Hevrøy O. Left ventricular dysfunction following rewarming from experimental hypothermia. Journal of Applied Physiology. 1998; 85 (6):2135-2139 - 80.
Scaravilli V, Bonacina D, Citerio G. Rewarming: Facts and myths from the systemic perspective. Critical Care. 2012; 16 :A25 - 81.
Alam HB, Rhee P, Honma K, Chen H, Ayuste EC, Lin T, et al. Does the rate of rewarming from profound hypothermic arrest influence the outcome in a swine model of lethal hemorrhage? The Journal of Trauma. 2006; 60 :134-146 - 82.
Rösli D, Schnüriger B, Candinas D, Haltmeier T. The impact of accidental hypothermia on mortality in trauma patients overall and patients with traumatic brain injury specifically: A systematic review and meta-analysis. World Journal of Surgery. 2020; 44 (12):4106-4117 - 83.
Rosenthal LM, Tong G, Wowro S, Walker C, Pfitzer C, Böttcher W, et al. A prospective clinical trial measuring the effects of cardiopulmonary bypass under mild hypothermia on the inflammatory response and regulation of cold-shock protein RNA-binding motif 3. Therapeutic Hypothermia and Temperature Management. 2020; 10 (1):60-70 - 84.
Caminos Eguillor JF, Ferrara G, Kanoore Edul VS, Buscetti MG, Canales HS, Lattanzio B, et al. Microcirculatory effects of rewarming in experimental hemorrhagic shock. Microvascular Research 2023; 147 :104490. Epub 2023 January 31. DOI: 10.1016/j.mvr.2023.104490 - 85.
Kim SH, Stezoski SW, Safar P, Fisherman SA. Hypothermia, but not 100% oxygen breathing, prolongs survival time during lethal uncontrolled hemorrhagic shock in rats. The Journal of Trauma. 1998; 44 :485-491 - 86.
Takasu A, Stezoski SW, Stezoski J, Safar P, Tisherman SA. Mild or moderate hypothermia, but not increased oxygen breathing, increases long-term survival after uncontrolled hemorrhagic shock in rats. Critical Care Medicine. 2000a; 28 :2465-2474 - 87.
Takasu A, Iwamoto S, Ando S, Minagawa Y, Kashiba M, Yamamoto Y, et al. Effects of various concentrations of inhaled oxygen on tissue disoxia, oxidative stress, and survival in a rat hemorrhagic shock model. Resuscitation. 2009; 80 :826-831 - 88.
Takasu A, Prueckner S, Tisherman SA, Stezoski SW, Stezoski J, Safar P. Effects of increased oxygen breathing in a volume controlled hemorrhagic shock outcome model in rats. Resuscitation. 2000; 45 :209-220 - 89.
Bickell WH, Wall MJ, Pepe PE, Martin RR, Ginger VF, Allen MK, et al. Immediate versus delayed fluid resuscitation for hypotensive patients with penetrating torso injuries. The New England Journal of Medicine. 1994; 331 :1105-1109 - 90.
Carden DL, Granger DN. Pathophysiology of ischemia-reperfusion injury. The Journal of Pathology. 2000; 190 :255-266 - 91.
Bonanno FG. Clinical pathology of the shock syndromes. Journal of Emergencies, Trauma, and Shock. 2011; 4 :233-235 - 92.
Bonanno FG. Physiopathology of shock. Journal of Emergencies, Trauma, and Shock. 2011; 4 (2):222-232. DOI: 10.4103/0974-2700.82210 - 93.
Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, et al. Current mechanistic concepts in ischemia and reperfusion injury. Cellular Physiology and Biochemistry. 2018; 46 :1650-1667 - 94.
Douzinas EE, Andrianakis I, Livaditi O, Paneris P, Tasoulis M, Pelekanou A, et al. The level of hypotension during hemorrhagic shock is a major determinant of the post-resuscitation systemic inflammatory response: An experimental study. BMC Physiology. 2008; 8 :15 - 95.
Santibanez-Gallerani AS, Barber AE, Williams SJ, ZhaoB SY, Shires GT. Improved survival with early fluid resuscitation following hemorrhagic shock. World Journal of Surgery. 2001; 25 (5):592-597. DOI: 10.1007/s002680020115 - 96.
Lee CC, Chang IJ, Yen ZS, Hsu CY, Chen SY, Su CP, et al. Delayed fluid resuscitation in hemorrhagic shock induces proinflammatory cytokine response. Annals of Emergency Medicine. 2007; 49 (1):37-44. DOI: 10.1016/j.annemergmed.2006.05.031 - 97.
Malinoski DJ, Slater MS, Mullins RJ. Crush injury and rhabdomyolysis. Critical Care Clinics. 2004; 20 (1):171-192. DOI: 10.1016/s0749-0704(03)00091-5 - 98.
Bonanno FG. Shock - a reappraisal: The holistic approach. Journal of Emergencies, Trauma, and Shock. 2012; 5 (2):167-177. DOI: 10.4103/0974-2700.96487 - 99.
Deitch EA. Gut-origin sepsis: Evolution of a concept. The Surgeon. 2012; 10 (6):350-356. DOI: 10.1016/j.surge.2012.03.003. Epub 2012 Apr 23 - 100.
Assimakopoulos SF, Triantos C, Thomopoulos K, Fligou F, Maroulis I, Marangos M, et al. Gut- origin sepsis in the critically ill patient: Pathophysiology and treatment. Infection. 2018; 46 (6):751-760. DOI: 10.1007/s15010-018-1178-5 - 101.
Bonanno F. Extending damage control philosophy to not-haemorrhagic situations: Implications for a reclassification of shock states. ANZ Journal of Surgery. 2008; 78 :634-637 - 102.
Pape HC, Moore EE, McKinley T, Sauaia A. Pathophysiology in patients with polytrauma. Injury. 2022; 53 (7):2400-2412 - 103.
Callcut RA, Kornblith LZ, Conroy AS, Robles AJ, Meizoso JP, Namias N, et al. The why and how our trauma patients die: A prospective Multicenter Western trauma association study. Journal of Trauma and Acute Care Surgery. 2019; 86 (5):864-870. DOI: 10.1097/TA.0000000000002205 - 104.
Minei JP, Cuschieri J, Sperry J, Moore EE, West MA, Harbrecht BG, et al. Inflammation and the host response to injury collaborative research program. The changing pattern and implications of multiple organ failure after blunt injury with hemorrhagic shock. Critical Care Medicine. 2012; 40 (4):1129-1135. DOI: 10.1097/CCM.0b013e3182376e9f - 105.
Efron P, Brakenridge SC, Mohr AM, Barrios EL, Polcz VE, Anton S, et al. The persistent inflammation, immunosuppression, and catabolism syndrome (PICS) ten years later. Journal of Trauma and Acute Care Surgery. 2023; 95 (5):790-799. DOI: 10.1097/TA.0000000000004087 - 106.
Douzinas EE. Hemorrhagic shock resuscitation: A critical issue on the development of post- traumatic multiple organ failure. Critical Care Medicine. 2012; 40 :1348-1349 - 107.
Tachon G, Harrois A, Tanaka S, Kato H, Huet O, Pottecher J. Microcirculatory alterations in traumatic hemorrhagic shock. Critical Care Medicine. 2014; 42 :1433-1441 - 108.
Ince C. Hemodynamic coherence and rationale for monitoring the microcirculation. Critical Care. 2015; 19 (Suppl. 3):S8 - 109.
Libert N, Harrois A, Duranteau J. Haemodynamic coherence in haemorrhagic shock. Best Practice & Research. Clinical Anaesthesiology. 2016; 30 :429-435 - 110.
Bonanno FG. Management of hemorrhagic shock: Physiology approach, timing and strategies. Journal of Clinical Medicine. 2022; 12 :260. DOI: 10.3390/jcm12010260 - 111.
Bonanno FG. Debunking the lethal triad and delineating damage control surgery. Minerva Surgery. Dec 2023; 78 (6):692-709. DOI: 10.23736/S2724-5691.23.09916-1. Epub 2023 Sep 13 - 112.
De Backer D, Ospina-Tascon G, Salgado D, Favory R, Creteur J, Vincent J-L. Monitoring the microcirculation in the critically ill patient: Current methods and future approaches. Intensive Care Medicine. 2010; 36 :1813-1825 - 113.
De Backer D, Donadello K, Sakr Y, Ospina-Tascon G, Salgado D, Colletta S. Microcirculatory alterations in patients with severe sepsis: Impact of time of assessment and relationship with outcome. Critical Care Medicine. 2013; 41 :791-799 - 114.
Jobsis FF. Noninvasive, infrared monitoring of cerebral and myocardial oxygen sufficiency and circulatory parameters. Science. 1977; 198 (4323):1264-1267 - 115.
Creteur J, De Backer D, Sakr Y, Koch M. Sublingual capnometry tracks microcirculatory changes in septic patients. Intensive Care Medicine. 2006; 32 :516-523 - 116.
Heringlake M, Garbers C, Kabler JH, Anderson I, Heinze H, Schon J, et al. Preoperative cerebral oxygen saturation and clinical outcomes in cardiac surgery. Anesthesiology. 2011; 114 (1):58-69 - 117.
Duret J, Pottecher J, Bouzat P, Brun J, Harrois A, Payen JF, et al. Skeletal muscle oxygenation in severe trauma patients during haemorrhagic shock resuscitation. Critical Care. 2015; 19 :141 - 118.
Hunt MF, Clark KT, Whitman G, Choi CW, Geocadin RG, Cho SM. The use of cerebral NIRS monitoring to identify acute brain injury in patients with VA-ECMO. Journal of Intensive Care Medicine. 2021; 36 (12):1403-1409 - 119.
Mayevsky A, Rogatsky GG. Mitochondrial function in vivo evaluated by NADH fluorescence: From animal models to human studies. American Journal of Physiology. Cell Physiology. 2007; 292 (2):C615-C640 - 120.
Hilty MP, Ince C. Automated quantification of tissue red blood cell perfusion as a new resuscitation target. Current Opinion in Critical Care. 2020; 26 (3):273-280 - 121.
Hilty MP, Akin S, Boerma C, Donati A, Erdem O, Giaccaglia P, et al. Automated algorithm analysis of sublingual microcirculation in an international multicentral database identifies alterations associated with disease and mechanism of resuscitation. Critical Care Medicine. 2020; 48 :e864-e875 - 122.
Duranteau J, De Backer D, Donadello K, et al. The future of intensive care: The study of the microcirculation will help to guide our therapies. Critical Care. 2023; 27 :190 - 123.
Shoemaker WC, Appel PL, Kram HB. Role of oxygen debt in the development of organ failure sepsis, and death in high-risk surgical patients. Chest. 1992; 102 (1):208-215 - 124.
Fiddian-Green RG, Haglund U, Gutierrez G, Shoemaker WC. Goals for the resuscitation of shock. Critical Care Medicine. 1993; 21 (2 Suppl.):S25-S31 - 125.
Kipnis E, Vallet B. Early norepinephrine resuscitation of life-threatening hypotensive septic shock: It can do the job, but at what cost? Critical Care. 2010; 14 :450 - 126.
Cauwels A, Brouckaert P. Nitrite regulation of shock. Cardiovascular Research. 2011; 89 :553-559 - 127.
Den Uil CA, Caliskan K, Lagrand WK, van der Ent M, Jewbali LS, van Kuijk JP, et al. Dose-dependent benefit of nitroglycerin on microcirculation of patients with severe heart failure. Intensive Care Medicine. 2009; 35 :1893-1899 - 128.
Tsai AG, Intaglietta M. Exogenous nitric oxide induces protection during hemorrhagic shock. Resuscitation. 2009; 80 :707-712 - 129.
Cabrales P. Low dose nitrite enhances perfusion after fluid resuscitation from hemorrhagic shock. Resuscitation. 2009; 80 :1431-1436 - 130.
Nachuraju P, Friedman AJ, Friedman JM, Cabrales P. Exogenous nitric oxide prevents cardiovascular collapse during hemorrhagic shock. Resuscitation. 2011; 82 :607-613 - 131.
Wang Y, You G, Kan X, Chen G, Zhao L, Zhou H. Nitric oxide inhalation, a proposed strategy for early treatment of hemorrhagic shock. Medical Hypotheses. 2011; 77 :182-184 - 132.
Samana CM, Diaby M, Fellahi JM, Mdhafar M, Eyraud D, Arock A, et al. Inhibition of platelets aggregation by inhaled nitric oxide in patients with acute respiratory distress syndrome. Anesthesiology. 1995; 83 :56-65 - 133.
Bloomfield GL, Holloway S, Ridings PC, Fisher BJ, Blocher CR, Sholley M, et al. Pretreatment with inhaled nitric oxide inhibits neutrophil migration and oxidative activity, resulting in attenuated sepsis-induced acute lung injury. Critical Care Medicine. 1997; 25 :584-593 - 134.
Waisman D, Brod V, Dickstein R, Abramovich A, Rotschild A, Bitterman H. Effects of inhaled nitric oxide on lung injury after intestinal ischemia-reperfusion in rats. Shock. 2005; 23 :150-155 - 135.
Hines IN, Grisham MB. Divergent roles of superoxide and nitric oxide in liver ischemia and reperfusion injury. Journal of Clinical Biochemistry and Nutrition. 2011; 48 :50-56 - 136.
Leeper CM, Yazer MH, Cladis FP, Saladino R, Triulzi DJ, Gaines BA. Cold-stored whole blood platelet function is preserved in injured children with hemorrhagic shock. Journal of Trauma and Acute Care Surgery. 2019; 87 (1):49-53 - 137.
Hazelton JP, Cannon JW, Zatorski C, Roman JS, Moore SA, Young AJ, et al. Cold-stored whole blood: A better method of trauma resuscitation? Journal of Trauma and Acute Care Surgery. 2019; 87 (5):1035-1041 - 138.
Assen S, Cardenas J, George M, Wang YW, Wade CE, Meyer D, et al. Hemostatic potential of cold-stored non-leukoreduced whole blood over time: An assessment of platelet function and thrombin generation for optimal shelf life. Journal of Trauma and Acute Care Surgery. 2020; 89 :429-434 - 139.
Clements T, McCoy C, Assen S, Cardenas J, Wade C, Meyer D, et al. The prehospital use of younger age whole blood is associated with an improved arrival coagulation profile. Journal of Trauma and Acute Care Surgery. 2021; 90 (4):607-614 - 140.
Fecher A, Stimpson A, Ferrigno L, Pohlman TH. The pathophysiology and management of hemorrhagic shock in the poly-trauma patient. Journal of Clinical Medicine. 2021; 10 :479 - 141.
Ichikawa J, Kouta M, Oogushi M, Komori M. Effects of room temperature and cold storage on the metabolic and haemostatic properties of whole blood for acute normovolaemic haemodilution. PLoS One. 2022; 17 (5):e0267980 - 142.
Yanagawa Y, Sakamoto T, Okada Y. Therapeutic hypothermia limited to the resuscitation period does not prolong survival after severe hemorrhagic shock in rats. Resuscitation. 2005; 67 :119-126 - 143.
Wang P, Cioffi WG, Bland KI, Chaudry IH. Should normothermia be restored and maintained during resuscitation after trauma and hemorrhage? The Journal of Trauma. 2000; 48 :58-65 - 144.
Takasu A, Norio H, Gotoh Y, Sakamoto T, Okada Y. Effect of induced-hypothermia on short-term survival after volume-controlled hemorrhage in pigs. Resuscitation. 2003; 56 :319-328 - 145.
Douzinas EE. Progressive hemorrhage: Administer oxygen or early resuscitation? Intensive Care Medicine. 2009; 35 :1664-1666 - 146.
Murry C, Richard V, Reimer K, Jennings RB. Ischemic preconditioning slows energy metabolism and delays ultrastructural damage during sustained ischemia. Circulation Research. 1990; 66 :913-993 - 147.
Zhao ZQ , Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. American Journal of Physiology. Heart and Circulatory Physiology. 2003; 285 (2):mH579-88 - 148.
Crimi G, Pica S, Raineri C, Bramucci E, De Ferrari GM, Klersy C, et al. Remote ischemic post-conditioning of the lower limb during primary percutaneous coronary intervention safely reduces enzymatic infarct size in anterior myocardial infarction: A randomized controlled trial. JACC Cardiovascular Interventions. 2013; 6 (10):1055-1063 - 149.
Tasoulis MK, Livaditi O, Stamatakos M, Stefanaki C, Paneris P, Prigouris P, et al. High concentrations of reactive oxygen species in the BAL fluid are correlated with lung injury in rabbits after hemorrhagic shock and resuscitation. The Tohoku Journal of Experimental Medicine. 2009; 219 (3):193-1999. DOI: 10.1620/tjem.219.193 - 150.
Sun HY, Wang NP, Kerendi F, Halkos M, Kin H, Guyton RA, et al. Hypoxic postconditioning reduces cardiomyocyte loss by inhibiting ROS generation and intracellular Ca2+ overload. American Journal of Physiology Heart and Circulatory Physiology. 2005; 288 (4):H1900-H1908 - 151.
Douzinas EE, Livaditi O, Andrianakis I, Prigouris P, Paneris P, Villiotou V, et al. The effect of hypoxemic resuscitation from hemorrhagic shock on blood pressure restoration and on oxidative and inflammatory responses. Intensive Care Medicine. 2008; 34 :1133-1141 - 152.
Douzinas EE, Orfanos SE, Livaditi O, Augustatou K, Villiotou V, Kavantzas N, et al. Hypoxemic resuscitation prevents pulmonary capillary endothelial dysfunction induced by normoxemic resuscitation from hemorrhagic shock. Critical Care Medicine. 2009; 37 :869-875 - 153.
Younkin DP, Wagerle LC, Chance B, Maria J, Delivoria-Papadopoulos M. 31P-NMR studies of cerebral metabolic changes during graded hypoxia in newborn lambs. Journal of Applied Physiology. 1987; 62 (4):1569-1574 - 154.
Tasoulis MK, Douzinas EE. Hypoxemic reperfusion of ischemic states: An alternative approach for the attenuation of oxidative stress mediated reperfusion injury. Journal of Biomedical Science. 2016; 23 :7. DOI: 10.1186/s12929-016-0220-0 - 155.
Dyson A, Stidwill R, Taylor V, Singer M. The impact of inspired oxygen concentration on tissue oxygenation during progressive haemorrhage. Intensive Care Medicine. 2009; 35 :1783-1791 - 156.
Hafner C, Wu J, Tiboldi A, Hess M, Mitulovic G, Kaun C, et al. Hyperoxia induces inflammation and cytotoxicity in human adult cardiac myocytes. Shock. 2017; 47 :436-444 - 157.
Muller CR, Courelli V, Govender K, Omert L, Yoshida T, Cabrales P. Hypoxically stored RBC resuscitation in a rat model of traumatic brain injury and severe hemorrhagic shock. Life Sciences. 2024; 340 :122423. DOI: 10.1016/j.lfs.2024.122423 - 158.
Naumann DN, Hazeldine J, Midwinter MJ, Hutchings SD, Harrison P. Poor microcirculatory flow dynamics are associated with endothelial cell damage and glycocalyx shedding after traumatic hemorrhagic shock. Journal of Trauma and Acute Care Surgery. 2018; 84 :81-88 - 159.
Cardenas J, Dong JF, Kozar RA. Injury-induced endotheliopathy: What you need to know. Journal of Trauma and Acute Care Surgery. 2023; 95 (4):454-463 - 160.
Milford EM, Reade MC. Resuscitation fluid choices to preserve the endothelial Glycocalyx. Critical Care. 2019; 23 :77 - 161.
Reitz KM, Moore HB, Guyette FX, Sauaia A, Pusateri AE, Moore EE, et al. Prehospital plasma in injured patients is associated with survival principally in blunt injury: Results from two randomized prehospital plasma trials. Journal of Trauma and Acute Care Surgery. 2020; 88 (1):33-41. DOI: 10.1097/TA.0000000000002485 - 162.
Diebel LN, Liberati DM. Effect of tranexamic acid on endothelial von Willebrand factor/ADAMTS-13 response to in vitro shock conditions. Journal of Trauma and Acute Care Surgery. 2023; 94 (2):273-280 - 163.
Bitterman H. Bench-to-bedside review: Oxygen as a drug. Critical Care. 2009; 13 (1):205 - 164.
Weenink RP, de Jonge SW, van Hulst RA, Wingelaar TT, van Ooij PAM, Immink RV, et al. Perioperative hyperoxyphobia: Justified or not? Benefits and harms of hyperoxia during surgery. Journal of Clinical Medicine. 2020; 9 :642 - 165.
Hunt TK. Normal repair. In: Hunt TK, Dunphy JE, editors. Fundamentals of Wound Management. New York: Appleton Century Crofts; 1979. pp. 53-63 - 166.
Hunt TK. Disorders of repair and their management. In: Hunt TK, Dunphy JE, editors. Fundamentals of Wound Management. New York: Appleton Century Crofts; 1979. pp. 96-103 - 167.
Gutierrez G, Marini C, Acero AL, Lund N. Skeletal muscle PO2 during hypoxemia and isovolemic anemia. Journal of Applied Physiology (1985). 1990; 68 (5):2047-2053. DOI: 10.1152/jappl.1990.68.5.2047 - 168.
Aggarwal NR, Brower RG, Hager DN, Thompson BT, Netzer G, Shanholtz C, et al. Oxygen exposure resulting in arterial oxygen tensions above the protocol goal was associated with worse clinical outcomes in acute respiratory distress syndrome. Critical Care Medicine. 2018; 46 (4):517-524 - 169.
Convertino VA, Lye KR, Koons NJ, Joyner MJ. Physiological comparison of hemorrhagic shock and VO2max: A conceptual framework for defining the limitation of oxygen delivery. Experimental Biology and Medicine (Maywood, N.J.). 2019; 244 (8):690-701