Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
Cancer is the leading cause of death, accounting for nearly 1 in 6 deaths globally. Surgical removal of tumors, radiation therapy, chemotherapy, small molecular inhibitors, and tumor-specific antibodies are standard treatments for cancer. However, these treatments can have adverse off-target effects and poor tumor penetrance and can be inefficient in control and prevention of cancer. In the past several years, cell-based immunotherapies have been developed and applied in the clinic. These cell-based therapies are engineered to be tumor-specific, persistent and have reduced off-target complications. For cell-based immunotherapy, patient-derived leukocytes are harvested, manipulated ex vivo, and reintroduced into patients for treatment. This chapter will describe the molecular techniques used to generate engineered leukocytes, such as T cells, natural killer (NK) cells, and dendritic cells (DCs) and their applications in cancer immunotherapy.
Seattle Children’s Research Institute, Seattle, United States
*Address all correspondence to: nikita.trivedi@seattlechildrens.org
1. Introduction
According to the National Cancer Institute statistics, 40 percent of adults have been diagnosed with cancer at least once in their lives [1]. Surgery, radiation, and chemotherapy are major treatment strategies for cancer [2]. Over the past several years, tumor-specific cell-based therapies have been developed and have demonstrated efficacy in the clinic [3, 4, 5, 6, 7]. This chapter will focus on the molecular methods used to ex vivo manipulate immune cells and the application and effectiveness of engineered leukocytes as cancer therapeutics.
2. Ex vivo manipulation of patient-derived cells for cancer immunotherapy
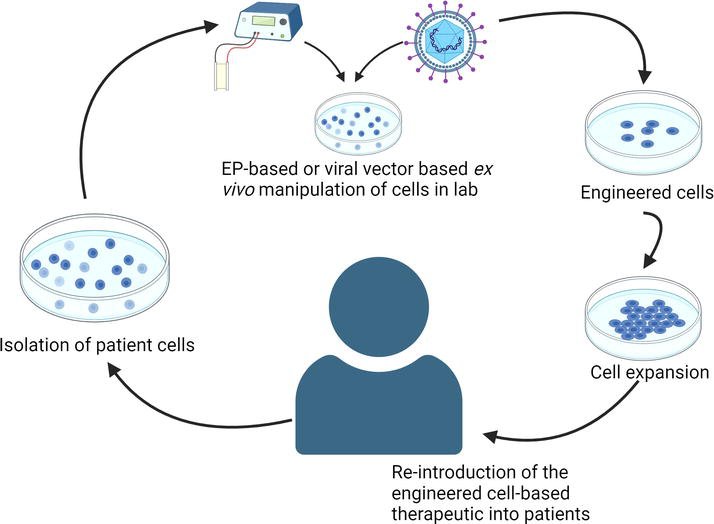
Patient leukocytes can be derived from the peripheral blood mononuclear cells (PBMCs), cord blood cells, induced pluripotent stem cells (iPSCs), and hematopoietic stem and progenitor cells (HSPCs) [5, 6, 7]. The cells are subjected to at least one or more of the following manipulations in a laboratory: (1) induced expression of one or more exogenous genes that confer anti-cancer properties to the cells, (2) gene knockdown or knockout to increase the efficacy of the cell therapy in evoking an immune response to the cancer, (3) exposure to factors and cytokines that expand and differentiate the donor cells into an effector population with potent anti-cancer capabilities, (4) exposure to tumor-associated antigens (TAAs) to generate an effector population capable of evoking anti-tumor immune responses [3, 4, 5, 6, 7]. These ex vivo-manipulated cells are then reintroduced into patients as autologous cell-based therapeutics [3, 4, 5, 6, 7]. Alternately, allogeneic engineered cells from a different donor can be introduced into cancer patients for treatment [7]. During the process of generating the anti-cancer therapeutic, the cells are subjected to several molecular methods (Figure 1).
Figure 1.
Schematic demonstrating ex vivo manipulation of patient-derived cells for generating cell-based therapeutics.
Ex vivo engineering of leukocytes is performed by the efficient transfer of ribonucleic acid (RNA) and deoxyribonucleic acid (DNA) cargo molecules into the cells [8]. This involves electroporation (EP), or viral vector-mediated delivery of genome editing reagents into the cells, or a combination of both techniques [8].
3.1 Molecular events of EP
EP is the application of external electric fields to cause reversible permeabilization of a cell membrane for the purpose of introducing molecules into the cell that are otherwise impermeable [9]. Patient or donor-derived cells are manipulated in a laboratory via EP-based transfer of RNA and DNA to induce gene disruption or exogenous gene expression. While EP is often used in the transfer of small RNA, DNA, and endonuclease enzymes into cells, its use in the transfer of large DNA vectors has been limited to a few studies because of its low efficiency [10, 11]. Large DNA constructs can be efficiently transferred by means of viral vectors [12].
3.2 mRNA-mediated gene expression
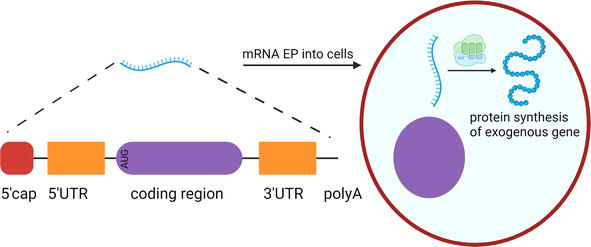
EP of messenger RNA (mRNA) is a method of gene transfer that induces transient expression of the gene of interest in the targeted cells. The mRNA is generated from a linear DNA template in a laboratory by a process known as in vitro transcription [13, 14]. RNA polymerase is used to generate a single-stranded mRNA transcript from a complementary DNA template [13, 14]. The mRNA undergoes several modifications to resemble a mature mRNA formed in eukaryotic cells [13, 14]. Post-transcription mRNA has a 5′ triphosphate, which is highly immunogenic [13, 14]. So, an inverted 7-methyl guanosine or 5’ cap gets added [13, 14]. The 5’ cap is crucial for the initiation of protein translation [13, 14]. mRNA is subject to degradation by RNA exonucleases. The addition of a polyA tail reduces mRNA degradation and increases its stability [13, 14]. The 3′ untranslated region (UTR) preceding the polyA tail has also been shown to enhance the stability of the mRNA (Figure 2) [13, 14].
Figure 2.
Gene expression via mRNA EP. Single stranded mRNA generated by in vitro transcription consists of 5′cap, 5′UTR, coding region, 3′UTR, and a polyA tail. Upon EP into the cells, the mRNA gets translated by host machinery to form functional proteins.
The synthetic mRNA can be delivered to the cells as naked mRNA, or as nanoparticle formulations, or via EP [14]. Upon entry into the cell, translation occurs in the cytosol, and the resulting proteins structurally and functionally resemble cellular proteins [15]. The synthetic mRNA eventually undergoes degradation, and the translation of the exogenous gene halts [15]. The transient and non-integrative nature of mRNA is a crucial safety feature of mRNA therapeutics [15].
3.3 Endonucleases for gene editing
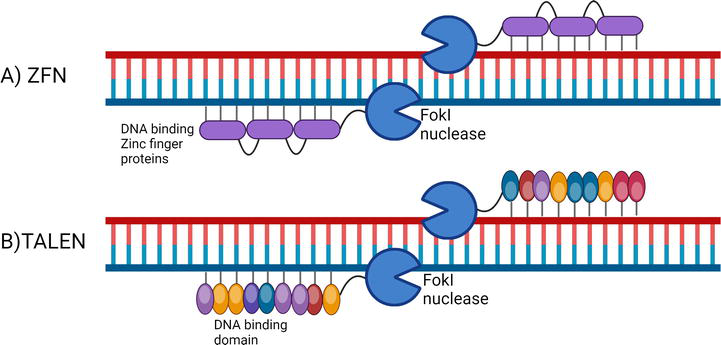
While mRNA delivery can be used for inducing transient gene expression, gene disruption and long-term expression of foreign genes can be carried out by genome editing [16]. Genome editing refers to modifications in the DNA sequence of a cell’s genome in a targeted and efficient manner [16]. The two major gene editing nucleases that have been developed are zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) [16]. The delivery of these endonucleases to the cells can be carried out via EP, although direct delivery and delivery via viral vectors have also been tested in various studies (Figure 3) [17, 18].
Figure 3.
Molecular mechanisms of gene editing nuclease. ZFN and TALEN consist of DNA binding proteins and Fokl endonuclease. The Fokl nuclease must form a dimer to make a site-specific cleavage of the target DNA.
ZFNs are chimeric proteins that constitute three to five zinc finger DNA-binding domains and a FokI endonuclease domain [19, 20]. The DNA-binding zinc finger domains are derived from transcription factors (TFs) [19, 20]. The DNA-binding domains are approximately 30 amino acids long, and they bind to three bases on the DNA [19, 20]. At least three consecutive DNA-binding domains are required for sufficient recognition [19, 20]. Two ZFNs are used to cut a single site, which is facilitated by the dimerization of the nuclease domains [21, 22, 23]. The ZFN monomer is not active until the two ZFNs assemble at the target site and form a dimer [21, 22, 23].
TALENs are proteins that are made up of DNA-binding modules linked to a FokI endonuclease cleavage domain [24]. The TALE DNA-binding repeat modules are derived from TALEs encoded by Xanthomonas proteobacteria [25]. The DNA-binding modules are made up of tandem repeats of approximately 34 amino acids in length [26, 27]. Like ZFNs, TALENs require dimerization of the endonuclease domain for cleavage of the target site [24].
ZFNs and TALENs are useful tools for the site-specific cleavage of DNA [21, 22, 23, 24, 25]. However, these techniques require expertise in protein biochemistry to generate the DNA-binding proteins for every new target site [21, 22, 23, 24, 25]. In comparison, clustered regularly interspaced short palindromic repeats (CRISPR)- CRISPR-associated protein 9 (Cas9) emerged as a simpler and more efficient tool for genome editing [17, 28].
3.4 Molecular mechanisms of CRISPR-Cas9 editing
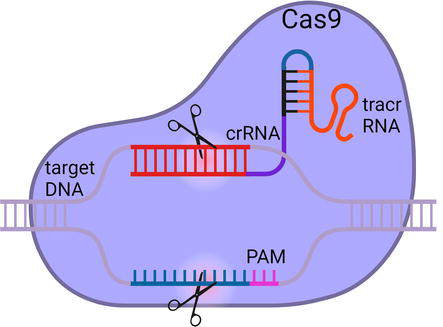
CRISPR-Cas9 was discovered as a part of adaptive immunity in prokaryotic cells for the detection and cleavage of foreign nucleic acids [28, 29]. Since its discovery, it has been widely applied for genome editing (Figure 4) [12].
Figure 4.
CRISPR-Cas9 mediated genome editing. crRNA-tracrRNA hybrids along with the Cas9 complex are delivered to the cells via EP. Upon recognition of the target site, Cas9 nuclease makes a double stranded break in the target DNA sequence.
The CRISPR-Cas9 machinery consists of a CRISPR RNA (crRNA) with a spacer motif complementary to a target DNA sequence [28, 29]. The crRNA hybridizes with the target sequence adjacent to a protospacer adjacent motif (PAM) [28, 29]. PAM sites are made of the sequence NGG, where N represents any nucleotide [28, 29]. The crRNA also hybridizes with a transactivating RNA (tracrRNA), and in turn, tracrRNA forms a complex with Cas9 [28, 29]. Alternately, single-guide RNA can also be synthesized instead of crRNA and tracrRNA hybrids [30]. The CRISPR-Cas9 complexes are delivered into the cells via EP [31]. Upon hybridization of the spacer motif of the crRNA to a target sequence, the Cas9 protein makes a blunt double-stranded break [28, 29]. In this method, Cas9 generates a site-specific cleavage with high efficiency [28, 29].
3.5 DNA repair pathways
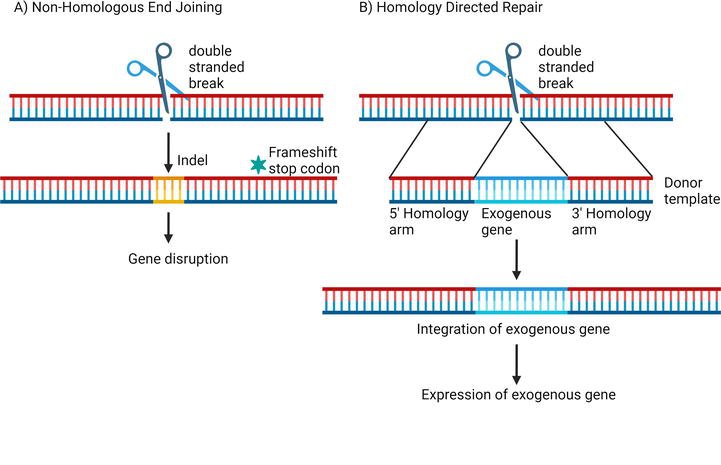
Endonucleases have been used to induce double-stranded DNA breaks into the genome of cells [17, 18]. These DNA breaks trigger the DNA repair pathway in the cells. In eukaryotic cells, DNA breaks can be repaired by the error-prone process of nonhomologous end joining (NHEJ) [32]. During NHEJ, nucleotide insertions or deletions (indels) can happen at the site of the cleavage [32]. These indels can cause frame shifts or introduce premature stop codons, causing disruption in the expression of the target gene [32].
Another method of DNA repair is known as homology-directed repair (HDR). It can occur naturally in cells by using sister chromatid or can be induced experimentally by providing repair templates that are complementary to the site of DNA break [33, 34]. HDR can be used to integrate exogenous genes into the genome of a cell and drive the expression of foreign genes using the host transcription and translation machinery [35].
HDR repair templates or donor templates are either single-stranded oligodeoxynucleotides or double-stranded DNA vectors that have two homology arms with sequences complementary to the region on either side of the targeted DNA break site [36]. The 5’ homology arm is followed by a promoter to drive expression of the inserted gene [37]. The promoter is adjacent to the exogenous gene, which gets inserted at the site of the double-stranded DNA break [37]. Because of the large size of the DNA repair templates, their delivery into the cells is challenging [10, 11]. In most genome editing studies, viral vectors are a preferred method for the delivery of DNA repair templates (Figure 5) [12].
Figure 5.
DNA repair pathways following double-stranded DNA break. (A) NHEJ is error prone and it leads to indels in the sequence which can cause gene disruption. (B) HDR can be induced by providing repair templates for the expression of exogenous gene at the targeted loci.
3.6 Viral vectors for genome engineering
Following endonuclease-mediated double-stranded DNA breaks, viral vectors such as adeno-associated viral vectors can be provided as a repair template to integrate the gene of interest at the targeted location [37]. On the other hand, vectors such as lentiviral vectors do not integrate at a specific target site but instead integrate at various sites in the actively transcribed gene loci of the transduced cell [38]. The most commonly used viral vectors for ex vivo genome engineering of leukocytes are adenovirus vectors, adeno-associated virus vectors, retrovirus vectors, and lentivirus vectors [39]. Each viral vector has several different serotypes that demonstrate tropism to certain cell types and therefore can be used for editing specific cell types [39]. The viral vectors have specific capsid or envelope proteins that can bind to receptors on the cell surface, causing internalization of the viral vector and the delivery of the DNA cargo into the cytosol [12]. The DNA cargo migrates into the nucleus and integrates into the genome. Alternately, certain viral genomes reside episomally within the host cell [39]. These viral vectors are a means to incorporate and induce the expression of exogenous genes in donor cells (Table 1) [39].
Method
Purpose
Advantages
Drawbacks
References
mRNA-mediated gene expression
Exogenous gene expression
High transfection efficiency Low-risk non-integrative method
Transient secretion of exogenous proteins because of low mRNA stability
Long-term secretion of exogenous protein by the transduced cells Delivery of large DNA constructs at the targeted loci
Possible risk of insertional mutagenesis Cells must express surface receptors capable of binding viral vectors Potential immunogenicity from vector component
4. Preclinical and clinical outcomes of engineered leukocytes in cancer immunotherapy
The advent of molecular techniques has facilitated the development of cell-based cancer immunotherapy. This section will discuss the leukocyte populations that have been engineered and are clinically relevant for cancer immunotherapy.
4.1 Engineered T cells for cancer immunotherapy
Immune cells play a crucial role in providing defense against tumors by specifically targeting and killing cancer cells [41]. White blood cells known as T lymphocytes can recognize tumor-associated antigens when presented by proteins known as the Major Histocompatibility Complex (MHC) [42]. Upon recognition of the tumor-associated antigens, T cells undergo activation and release cytotoxic proteins that target the tumor cells [43]. Moreover, T cells can release inflammatory cytokines that can activate other immune cells and promote tumor clearance [43]. For efficient recognition of cancer cells, T cells have been engineered to express tumor antigen-specific T cell receptors (TCR) [44, 45]. However, tumor cells can downregulate the expression of MHC and therefore reduce the presentation of the antigens and evade immune recognition [43].
4.1.1 Chimeric antigen receptor T cells
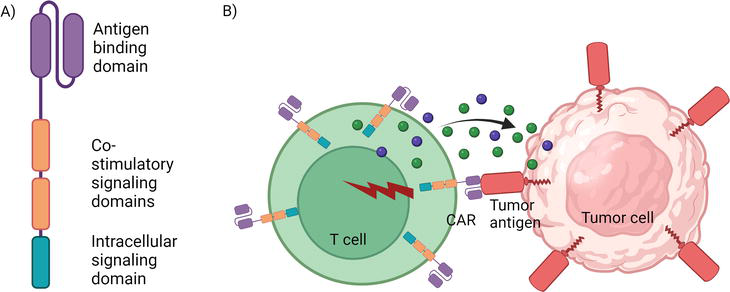
To facilitate T cells to recognize tumor cells in the absence of MHC proteins, chimeric antigen receptor T cells (CAR T cells) were designed [46]. The CAR is a synthetic receptor that directs the T cells to recognize antigen-expressing tumor cells directly without the need for MHC proteins [46]. The CAR consists of an antigen-binding domain on the extracellular side, which is derived from antigen-binding regions of antibodies [47]. The antigen-binding domain is linked to intracellular signaling domains (for example, CD3 zeta), which induces T cell activation [47]. There are also certain co-stimulatory receptor intracellular domains that enhance signaling post-activation [47].
The CAR can be expressed via genomic integration using viral vectors or by the EP of CAR mRNA into the T cells [3, 4, 48, 49, 50]. These engineered CAR T cells can be reintroduced into patients as therapeutics [3, 4, 48, 49, 50]. The T lymphocytes get activated upon CAR-mediated recognition of the tumor antigen and release cytotoxic and inflammatory molecules that target the tumor cells [47]. CAR T cell therapy has shown high efficacy in the treatment of B cell malignancies (Figure 6) [51, 52, 53, 54].
Figure 6.
CAR T cell structure and function. (A) The structure of a CAR. (B) Engineered T cell expressing CAR bind to an antigen on the surface of a tumor cell. Upon antigen recognition, CAR T cell becomes activated and releases cytokines and cytotoxic molecules to kill the tumor cell.
4.1.2 Engineered TCR-based therapeutics
Unlike hematologic malignancies, CAR T cell treatment in solid tumors has not yielded encouraging results [55]. Engineered T cell receptor therapy is an alternative to CAR T cell therapy [56]. CAR T cells are restricted to the recognition of surface antigens, whereas TCR can recognize surface and intracellular antigens presented by APCs [56]. Moreover, TCR requires a lower epitope density for activation than a CAR, which can lead to efficient T cell responses in tumors with low antigen expression [56]. Because of these reasons, engineered TCRs have been developed for the treatment of various cancers.
Engineered T cells that are specific to different cancer antigens such as melanoma antigen recognized by T cells-1 (MART-1), glycoprotein-100, carcinoembryonic antigen (CEA), cancer testis antigen called NY-ESO-1, and melanoma antigen gene protein family (MAGE) have been generated [57, 58, 59, 60, 61]. These engineered T cells have also been tested in clinical trials and have produced encouraging results [57, 58, 59, 60, 61]. For instance, in patients with metastatic synovial sarcoma, T cells engineered to express TCR specific to the cancer antigen NY-ESO-1 were infused into patients [60]. The treatment showed persistence of edited cells for about 6 months, and antitumor responses were observed in 50 percent of the patients [60]. Currently, more work is being done to find cancer neoantigens to develop neo-TCR-engineered T cells with enhanced tumor-specificity [62].
4.1.3 T cell gene disruption in cancer immunotherapy
A major challenge in the treatment of cancer is the immunosuppressive tumor microenvironment (TME). To evade detection and clearance, tumor cells express surface proteins that can suppress the immune cells [63]. One such surface protein is known as programmed death ligand 1 (PD-L1), which is highly expressed by several cancer cells [63]. PD-L1 interacts with another protein expressed on immune cells, especially T lymphocytes, known as programmed cell death protein-1 (PD-1) [63]. The PD-L1-PD-1 interaction limits T cell activation and prevents effective tumor clearance [63]. Elimination of PD-1 in T cells enhances T cell activation and the killing of cancer cells. EP of CRISPR-Cas9 or TALEN reagents have been used to disrupt the expression of the PD-1 gene in T cells [50, 64].
Recent studies in cancer immunotherapy have now focused on multiplex genome editing [3, 4]. Patient-derived T cells are harvested and are manipulated to induce the expression of a CAR or tumor antigen-specific T cell receptor along with disruption of immunosuppressive genes such as PD-1 [3, 4]. The multiplex genome editing approach allows the generation and delivery of novel cell-based therapies that are more effective than conventional cell-based therapies.
4.2 Engineered natural killer (NK) cells for cancer immunotherapy
Engineered T cells have shown efficacy in cancer immunotherapy; however, generating autologous engineered T cells is an expensive and time-consuming process [65]. NK cells can be an allogeneic donor for CAR therapy that does not cause graft versus host disease [65]. NK cells are lymphocytes that, like T cells, also have the capacity to target tumor cells through the release of cytotoxic and inflammatory molecules [66]. NK cells have multiple mechanisms to carry out tumor killing, which include activation via surface receptors, engineered CARs, and antibodies [66]. Upon activation, NK cells release cytotoxic granules consisting of perforin and granzyme that perforate and kill the target cells [66].
Another advantage of developing NK cells as an allogeneic therapeutic product is the source of the donor NK cells [67, 68]. NK cells can be generated from PBMCs, iPSCs, and cord blood cells [7, 67, 68]. The cord blood-derived NK cells provide a rich source that can be expanded in vitro by the addition of recombinant factors and cytokines [67, 68]. These in vitro-generated NK cells can be engineered to produce an NK-CAR product [68, 69].
4.2.1 CAR-NK therapy
Several studies have been done on the development of NK-CAR [70, 71, 72]. Activation of NK cells occur through the engagement of the surface receptor known as NKG2D [73, 74, 75]. The ligand for NKG2D is expressed on cancer cells [73, 74, 75]. NK-CAR is made up of the NKG2D receptor protein and intracellular activating domains [73, 74, 75]. The NK-CAR is delivered into NK cells via EP, and the NK-CAR-expressing cells are reintroduced into patients for tumor clearance [73, 74, 75].
NK cells can also be engineered to express tumor-antigen-specific CARs, similar to CAR T cells [69, 76]. Cord blood-derived NK cells expressing anti-CD19 CAR have been administered in phase 1 and 2 clinical trials for lymphoma and leukemia patients [76]. There was a 73 percent response rate among patients without the development of any adverse effects [76]. CAR-NK treatments that target the tumor antigen CD70 are being developed for non-Hodgkin lymphoma [69]. This NK cell therapy is currently being tested in a clinical trial (NCT05092451) for patients with CD70-expressing hematological malignancies [77].
Over the past several years, NK cell therapies have evolved to become long-lasting and efficacious anti-cancer therapies. Interleukin 15 was incorporated as part of the CAR construct to maintain the metabolic fitness and effector functions of the CAR NK cells in the TME [78, 79]. In another study, an NK self-recognizing inhibitory CAR was co-expressed with a tumor-targeting activating CAR to prevent NK cells from killing sibling NK cells [79]. Both studies demonstrate that NK cell therapies can be subjected to innovative engineering to generate a persistent and efficient therapeutic product [78, 79].
4.3 Engineered dendritic cells (DCs) for cancer immunotherapy
DCs are specialized immune cells that reside in lymphoid and nonlymphoid compartments and act as sentinels [80]. Upon antigen encounter, DCs can efficiently capture antigen, process it, and present it in the context of MHCI and MHCII molecules [5, 81]. DC-mediated antigen presentation is a major pathway that facilitates the activation and expansion of antigen-specific CD4 and CD8 T cells [5, 81]. DCs also display the capacity to migrate between lymphoid and nonlymphoid tissues, causing antigen spreading [5, 81]. DCs can modulate inflammatory signals and control lymphocyte homing to various tissues [5, 81]. Because of these properties, manipulating DCs can mediate the regulation of the TME [5, 81]. For the purposes of immunotherapy, patient-derived peripheral blood monocytes or HSPCs can be treated with recombinant cytokines or factors to induce differentiation and maturation into a DC population [5, 82, 83].
4.3.1 DCs as APCs in cancer
To exploit the APC function of DCs in cancer immunotherapy, patient-derived DCs can be loaded with TAA and injected back into patients [83]. The TAA can be introduced into DCs by incubation with tumor lysates, or by incubation with tumor-derived or recombinant peptides, or via EP of mRNA encoding for tumor antigens into the DCs [83]. For metastatic prostate cancers, a DC-based therapy known as sipuleucel-T is approved by the Food and Drug Administration [84]. In this treatment, patient-derived immune cells are incubated with tumor antigen prostate-specific phosphatase to generate potent tumor-specific APCs that are reintroduced into patients to activate anti-tumor immune responses [84]. In ovarian cancer, incubation of patient-derived DCs with autologous whole tumor lysates demonstrated efficient generation of a DC-based vaccine [85]. Vaccination induced a T cell response against known tumor epitopes as well as previously unrecognized neoepitopes [85]. The potent T cell response against autologous tumor antigens was associated with prolonged patient survival [85]. In a phase 2 study of acute myeloid leukemia, DCs electroporated with the tumor antigen known as Wilm’s tumor 1 mRNA were given as post-remission treatment [86]. This DC vaccination was demonstrated as an effective strategy to prevent or delay the chance of relapse in patients that are in remission [86]. These studies have highlighted the importance of DCs in driving a tumor antigen-specific T cell response, tumor control, and prevention.
4.3.2 Engineered DCs in the TME
DCs can also be edited to knock out certain proteins that reduce their effector functions [87, 88, 89]. Deletion of protein associated with the antigen processing pathways, such as YTHDF1, has demonstrated improved antigen presentation and anti-tumor T cell responses [89]. To enhance the immune stimulatory capacity of DCs, overexpression of proteins that increase DC effector function has been employed [87, 88]. In patients with non-small cell lung carcinoma, peripheral blood-derived DCs were transduced to express the C-C motif chemokine ligand 21 (CCL21) using an adenoviral vector [90]. Intra-tumor vaccination with CCL21-expressing DCs demonstrated a systemic increase in anti-tumor responses along with a localized increase in the infiltration of CD8 T cells in the TME [90].
These studies showcase a range of functions performed by DCs and a central role for DCs in driving antigen-specific T cell responses in cancer.
Cell-based immunotherapies have revolutionized cancer treatments through the development of tumor-specific and personalized regimens for patients. However, there are several areas of cell-based cancer immunotherapy that need improvement.
5.1 Low persistence of engineered cells in patients
A major reason for the poor outcome of cell-based cancer immunotherapy is the limited persistence of the engineered T cells [91]. Recent studies have shown that a T cell memory-like transcriptomic profile of CAR T cells is associated with good treatment outcomes and the long-term persistence of cells [92]. A TF known as FOXO1 is involved in the memory programming of CAR T cells [93]. In future studies, CAR T cells with memory reprogramming can be generated to increase the persistence and efficacy of CAR T cell therapy.
5.2 Toxicity from cell-based immunotherapy
CAR T cells have been effective in hematological malignancies; however, infusion of CAR T cells has been associated with severe adverse effects [94]. Cytokine release syndrome (CRS) occurs because of systemic inflammation and a rise in the level of cytokines upon CAR T injection [94]. An antibody that blocks the interleukin-6 receptor is used as a treatment measure for CRS [94]. Patients are also at risk of developing immune effector cell-associated neurotoxicity syndrome (ICANS) and need to be in the care of a trained professional that can recognize the signs of ICANS [94]. Intensive care support and corticosteroids are used to treat ICANS [94]. These adverse effects last from hours to several days post-infusion of CAR T cells [94]. Another challenge is the application of CAR T cells in the treatment of solid tumors [95]. The CAR T cells can target nonmalignant tissue that expresses the tumor antigen and cause substantial toxicity [95]. Safe cell-based therapies with reduced systemic adverse effects need to be developed. NK cell therapies have shown efficacy in tumor control without the adverse effects associated with T cell therapies [65].
5.3 Off-the-shelf engineered cell-based therapy
Cell-based treatments are generated from autologous cells that are harvested from individual patients, manipulated ex vivo, and reintroduced into the same patient [3, 4, 5, 6, 7]. While this method ensures personalized treatment, the process of generating patient-derived engineered cells is expensive, time-consuming, labor-intensive, and requires expertise [3, 4, 5, 6, 7]. Also, this method is dependent on the patient having a sufficient population of donor cells. Therefore, having an alternative allogeneic cell-based product or off-the-shelf product will be cost-effective, rapid, and universally applicable to more patients [65, 69, 76]. The generation of engineered NK cells is a step in the direction of generating off-the-shelf products and should be further developed [65, 69, 76].
5.4 In vivo engineering of leukocytes for cancer immunotherapy
Another drawback of the current state of cell-based therapies is the duration and feasibility of ex vivo cell manufacturing [96, 97]. Biotechnology companies are studying ways to deliver viral vectors to engineer cells in vivo rather than manipulating cells ex vivo [96, 97]. VivoVec, a replication-incompetent lentiviral vector platform, has been developed by Umoja Biopharma to generate CAR T cells [97]. The VivoVec particle expresses an anti-CD3 single-chain variable fragment that allows it to bind and transduce T cells [97]. Upon transduction, a payload transgene consisting of anti-CD19 CAR gets expressed by the T cells [97]. Administration of VivoVec particles to humanized mice with CD19-expressing tumors demonstrated expansion of CAR T cells and clearance of B cell tumors [97]. Interius Biotherapeutics has also developed a non-replicating, self-inactivating lentiviral vector delivery platform to generate anti-CD20 CAR T cells and CAR NK cells [96]. The lentiviral vector expresses surface anti-CD7, which allows it to bind and transduce T cells and NK cells [96]. The T cells and NK cells express the functional anti-CD20 CAR, and this strategy has been shown to be efficacious in tumor clearance in preclinical models [96]. Taken together, these studies demonstrate the advances in gene therapy for cancer treatments.
Engineered leukocytes have been a major advance in cancer therapeutics because they generate tumor-specific immune responses in an autologous manner for patients. The efficacy of CAR T cells has been very encouraging for the further development of engineered leukocytes. However, the personalized treatment approach using autologous cells is expensive and not feasible to administer to a large cohort of patients. There is a need to develop allogeneic engineered products such as NK CARs that are well tolerated, efficacious, and can be applied universally to patients. Another approach that does not require donor cells is gene therapy platforms that can generate tumor-specific engineered cells in vivo by delivering viral vectors. However, treatment with viral vectors always presents a risk of immunogenicity and insertional mutagenesis. Overall, cell-based immunotherapies have revolutionized cancer treatments and will continue to evolve in the future.
1.National Cancer Institute. Cancer Statistics. 2020. Available from: https://www.cancer.gov/about-cancer/understanding/statistics
2.Saini KS, Twelves C. Determining lines of therapy in patients with solid cancers: A proposed new systematic and comprehensive framework. British Journal of Cancer. 2021;125(2):155-163
3.Ren J et al. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clinical Cancer Research. 2017;23(9):2255-2266
4.Stadtmauer EA et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367(6481):eaba7365
5.Perez CR, De Palma M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nature Communications. 2019;10(1):5408
6.Abou-El-Enein M et al. Scalable manufacturing of CAR T cells for cancer immunotherapy. Blood Cancer Discovery. 2021;2(5):408-422
7.Shin MH et al. NK cell-based immunotherapies in cancer. Immune Network. 2020;20(2):e14
8.Lukjanov V, Koutna I, Simara P. CAR T-cell production using nonviral approaches. Journal of Immunology Research. 2021;2021:6644685
9.Kotnik T et al. Cell membrane electroporation-part 1: The phenomenon. IEEE Electrical Insulation Magazine. 2012;28(5):14-23
10.Lesueur LL, Mir LM, Andre FM. Overcoming the specific toxicity of large plasmids electrotransfer in primary cells in vitro. Molecular Therapy - Nucleic Acids. 2016;5(3):e291
11.Ribeiro S et al. Plasmid DNA size does affect nonviral gene delivery efficiency in stem cells. Cellular Reprogramming. 2012;14(2):130-137
12.Lino CA et al. Delivering CRISPR: A review of the challenges and approaches. Drug Delivery. 2018;25(1):1234-1257
13.Beck JD et al. mRNA therapeutics in cancer immunotherapy. Molecular Cancer. 2021;20(1):69
14.Deng Z et al. mRNA vaccines: The dawn of a new era of cancer immunotherapy. Frontiers in Immunology. 2022;13:887125
15.Qin S et al. mRNA-based therapeutics: Powerful and versatile tools to combat diseases. Signal Transduction and Targeted Therapy. 2022;7(1):166
16.Shamshirgaran Y et al. Tools for efficient genome editing; ZFN, TALEN, and CRISPR. Methods in Molecular Biology. 2022;2495:29-46
17.Bhardwaj A, Nain V. TALENs-an indispensable tool in the era of CRISPR: A mini review. Journal, Genetic Engineering & Biotechnology. 2021;19(1):125
18.Bobis-Wozowicz S et al. Non-integrating gamma-retroviral vectors as a versatile tool for transient zinc-finger nuclease delivery. Scientific Reports. 2014;4:4656
19.Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011;188(4):773-782
20.Pabo CO, Peisach E, Grant RA. Design and selection of novel Cys2His2 zinc finger proteins. Annual Review of Biochemistry. 2001;70:313-340
21.Bitinaite J et al. FokI dimerization is required for DNA cleavage. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(18):10570-10575
22.Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(3):1156-1160
23.Smith J et al. Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Research. 2000;28(17):3361-3369
24.Joung JK, Sander JD. TALENs: A widely applicable technology for targeted genome editing. Nature Reviews. Molecular Cell Biology. 2013;14(1):49-55
25.Bogdanove AJ, Voytas DF. TAL effectors: Customizable proteins for DNA targeting. Science. 2011;333(6051):1843-1846
26.Boch J et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326(5959):1509-1512
27.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326(5959):1501
28.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816-821
29.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819-823
30.Chen B et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155(7):1479-1491
31.Horodecka K, Duchler M. CRISPR/Cas9: Principle, applications, and delivery through extracellular vesicles. International Journal of Molecular Sciences. 2021;22(11):6072
32.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annual Review of Biochemistry. 2010;79:181-211
33.Carroll D. Genome engineering with targetable nucleases. Annual Review of Biochemistry. 2014;83:409-439
34.Wang J et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nature Biotechnology. 2015;33(12):1256-1263
35.Schubert MS et al. Optimized design parameters for CRISPR Cas9 and Cas12a homology-directed repair. Scientific Reports. 2021;11(1):19482
36.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 2013;8(11):2281-2308
38.Poletti V, Mavilio F. Designing lentiviral vectors for gene therapy of genetic diseases. Viruses. 2021;13(8):1526
39.Bulcha JT et al. Viral vector platforms within the gene therapy landscape. Signal Transduction and Targeted Therapy. 2021;6(1):53
40.Yip BH. Recent advances in CRISPR/Cas9 delivery strategies. Biomolecules. 2020;10(6):839
41.Hiam-Galvez KJ, Allen BM, Spitzer MH. Systemic immunity in cancer. Nature Reviews. Cancer. 2021;21(6):345-359
42.Geiger TL, Jyothi MD. Development and application of receptor-modified T lymphocytes for adoptive immunotherapy. Transfusion Medicine Reviews. 2001;15(1):21-34
43.Shah K et al. T cell receptor (TCR) signaling in health and disease. Signal Transduction and Targeted Therapy. 2021;6(1):412
44.Schober K et al. Orthotopic replacement of T-cell receptor alpha- and beta-chains with preservation of near-physiological T-cell function. Nature Biomedical Engineering. 2019;3(12):974-984
45.Rapoport AP et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nature Medicine. 2015;21(8):914-921
46.Sterner RC, Sterner RM. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer Journal. 2021;11(4):69
47.Jayaraman J et al. CAR-T design: Elements and their synergistic function. eBioMedicine. 2020;58:102931
48.Cherkassky L et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. The Journal of Clinical Investigation. 2016;126(8):3130-3144
49.Serganova I et al. Enhancement of PSMA-directed CAR adoptive immunotherapy by PD-1/PD-L1 blockade. Molecular Therapy - Oncolytics. 2017;4:41-54
50.Rupp LJ et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific Reports. 2017;7(1):737
51.Brentjens RJ et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Science Translational Medicine. 2013;5(177):177ra38
52.Kalos M et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Science Translational Medicine. 2011;3(95):95ra73
53.Maude SL et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. The New England Journal of Medicine. 2014;371(16):1507-1517
54.Turtle CJ et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. The Journal of Clinical Investigation. 2016;126(6):2123-2138
55.Albelda SM. CAR T cell therapy for patients with solid tumours: Key lessons to learn and unlearn. Nature Reviews. Clinical Oncology. 2024;21(1):47-66
56.Baulu E et al. TCR-engineered T cell therapy in solid tumors: State of the art and perspectives. Science Advances. 2023;9(7):eadf3700
57.Johnson LA et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535-546
58.Parkhurst MR et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Molecular Therapy. 2011;19(3):620-626
59.Robbins PF et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clinical Cancer Research. 2015;21(5):1019-1027
60.D'Angelo SP et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 (c259)T cells in synovial sarcoma. Cancer Discovery. 2018;8(8):944-957
61.Ramachandran I et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. Journal for Immunotherapy of Cancer. 2019;7(1):276
62.Pang Z et al. Neoantigen-targeted TCR-engineered T cell immunotherapy: Current advances and challenges. Biomarker Research. 2023;11(1):104
63.Ai L, Xu A, Xu J. Roles of PD-1/PD-L1 pathway: Signaling, cancer, and beyond. Advances in Experimental Medicine and Biology. 2020;1248:33-59
64.Menger L et al. TALEN-mediated inactivation of PD-1 in tumor-reactive lymphocytes promotes intratumoral T-cell persistence and rejection of established tumors. Cancer Research. 2016;76(8):2087-2093
65.Daher M et al. CAR-NK cells: The next wave of cellular therapy for cancer. Clinical & Translational Immunology. 2021;10(4):e1274
66.Yoon SR, Kim TD, Choi I. Understanding of molecular mechanisms in natural killer cell therapy. Experimental & Molecular Medicine. 2015;47(2):e141
67.Damele L et al. Cord blood-derived natural killer cell exploitation in immunotherapy protocols: More than a promise? Cancers (Basel). 2022;14(18):4439
68.Mehta RS, Shpall EJ, Rezvani K. Cord blood as a source of natural killer cells. Frontiers in Medicine (Lausanne). 2015;2:93
69.Rafei H, Basar R, Acharya S, Zhang P, Liu P, Moseley SM, et al. Targeting T-cell lymphoma using CD70-directed cord blood-derived CAR-NK cells. Blood. 2023;142:4811
70.Wilk AJ et al. Charge-altering releasable transporters enable phenotypic manipulation of natural killer cells for cancer immunotherapy. Blood Advances. 2020;4(17):4244-4255
71.Liu S et al. NK cell-based cancer immunotherapy: From basic biology to clinical development. Journal of Hematology & Oncology. 2021;14(1):7
72.Schmidt P, Raftery MJ, Pecher G. Engineering NK cells for CAR therapy-recent advances in gene transfer methodology. Frontiers in Immunology. 2020;11:611163
73.Chang YH et al. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Research. 2013;73(6):1777-1786
74.Ng YY, Tay JCK, Wang S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Molecular Therapy - Oncolytics. 2020;16:75-85
75.Xiao L et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Molecular Therapy. 2019;27(6):1114-1125
76.Liu E et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. The New England Journal of Medicine. 2020;382(6):545-553
77.M.D. Clinical trial information. Anderson Cancer Center; Available from: https://clinicaltrials.gov/study/NCT05092451
78.Li L et al. Loss of metabolic fitness drives tumor resistance after CAR-NK cell therapy and can be overcome by cytokine engineering. Science Advances. 2023;9(30):eadd6997
79.Li Y et al. KIR-based inhibitory CARs overcome CAR-NK cell trogocytosis-mediated fratricide and tumor escape. Nature Medicine. 2022;28(10):2133-2144
80.Wculek SK et al. Dendritic cells in cancer immunology and immunotherapy. Nature Reviews. Immunology. 2020;20(1):7-24
81.Santos PM, Butterfield LH. Dendritic cell-based cancer vaccines. Journal of Immunology. 2018;200(2):443-449
82.Lin MJ et al. Cancer vaccines: The next immunotherapy frontier. Nature Cancer. 2022;3(8):911-926
83.Fu C et al. Dendritic cell-based vaccines against cancer: Challenges, advances and future opportunities. Immunological Investigations. 2022;51(8):2133-2158
84.Handy CE, Antonarakis ES. Sipuleucel-T for the treatment of prostate cancer: Novel insights and future directions. Future Oncology. 2018;14(10):907-917
85.Tanyi JL et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Science Translational Medicine. 2018;10(436):eaao5931
86.Anguille S et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood. 2017;130(15):1713-1721
87.Wilgenhof S et al. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patients with pretreated advanced melanoma. Journal of Clinical Oncology. 2016;34(12):1330-1338
88.Sundarasetty BS et al. Lentivirus-induced 'Smart' dendritic cells: Pharmacodynamics and GMP-compliant production for immunotherapy against TRP2-positive melanoma. Gene Therapy. 2015;22(9):707-720
89.Han D et al. Anti-tumour immunity controlled through mRNA m(6)a methylation and YTHDF1 in dendritic cells. Nature. 2019;566(7743):270-274
90.Lee JM et al. Phase I trial of intratumoral injection of CCL21 gene-modified dendritic cells in lung cancer elicits tumor-specific immune responses and CD8(+) T-cell infiltration. Clinical Cancer Research. 2017;23(16):4556-4568
91.Pietrobon V et al. Improving CAR T-cell persistence. International Journal of Molecular Sciences. 2021;22(19):10828
92.Chen GM et al. Integrative bulk and single-cell profiling of premanufacture T-cell populations reveals factors mediating long-term persistence of CAR T-cell therapy. Cancer Discovery. 2021;11(9):2186-2199
93.Doan A et al. FOXO1 is a master regulator of CAR T memory programming. Research Square. 2023
94.Tallantyre EC et al. Neurological updates: Neurological complications of CAR-T therapy. Journal of Neurology. 2021;268(4):1544-1554
95.Flugel CL et al. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nature Reviews. Clinical Oncology. 2023;20(1):49-62
96.Andorko JI et al. Targeted in vivo generation of CAR T and NK Cells utilizing an engineered lentiviral vector platform. Blood. 2023;142:763
97.Michels KR et al. Preclinical proof of concept for VivoVec, a lentiviral-based platform for in vivo CAR T-cell engineering. Journal for Immunotherapy of Cancer. 2023;11(3)
Written By
Nikita Trivedi
Submitted: 27 December 2023Reviewed: 03 January 2024Published: 10 April 2024