
Abstract
This chapter reviews the important neurophysiological mechanisms that drive symptoms characteristic of comorbid depression and metabolic disease. It outlines how insulin impairment in the periphery
Keywords
- serotonin
- insulin
- tryptophan
- diabetes
- depression
- complex adaptive systems
- psychological resilience
1. Introduction
Serotonin (5-hydroxytryptamine or 5-HT) is an important neurotransmitter and hormone involved in various functions, such as emotion, cognition, learning, metabolism, sleep, platelet function, and gastrointestinal motility [1, 2, 3, 4, 5]. These functions depend on tight regulation of available 5-HT and other indole metabolites of Tryptophan (Trp) throughout the body and brain [1]. Research continues to uncover how imbalances of these molecules is associated with insulin impairment, and a cascade of disruption via complex interactions between the mind and body. This review highlights bidirectional feedbacks between 5-HT and insulin imbalances that are associated with energy and emotion [6, 7, 8, 9]. It attempts to explain some of the underlying limitations to psychological resilience commonly experienced by persons with compromised insulin function, and to explain potential mechanistic links behind highly comorbid depression and metabolic disease.
1.1 Comorbid depression and metabolic disease
Diabetes mellitus is a metabolic condition characterized by chronic high blood sugar levels due to an underlying impairment in insulin secretion (Type 1, T1D), action (Type 2, T2D) or both [10]. Metabolic syndrome – the co-occurrence of insulin resistance, obesity, atherogenic dyslipidemia, and hypertension – is a high-risk factor for the development of T2D [11]. Diabetes is commonly comorbid with mood disorders linked to altered central 5-HT, and most often with disorders involving depression [12, 13, 14]. Diabetics are two times more likely to have depression compared to non-diabetics [15, 16, 17]. This comorbid relationship between mental health disorders and both types of diabetes appears to be bidirectional, and their co-occurrence tends to exacerbate symptom severity [12, 18, 19].
Depression refers to any of several depressive or mood disorders outlined in the DSM3 handbook, broadly involving extended periods of sadness, emptiness and depressed mood, and often accompanied by disturbances in cognition, sleep and energy levels [18, 20, 21]. There is less clarity and consensus on underlying impairments involved in depression. Since the 1960s, depression has most commonly been explained by a deficiency in brain 5-HT activity [22, 23]. This paralleled industry’s need to allocate pharmaceutical prescriptions according to symptomology [24] and thus their marketing of selective serotonergic medications [25, 26]; most commonly, 5-HT reuptake inhibitors (SSRIs) which function to increase synaptic 5-HT and upregulate 5-HT neurotransmission [7].
The neurobiological basis and treatment of depression is still the predominant narrative accepted by the Western public and endorsed by leading research and educational materials [27, 28]. However, the idea that depression is caused strictly by low 5-HT activity is mostly rejected by experts [25, 28, 29]. Some even argue that high 5-HT activity is behind depressive states, and that this is a functional response [7]. What remains largely undisputed is the extensive evidence that changes in brain 5-HT has some role in depressive states and energy regulation.
1.2 Mechanisms behind diagnostic labels
Medical research on diabetes and depression now consider the environmental and sociocultural contexts in which these neurophysiological systems interact, and how these contexts shape our organization of disease and disorder [26, 29, 30, 31]. Sociocultural factors influence how these states are generated, whether their symptoms and syndromes are interpreted as adaptive or maladaptive, and at what point these symptoms become categorized as diseases, syndromes or disorders [24, 30, 31, 32, 33]. Psychological disorders are subjectively categorized by non-universal, Western constructs of self, normality and value [34]. Moreover, our scientific understandings of the neurological basis for cognition, emotion and behavior have been predominantly measured on Western populations, despite the evidence that changes in neurobiology lead to different expressions in different cultures [35].
The RDoC system from the National Institute of Mental Health is a more recent and systemic tool for categorizing mental health by functional constructs that represent a specified functional dimension of behavior [36]. The constructs are systems of response to the environment (e.g., positive valence, or reward system) which are then characterized in aggregate by the genes, molecules, circuits and behaviors involved. The RDoC thus examines the mechanism that “drive psychiatric symptoms” [37].
1.3 Peripheral insulin impairment and central serotonin availability
In light of these evolving explanations, this chapter focuses on mechanistic links between insulin and 5-HT systems that
We thus use the unconventional term,
Experimental studies over several decades have investigated the mechanisms of Trp metabolism and the impacts of induced-diabetes on the distribution of 5-HT in distinct central and peripheral systems. Yet this literature remains largely fragmented, each representing isolated pieces of complex relationships involved in insulin-5-HT imbalance [38, 39, 40, 41, 42]. Martin and colleagues [43] addressed this gap in a novel review examining the links between central insulin impairment and central serotonergic activity. They highlight several mechanisms potentially linking these systems, including increased oxidative stress, inflammation, and hypothalamic-pituitary–adrenal axis activation. However, the bulk of this research emphasizes the downstream processes of insulin signaling in the brain. Martin and colleagues [43] show that insulin and 5-HT coregulate processes within the brain, including neurogenesis. Thus, the impairment of
In contrast, the present chapter examines the role of
In summary, this chapter aims to highlight the significant role of normal peripheral insulin systems in supporting 5-HT availability to the brain, and the multitude of pathways that impair normal 5-HT availability to the brain when this system is not functioning well. This can occur at varying degrees among individuals across these metabolic diseases and syndromes, and in non-diseased states.
2. Tryptophan metabolism
Tryptophan (Trp) is an essential amino acid that serves as a building block for 5-HT (5-HT), kynurenic acid, melatonin, and quinolinic acid, among other compounds. Imbalances in Trp levels have been associated with various diseases and disorders, including cancer, dementia, diabetes, and depression [44, 45]. Most notably, individuals with diabetes, depression, or both conditions tend to have lower levels of Trp in their blood [46, 47]. Trp is primarily metabolized through three pathways: the indole pathway, the 5-hydroxyindole pathway (which is involved in 5-HT production), and the kynurenine pathway (which converts 90% of Trp into other compounds) [2, 48].
2.1 Central tryptophan regulation
The central and peripheral pools of 5-HT are separated by the blood-brain barrier (BBB): a protective layer between the brain and blood vessels. Unlike 5-HT, Trp can cross the BBB (Figure 1) through a specific transporter called the large neutral amino acid transporter (LAT1) [49]. However, other large neutral amino acids (LNAA), such as tyrosine, phenylalanine, leucine, isoleucine, and valine, also compete for entry into the brain using the same transporter [50, 51, 52]. When there are high levels of these competing LNAA in the blood, the ratio of Trp to LNAA decreases, leading to reduced availability of Trp in the central nervous system [50, 51, 52, 53, 54]. Since Trp and other LNAAs4 are essential amino acids, the composition of our diet strongly influences the ratio of Trp to LNAA in the blood and subsequently affects Trp uptake in the brain [53, 55, 56]. Studies have shown that elevated levels of Trp in the blood after a meal can increase peripheral Trp:LNAA [57], which is followed by elevated levels of Trp and 5-HT in the central nervous system [58, 59].
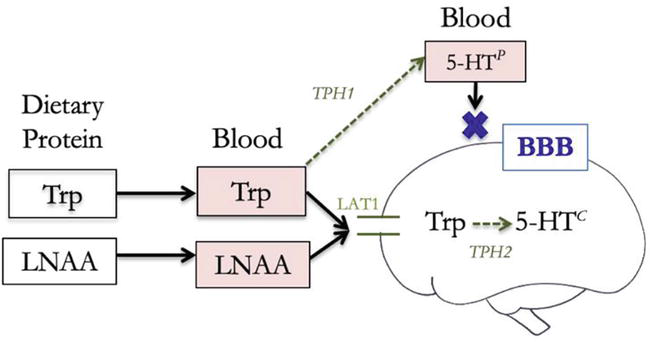
Figure 1.
Tryptophan transport past the blood brain barrier determines tryptophan availability for central serotonin synthesis (5-HT
2.2 Peripheral tryptophan regulation
The ratio of Trp to other large neutral amino acids (LNAA) in our diet is particularly important because Trp is the most limited LNAA, usually comprising only 1–2% of total protein [60, 61, 62].
When high-Trp proteins are consumed alone, they significantly increase the levels of Trp in the blood and the Trp to LNAA ratio compared to low-Trp proteins [39, 63, 64]. However, due to the higher concentrations of other LNAA in most dietary proteins, a high-carbohydrate meal actually increases the Trp to LNAA ratio more than a high-protein meal [65, 66]. This unexpected effect is due to the role of insulin in modulating Trp in the peripheral blood, and the stronger insulinogenic capacity of carbohydrates.
Insulin plays a crucial role in facilitating Trp uptake into the brain (Figure 2). Insulin helps transport LNAA into skeletal tissues, which reduces competing LNAAs in the blood, increases Trp:LNAA, and reduces competition for Trp to enter the brain [40, 67]. Insulin also helps transport Trp into skeletal tissues, but a larger proportion of branched-chain amino acids (BCAA) including valine, isoleucine, and leucine are taken up by muscle tissues in response to insulin. As a result, there is an elevated Trp:LNAA in the blood that is proportional to the circulating insulin levels [57, 68].
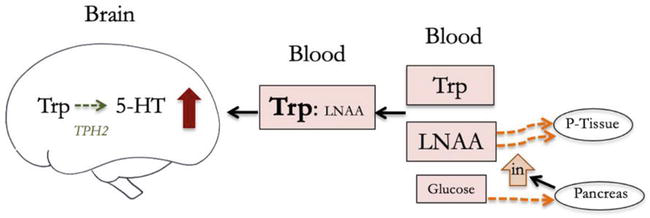
Figure 2.
Insulin facilitates tryptophan transport by increasing Trp:LNAA in the blood (5-HT = serotonin, Trp = tryptophan, LNAA = large neutral amino acid, BBB = blood brain barrier, TPH = tryptophan hydroxylase, P-tissue = skeletal tissues, in = insulin).
Insulin does impact central Trp uptake through another mechanism, but this process does not have a significant effect on Trp availability in the central nervous system. To cross the blood-brain barrier via the LAT1 transporter, Trp must be unbound [54]. Within the limited pool of Trp in the blood, most of it is reversibly bound to plasma albumin protein, with only about 10% of Trp being unbound. Insulin comes into play in this pathway by removing non-esterified fatty acids (NEFA) from albumin, creating space for Trp to bind to albumin and reducing the amount of free Trp available for transport [69]. However, the binding of Trp to albumin is transient, so the impact on Trp availability in the central nervous system is negligible [70]. As a result of this constant state of dissociation and binding with albumin, approximately 70–80% of Trp in the blood is available to cross the blood-brain barrier [70].
2.3 Insulin impairment and tryptophan
Research below demonstrates that impaired peripheral insulin function will alter Trp homeostasis and metabolic pathways in two ways: i) by redirecting Trp to regulate immune response in hyperglycemic conditions, reducing peripheral Trp availability and ii) by modifying insulin’s role in regulating amino acid ratios, limiting Trp transport into the brain. Multiple pathways leading to reduced Trp levels and altered Trp metabolism in diabetic states have been extensively studied in rodents and humans; insulin plays a crucial role in modulating these changes.
Chemical impairment of insulin production in rodents5 resulted in a significant decrease in total plasma Trp after 7 day [42], 28 days [40], and 35 days [71]. Similar findings have been reported in human studies. Three studies involving children with clinical type 1 diabetes (T1D) found lower plasma Trp and Trp:LNAA levels compared to non-diabetics [38, 72, 73]. Adolescents with metabolic syndrome also showed lower peripheral Trp levels compared to controls [74, 75]. In a study involving healthy controls and diabetic adults individuals with T1D had significantly lower serum Trp levels compared to the control group [76]. Chemically induced acute T1D in rodents also led to a significant increase in plasma branched-chain amino acids (BCAAs) valine, leucine, and isoleucine, resulting in a decrease in the overall Trp:LNAA [71, 77] and subsequently reduced central Trp levels [40, 71, 77, 78, 79]. Human studies have also found low Trp:LNAA in individuals with diabetes [77, 80].
The culmination of this research shows that impaired insulin function and insulin deficiency limit the uptake of Trp competitors into skeletal tissues, maintain low Trp:LNAA levels and LNAA competition for LAT1, and thus result in lower central Trp levels (Figure 3). Importantly, elevated peripheral BCAA concentrations have been associated with an increased risk of future diabetes and may indicate impaired insulin function even before the diagnosis of metabolic disorders [81, 82].
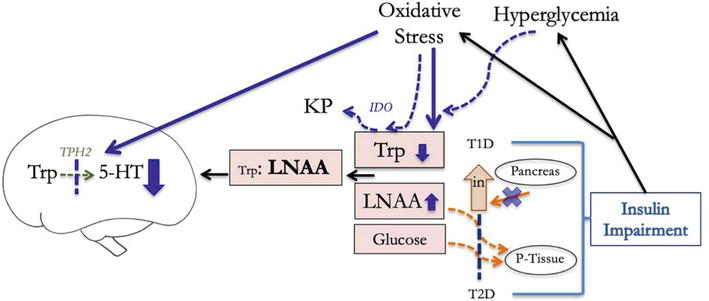
Figure 3.
Insulin impairment lowers serotonin production in the brain by impaired ability to elevate peripheral Trp:LNAA and facilitate central Trp uptake, and by alteraing TPH2 enzyme activity (5-HT = serotonin, Trp = tryptophan, LNAA = large neutral amino acid, TPH2 = tryptophan hydroxylase 2, P-tissue = skeletal tissues, in = insulin, KP = kyneurenine pathway, T1D = type 1 diabetes, T2D = type 2 diabetes, IDO = indoleamine 2,3-dioxygenase).
Studies in rodents and humans have shown that while initial insulin impairment increases unbound peripheral trp chronic stages of insulin impairment eventually led to a significant decrease in plasma Trp, further reducing the Trp:LNAA and central Trp levels [83, 84]. Chronic insulin impairment can affect peripheral Trp levels through changes in Trp metabolism and oxidation. Impaired insulin function disrupts normal glucose uptake, leading to hyperglycemic states that cause inflammation and increased production of reactive oxygen species by mitochondria [85, 86, 87]. Chronic inflammation and oxidative stress trigger an immune response that upregulates Trp metabolism in the kynurenine pathway, ultimately reducing peripheral Trp pools (Figure 3) [72, 88, 89, 90, 91]. This also has direct impacts on Trp levels because the highly reactive indole ring of Trp can be easily damaged by oxidizing species [73, 92].
New research suggests a connection between plasma Trp and peripheral insulin through the GPR142 receptor located on pancreatic islet cells. GPR142 is a G-protein coupled receptor that specifically binds to Trp and phenylalanine [77]. Binding of Trp to GPR142 stimulates the release of insulin, glucagon-like peptide-1 (GLP-1), and glucose-dependent insulinotropic polypeptide (GIP), helping to regulate glucose levels [93]. Administration of Trp in obese mice significantly increased glucose metabolism and insulin secretion [94]. In GPR142 knockout mice, Trp supplementation did not lead to increased insulin release or improved glucose tolerance compared to controls, highlighting the importance of GPR142 in the Trp-insulin link [94]. The action of Trp on GPR142 receptors in the pancreas creates a positive feedback loop where chronically reduced Trp levels in impaired insulin states may further decrease insulin secretion, disrupt glucose homeostasis, and subsequently lower plasma Trp levels.
3. Serotonin synthesis
5-HT is synthesized from tryptophan (Trp) through the 5-hydroxyindole metabolic pathway (Figure 3). This process is catalyzed by either Trp hydroxylase enzyme 1 (TPH1) or Trp hydroxylase enzyme 2 (TPH2). TPH1 is primarily found in peripheral tissues, while TPH2 is responsible for converting Trp to 5-HT in the brain. The rate of 5-HT synthesis is dependent on the activity of the TPH enzyme. In the brain, the synthesis of the intermediate molecule 5-hydroxytryptophan (5-HTP) is the limiting step, and this step is determined by the availability of Trp as a substrate.
3.1 Insulin impairment and serotonin
Insulin impairment not only affects central 5-HT synthesis by altering enzyme activity in the 5-hydroxyindole pathway, but it also impacts the catalytic function of TPH2. In rodents with chemically-induced insulin impairment for 7 days, TPH2 activity was reduced in the cerebral cortex and brainstem [89]. This decrease in activity was previously attributed solely to low central Trp availability as a result of the mechanisms described earlier. However, this same study found that the TPH2 enzyme in diabetic rodents had a significantly lower affinity for Trp and reduced phosphorylating capacity required to stimulate the enzyme. Another study with T1D rodents observed similar catalytic dysfunction of TPH2, as well as reduced expression of the enzyme [42]. These epigenetic and kinetic changes were attributed to hyperglycemic conditions: elevated peripheral glucose levels (hyperglycemia) led to increased brain glucose levels, which can trigger inflammation, reactive oxygen species, and oxidative stress, ultimately causing damage to TPH2 expression and function [95, 96, 97, 98].
Chronic elevation of glucocorticoid levels, commonly seen in diseases associated with insulin impairment, can also explain the reduced expression of TPH2 [99, 100]. Excess glucocorticoids are released in response to chronic stress through activation of the hypothalamic–pituitary–adrenal axis. Glucocorticoid receptors act as transcription factors that inhibit TPH2 expression and can significantly decrease 5-HT levels in the raphe nuclei after one week of excess exposure [101]. Elevated glucocorticoid levels are also independently linked to peripheral tissue insulin resistance, impaired insulin production, and high blood glucose levels [99, 100, 102]. Overall, chronic release of glucocorticoids appears to indirectly impair central 5-HT availability by altering Trp transport and conversion, as well as via hyperglycemic conditions. The correlation between elevated glucocorticoids and depressive symptoms highlights this stress response as a contributing factor in the disrupted regulation of Trp and 5-HT, which underlies psycho-metabolic comorbidities [103]. In summary, both reduced central Trp availability and impaired TPH2 activity disrupt the 5-hydroxyindole pathway in individuals with chronic insulin impairment.
The effects of altered Trp metabolism in response to peripheral insulin impairment, such as lower peripheral Trp, elevated peripheral LNAA, and lower central TPH2 expression and function, are expected to subsequently reduce central 5-HT pools available for neurotransmission. This prediction is supported by several rodent studies, where chemically induced insulin impairment led to a significant decrease in central 5-HT synthesis. This decrease was observed after 4 weeks in the whole brain [78], after 1 week in the whole brain [104], after 2 weeks in the whole brain [41] and the hypothalamus [89], and after 50 days in the striatum and pons medulla [105], as indicated by 5-HTP accumulation. Additionally, 5-HT synthesis measured by TPH2 activity was significantly lower after 7 days in the cerebral cortex and brainstem [89]. While this is beyond the scope of the present chapters, readers may consider that insulin impairment is also expected to compromise other systems dependent on Trp and 5-hydroxyindole metabolism of Trp. For example, the synthesis of the 5-HT metabolite, melatonin (MLT), appears to be significantly reduced in diabetic states [6].
3.2 Insulin treatment
Insulin treatments in diabetic rodents have a limited capacity to restore normal Trp and 5-HT levels in the brain. Prolonged periods of hyperglycemia place greater stress on both insulin secretion and insulin sensitivity, further reducing the potential for recovery [106]. A comprehensive study examined the effects of insulin treatment on rodents with 7 days of insulin impairment by measuring TPH2 activity, central 5-HT levels, central Trp levels, and blood glucose levels [42]. Following insulin treatment, rodents with induced T1D showed normal blood glucose and Trp levels in the brain, but TPH2 activity and 5-HT levels remained depressed after 7 and 14 days of insulin treatment [42]. However, These findings are contradicted by a different study of rodents with 7 days of insulin impairment, where insulin treatment after 14 days was able to restore near-normal central 5-HT levels [104].
A study from 1991 suggested that prolonged insulin impairment may result in irreversible reduction in central 5-HT levels [77]. Rodents with 10–30 days of insulin impairment were treated with insulin at various time intervals ranging from 15 to 135 days. Insulin treatment restored serum Trp levels throughout all time spans but could not restore central Trp levels beyond 15 days. It is important to note that this study induced insulin impairment using alloxan, which is known to cause higher cellular toxicity in non-target cells compared to streptozotocin used in other studies [42, 77, 104, 107].
Human studies also offer insights into the consequences long term insulin impairment, as in the case of clinically diagnosed diabetes. In comparison to normal controls, diabetics had higher levels of plasma BCAA [108], tyrosine and phenylalanine [109], and lower levels of plasma Trp [110]. The subsequently low plasma Trp:LNAA was not reversed by weight loss [111]. Diversion of Trp metabolism to the kynurenine pathway in response to stress persisted after bariatric surgery [112] and plasma Trp and Trp:LNAA were little changed by Trp dosing in obese persons compared to lean [109].
Both rodent and human studies indicate that chronic peripheral insulin impairment may have long-term effects on 5-HT availability that cannot be fully restored by insulin treatment, underscoring the complexity of identifying effective treatments.
4. Behavioral and psychotropic treatments
The dysregulation of insulin and serotonergic systems is further complicated by a positive feedback loop involving common behavioral responses and psychotropic medical treatments. For instance, central 5-HT depletion can lead to cravings for carbohydrates in an attempt to raise peripheral insulin levels [49, 113, 114, 115]. However, a high-glycemic diet increases the risk of insulin resistance, impaired insulin secretion, and depression [106, 116, 117, 118]. Carbohydrate cravings are also common in various disorders associated with mood changes and serotonergic modulation, such as atypical depression, seasonal affective disorder, late luteal phase dysphoric disorder, and binge eating disorder. Serotonergic medications that upregulate 5-HT neurotransmission are often prescribed to treat these disorders [119, 120].
However, these medications may exacerbate Trp and 5-HT imbalances in individuals with peripheral insulin impairment. As serotonergic medications are not specific to the central nervous system, they disrupt 5-hydroxyindole homeostasis in the periphery, where most 5-HT is synthesized [121]. This can lead to apoptosis of pancreatic cells and acute pancreatitis (inflammation and tissue damage), and thereby inhibit insulin secretion [122, 123]. It can also impair insulin receptor function [124]. Several studies have observed an increased risk of type 2 diabetes associated with SSRI use [125, 126, 127, 128], despite the effect of SSRIs on glucose homeostasis [118], and this risk seems to dose, concentration and duration dependent [127, 128]. Moreover, individuals with pre-existing low levels of peripheral Trp are more susceptible to anxious and depressive episodes when taking serotonergic antidepressants (Figure 4) [129, 130].
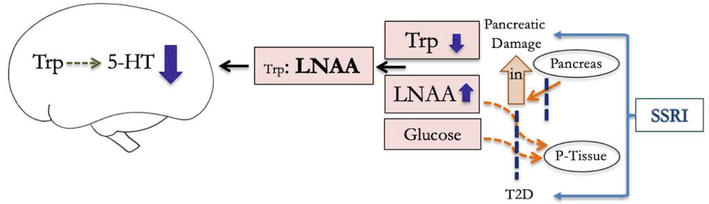
Figure 4.
Selective serotonin reuptake inhibitors (SSRI) disrupt homeostatic regulation of serotonin in the periphery, which can restrict peripheral insulin secretion and sensitivity, reducing available central 5-HT (5-HT = serotonin, Trp = tryptophan, LNAA = large neutral amino acid, P-tissue = skeletal tissues, in = insulin, KP = kyneurenine pathway, T1D = type 1 diabetes, T2D = type 2 diabetes).
In summary, the regulation of Trp and 5-HT homeostasis is crucial in both the periphery and brain. Insulin impairment and common attempts to regulate emotional changes caused by this impairment can disrupt this delicate balance. The evidence from rodent and human studies presented in this chapter suggests that behavioral and medical treatments that exacerbate insulin impairment also pose a risk of long-term and potentially irreversible impairment of Trp metabolism and transport, as well as central 5-HT availability.
5. Interpretations and applications
How do we interpret the consequences of low Trp levels and restricted central 5-HT in light of positive feedbacks with higher degree and duration of insulin impairment? Returning to ideas presented in the Introduction, what do these interactions and outcomes mean within a new paradigm that dissociates neurobiology from absolute diagnostic labels and questions the 5-HT ‘chemical imbalance’? Low and high 5-HT hypotheses, and the medications targeting neurotransmission upregulation, are not modeled on balance. Rather, they are modeled on extremes and deficits of a single chemical and blind targeting of multiple systems in the body [131], all in an attempt to chronically maintain ideal emotional states.
5.1 Complex adaptive living systems
These mechanisms can instead be viewed with a model of balance that better reflects the reality of functional living systems. Optimization of single brain chemicals is reflective of a linear cause-effect reductionist approach to health [132, 133]. Alternatively, we interpret the patterns of interaction among neurological and metabolic systems with the view that human beings are composed of, and operate within, complex adaptive systems (CAS) [132, 133, 134].
A complex adaptive system is a collection of specialized agents (components or parts) that can be understood within the context of the whole and the interconnecting systems they comprise [133, 134]. Diverse interactions between these systems and environmental stimuli will adapt to give rise to non-linear, unpredictable and ever-changing dynamics [26, 135, 136]. While defined by dynamic change and evolution, complex living systems conserve their conditions for renewability through self-regulating feedback loops that link interacting agents [134, 137]. Collectively, these traits allow CAS to maintain homeostatic equilibrium such that they maintain a dynamic form of stability over time [138] and develop resiliency in the face of disturbance and adversity [133, 134, 135, 137].
This CAS perspective is especially useful in understanding complex systems that are not easily measured, understood and predicted [132]. Applying the CAS perspective to the human brain and body, we view health as homeostatic equilibrium of essential interactions, systems and functions needed for sustaining the human as a self-organizing system. This lens can be applied to neurophysiological systems by viewing them as normally fluctuating between order and disorder, and by focusing on “state change” rather than static maintenance of absolute highs and lows [132]. We interpret positive feedback loops with their “tend[ency] towards chaos and decay” and identify a need to counterbalance by negative feedback [132]. The CAS model thus shifts our view of neurophysiological imbalance from the failure of a single component in a particular place, to the idea that imbalance arises from interaction between multiple systems that are not able to self-regulate and maintain dynamic equilibrium [132, 133]. It also recognizes that these interactions will generate emergent, unpredictable outcomes that manifest differently in unique individuals and sociocultural contexts [133, 137]. We apply these principles to our understanding of the insulin system as it interacts with 5-hydroxyindole system in the body and brain.
5.2 Systems of Insulin and 5-hydroxyindole metabolism
Functional homeostatic regulation occurs when components of the system are maintained within upper and lower bounds, and fluctuate dynamically in response to changes in their environment [24, 26, 135, 139]. Indeed, both an excess and deficiency of 5-HT and its precursor throughout the body can lead to health imbalances [2]: sufficiently low peripheral 5-HT is needed to maintain insulin sensitivity and prevent obesity [140, 141], yet sufficiently high levels of peripheral 5-HT are needed to maintain pancreatic insulin secretion [142] and normal glucose levels [143]. Similarly, a “high” Trp diet in human studies has been labeled both a protective [144] and risk factor [83] for later development of T2D.
A state of acute tryptophan depletion (ATD) can be experimentally induced to explore the behavioral and cognitive responses to low central 5-HT. Reduced central 5-HT from ATD led not only to negative affect bias (sad mood), but also better punishment prediction accuracy and more risk aversive behavior [145, 146, 147]; it also enhanced negative reciprocity, expressed as greater punishment or retaliation in response to perceived unfairness [148]. While these findings tend to be interpreted as explanations of maladaptive depressive states, we can revisit this from a CAS perspective. Low central 5-HT states may be an adaptive, functional response to avoid harm that is learned in environments where punishment is common and then recruited when complex social issues are interpreted as chronic danger. Other scholars predict that hypervigilance is an adaptive response for a child raised in an unpredictable environment [32], and that social avoidance is an adaptive response to volatile social experiences [149].
Evolutionary psychologists tend to view short periods of stress, anxiety or depression as adaptive responses that can increase resiliency in certain contexts if the triggers are addressed [7, 147, 149, 150]. For example, the
Both extremes of lowered central 5-HT availability and heightened 5-HT neurotransmission impair the brain’s ability to accurately interpret facial emotions; ATD inaccurately escalated the interpretation of fearful emotion to anger, while SSRI’s dissolved the ability to distinguish fearful and normal emotions [145]. These results support the idea that functional 5-hydroxyindole metabolism and 5-HT neurotransmission may require dynamic homeostatic regulation.
From a resiliency perspective, ATD alone does not seem to initiate the cascade of positive feedbacks in insulin and 5-hydroxyindole systems; it worsens or triggers them. In fact, Trp-deficiency induced episodes of anxiety and depression in people with a personal or familial history of mental health disorder, yet had little to no emotional effect on healthy controls [153, 154, 155]. Similarly, in comparisons of high-Trp versus low-Trp consumption (where high-Trp levels significantly elevated plasma Trp and Trp:LNAA), high Trp improved memory exclusively in individuals susceptible to high stress [64], and reduced vulnerability to experimental triggers for fatigue, negative affective bias, and diminished well-being [63].
It is important to recognize that these ATD tests represent acutes stress, while stress responses outside of the laboratory are often triggered by more complex, abstract threats and exist in a more pervasive form that requires reflection on previous experiences and learned behavior [146, 147]. The chronic nature of these real stressors can cause damage to the brain and body that correspond with mental unwellness and symptoms of non-communicable diseases [156, 157].
Interpreting these experiments through CAS perspective, we suggest that recovery from fluctuations in mood and depressive episodes may be supported by the adaptive capacity to fluctuate high and low neurotransmission in response to biosocial environmental disturbances. 5-HT availability is a limiting factor in the potential to upregulate neurotransmission at a given time and in response to a given situation. Availability also determines the potential to downregulate in a time-responsive manner relative to upregulation, as needed to re-establish equilibrium.
5.3 Serotonin as resilience molecule
This idea is supported by evolutionary studies of 5-hydroxyindole metabolism arguing that a tightly regulated balance of Trp content and availability is most ideal for all living organisms in order to maintain the adaptive function of its intermediates [158]. The human body has developed several mechanisms for maintaining a narrow range of Trp in different regions [158] and in relation to co-existing amino acids [2]. The 5-hydroxyindole pathway that produces 5-HT from Trp has been highly conserved from unicellular bacteria to mammals, and shares one common function across all organisms and tissues: adaptive response to environmental stress [158, 159, 160]. Despite the body having very low Trp concentrations compared to other amino acids, its metabolite 5-HT serves many crucial biological functions. 5-HT is considered the homeostatic regulator of the central nervous, neuroendocrine, gut and immune systems, and the biochemical connector of mind and body with the environment [8, 158].
The adaptive function of 5-HT as a moderator of stress response can be understood as a trait supporting psychological resilience: the ability to cope with or recover from adversity [161]. We argue that altered Trp metabolism as a result of chronic insulin impairment ultimately impairs psychological resiliency, particularly among already vulnerable individuals. By limiting availability of 5-HT in the brain, chronic insulin impairment disrupts the ability to finely regulate changes in 5-HT neurotransmission in response to changing environmental contexts. Since 5-HT is responsible for adaptive response to stress, this represents a loss of neurological options that will further increase vulnerability to emotional and energy disturbances.
Insulin impairment is one mechanism that impairs the adaptive capacity of Trp metabolites like 5-HT to mitigate impacts of environmental stress. The dynamic state of insulin impairment and associated glucose intolerance throughout the life course of prediabetic and diabetic individuals [10, 162, 163], as well as the diverse experiences of individuals themselves, must be considered in light of experimental studies. Moreover, our understanding of the complex and dynamic state of insulin impairment, diabetes and metabolic disorder, cannot be based on short-term rodent studies alone. In combination with human diabetic studies, these experiments do identify clear limits in physiological function during these complex states; most notably, during early states of insulin impairment that occurs long before changes to blood-glucose, weight, or energy levels are detected at diagnosis of metabolic disorder. Together, these studies demonstrate a strain in adaptive response to stress that will be affected by the severity and duration of insulin impairment and individual human characteristics.
6. Conclusion
Insulin impairment disrupts the homeostatic adaptive capacity to regulate central 5-HT and impairs psychological resilience to stress by altering normal 5-hydroxyindole metabolism of Trp. The small fraction of 5-HT in the brain is more vulnerable to insulin impairment than peripheral 5-HT since, in addition to being limited by low peripheral Trp stores, it is also restricted by impaired TPH2 activity and impaired central Trp uptake. Insulin impairment represents a loss of options for the many roles of central 5-HT, which are increasingly restricted by higher degree and duration of insulin impairment, as well as serotonergic medications and dietary cravings induced by the dysfunction itself.
Neurophysiological studies of high and low 5-HT might be better understood from the perspective homeostatic balance of 5-hydroxyindole metabolism, and how this is shaped by dynamic states of other molecules in the body, beyond insulin impairment. Future studies may elucidate our understanding of a bidirectional relationship between insulin and 5-HT function and explore the 5-hydroxyindole response to glycemic imbalance and oxidative stress under diverse conditions and locations in the brain and body (i.e., how different receptors in different areas of the brain respond to insulin impairment over time). Better integration of experimental studies that focus on the origins of neurophysiological imbalance may help identify treatment that supports adaptive capacity inherent to the 5-HT system. Most significantly, we encourage neurophysiological research to consider experimental design and interpretation with the resiliency model of complex adaptive living systems.
Acknowledgments
Thank you to the Trinity College, University of Toronto Independent Study Course for supporting this research.
References
- 1.
Azmitia E. Chapter 1.1 - evolution of serotonin: Sunlight to suicide. Handbook of Behavioral Neuroscience. 2010; 21 :3-22 - 2.
Sainio EL, Pulkki K, Young SN. L-tryptophan: Biochemical, nutritional and pharmacological aspects. Amino Acids. 1996; 10 (1):21-47 - 3.
Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annual Review of Medicine. 2009; 60 :355-366 - 4.
Kraus C, Castrén E, Kasper S, Lanzenberger R. Serotonin and neuroplasticity - links between molecular, functional and structural pathophysiology in depression. Neuroscience and Biobehavioral Reviews. 2017; 77 :317-326 - 5.
Yabut JM, Crane JD, Green AE, Keating DJ, Khan WI, Steinberg GR. Emerging roles for serotonin in regulating metabolism: New implications for an ancient molecule. Endocrine Reviews. 2019; 40 (4):1092-1107 - 6.
Amaral FG, Turati AO, Barone M, Scialfa JH, Buonfiglio D, Peres R, et al. Melatonin synthesis impairment as a new deleterious outcome of diabetes-derived hyperglycemia. Journal of Pineal Research. 2014; 57 (1):67-79 - 7.
Andrews PW, Bharwani A, Lee KR, Fox M, Thomson JA. Is serotonin an upper or a downer? The evolution of the serotonergic system and its role in depression and the antidepressant response. Neuroscience and Biobehavioral Reviews. 2015; 51 :164-188 - 8.
Azmitia EC. Modern views on an ancient chemical: Serotonin effects on cell proliferation, maturation, and apoptosis. Brain Research Bulletin. 2001; 56 (5):413-424 - 9.
Heisler LK, Kanarek RB, Gerstein A. Fluoxetine decreases fat and protein intakes but not carbohydrate intake in male rats. Pharmacology, Biochemistry, and Behavior. 1997; 58 (3):767-773 - 10.
ADA (American Diabetes Association). Diagnosis and classification of diabetes mellitus. Diabetes Care. 2014; 36 (1):S67-S74 - 11.
Huang PL. A comprehensive definition for metabolic syndrome. Disease Models & Mechanisms. 2009; 2 (5-6):231-237 - 12.
Balhara YPS. Diabetes and psychiatric disorders. Indian Journal of Endocrinology Metabolism. 2011; 15 (4):274-283 - 13.
Toalson P, Ahmed S, Hardy T, Kabinoff G. The metabolic syndrome in patients with severe mental illnesses. Primary Care Companion Journal of Clinical Psychiatry. 2004; 6 (4):152-158 - 14.
Canadian Diabetes Association Clinical Practice Guidelines Expert Committee, Robinson DJ, Luthra M, Vallis M. Diabetes and mental health. Can Journal of Diabetes. 2013; 37 (Suppl. 1):S87-S92 - 15.
Fisher L, Skaff MM, Mullan JT, Arean P, Glasgow R, Masharani U. A longitudinal study of affective and anxiety disorders, depressive affect and diabetes distress in adults with type 2 diabetes. Diabetic Medicine. 2008; 25 (9):1096-1101 - 16.
Egede LE, Zheng D, Simpson K. Comorbid depression is associated with increased health care use and expenditures in individuals with diabetes. Diabetes Care. 2002; 25 (3):464-470 - 17.
Hermanns N, Kulzer B, Krichbaum M, Kubiak T, Haak T. Affective and anxiety disorders in a German sample of diabetic patients: Prevalence, comorbidity and risk factors. Diabetic Medicine. 2005; 22 (3):293-300 - 18.
Pan A, Lucas M, Sun Q , van Dam RM, Franco OH, Manson JE, et al. Bidirectional association between depression and type 2 diabetes mellitus in women. Archives of Internal Medicine. 2010; 170 (21):1884-1891 - 19.
Musselman DL, Betan E, Larsen H, Phillips LS. Relationship of depression to diabetes types 1 and 2: Epidemiology, biology, and treatment. Biological Psychiatry. 2003; 54 (3):317-329 - 20.
Chand SP, Arif H. Depression. Treasure Island, FL: StatPearls Publishing; 2023. Available from: http://www.ncbi.nlm.nih.gov/books/NBK430847/ - 21.
Rose AL, Hopko DR, Lejuez CW, Magidson JF. Major depressive disorder. In: Functional Analysis in Clinical Treatment. 2nd ed. San Diego, CA, US: Elsevier Academic Press; 2020. pp. 339-373 - 22.
Albert PR, Benkelfat C, Descarries L. The neurobiology of depression—Revisiting the serotonin hypothesis. I. Cellular and molecular mechanisms. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 2012; 367 (1601):2378-2381 - 23.
Coppen A. The biochemistry of affective disorders. The British Journal of Psychiatry. 1967; 113 (504):1237-1264 - 24.
Kirmayer LJ, Gomez-Carrillo A, Veissière S. Culture and depression in global mental health: An ecosocial approach to the phenomenology of psychiatric disorders. Social Science & Medicine. 2017; 183 :163-168 - 25.
Cowen PJ, Browning M. What has serotonin to do with depression? World Psychiatry. 2015; 14 (2):158-160 - 26.
Kirmayer LJ. Re-visioning psychiatry: Toward an ecology of mind in health and illness. In: Kirmayer LJ, Lemelson R, Cummings CA, editors. Re-Visioning Psychiatry. Cambridge: Cambridge University Press; 2015. pp. 622-660. Available from: https://www.cambridge.org/core/product/identifier/9781139424745%23CT-bp-24/type/book_part - 27.
Pescosolido BA, Martin JK, Long JS, Medina TR, Phelan JC, Link BG. “A disease like any other”? A decade of change in public reactions to schizophrenia, depression, and alcohol dependence. AJP. 2010; 167 (11):1321-1330 - 28.
Moncrieff J, Cooper RE, Stockmann T, Amendola S, Hengartner MP, Horowitz MA. The serotonin theory of depression: A systematic umbrella review of the evidence. Molecular Psychiatry. 2023; 28 (8):3243-3256 - 29.
Pies R. Psychiatry’s New Brain-Mind and the Legend of the “Chemical Imbalance.” Psychiatric Times [Internet]. 2011 [cited 2023 Nov 7]. Available from: https://www.psychiatrictimes.com/view/psychiatrys-new-brain-mind-and-legend-chemical-imbalance - 30.
Chatterton C. Metabolic syndrome: The construction of a “New” medical problem and the socio-ethical consequences [Internet] [PhD]. Cardiff University. 2014 [cited 2023 Nov 5]. Available from: https://orca.cardiff.ac.uk/id/eprint/58973/ - 31.
Simmons RK, Alberti KGMM, Gale EAM, Colagiuri S, Tuomilehto J, Qiao Q , et al. The metabolic syndrome: Useful concept or clinical tool? Report of a WHO expert consultation. Diabetologia. 2010; 53 (4):600-605 - 32.
Haltigan J. Rethinking mental illness – and health. CAMH Discovers [Internet]. 2018 [cited 2020 Jun 30]. Available from: https://www.camh.ca/en/camh-news-and-stories/rethinking-mental-illness-and-health - 33.
Preskorn SH, Baker B. The overlap of DSM-IV syndromes: Potential implications for the practice of Polypsychopharmacology, psychiatric drug development, and the human genome project. Journal of Psychiatric Practice®. 2002; 8 (3):170 - 34.
La Roche MJ, Fuentes MA, Hinton D. A cultural examination of the DSM-5: Research and clinical implications for cultural minorities. Professional Psychology: Research and Practice. 2015; 46 (3):183-189 - 35.
Kim HS, Sherman DK, Taylor SE, Sasaki JY, Chu TQ , Ryu C, et al. Culture, serotonin receptor polymorphism and locus of attention. Social Cognitive and Affective Neuroscience. 2010; 5 (2-3):212-218 - 36.
National Institute of Mental Health. Research Domain Criteria (RDoC) [Internet]. 2015 [cited 2023 Feb 10]. Available from: https://www.nimh.nih.gov/research/research-funded-by-nimh/rdoc - 37.
Sanislow CA, Pine DS, Quinn KJ, Kozak MJ, Garvey MA, Heinssen RK, et al. Developing constructs for psychopathology research: Research domain criteria. Journal of Abnormal Psychology. 2010; 119 (4):631-639 - 38.
Herrera R, Manjarrez G, Nishimura E, Hernandez J. Serotonin-related tryptophan in children with insulin-dependent diabetes. Pediatric Neurology. 2003; 28 (1):20-23 - 39.
Fernstrom JD, Langham KA, Marcelino LM, Irvine ZLE, Fernstrom MH, Kaye WH. The ingestion of different dietary proteins by humans induces large changes in the plasma tryptophan ratio, a predictor of brain tryptophan uptake and serotonin synthesis. Clinical Nutrition. 2013; 32 (6):1073-1076 - 40.
MacKenzie RG, Trulson ME. Effects of insulin and streptozotocin-induced diabetes on brain tryptophan and serotonin metabolism in rats. Journal of Neurochemistry. 1978; 30 (1):205-211 - 41.
Crandall EA, Gillis MA, Fernstrom JD. Reduction in brain serotonin synthesis rate in streptozotocin-diabetic rats. Endocrinology. 1981; 109 (1):310-312 - 42.
Manjarrez-Gutierrez G, Neri-Gómez T, Herrera-Márquez R, Mondragón J, Oca A, Rodríguez J. Brain serotonergic disturbances caused by diabetes mellitus are not reversed by insulin treatment. 2015 - 43.
Martin H, Bullich S, Martinat M, Chataigner M, Di Miceli M, Simon V, et al. Insulin modulates emotional behavior through a serotonin-dependent mechanism. Molecular Psychiatry. 2022; 7 :1-10 - 44.
Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: Beyond IDO and tryptophan depletion. Cancer Research. 2012; 72 (21):5435-5440 - 45.
Lovelace MD, Varney B, Sundaram G, Lennon MJ, Lim CK, Jacobs K, et al. Recent evidence for an expanded role of the kynurenine pathway of tryptophan metabolism in neurological diseases. Neuropharmacology. 2017; 112 (Pt B):373-388 - 46.
Manjarrez-Gutierrez G, Marquez RH, Mejenes-Alvarez SA, Godinez-Lopez T, Hernandez-R J. Functional change of the auditory cortex related to brain serotonergic neurotransmission in type 1 diabetic adolescents with and without depression. The World Journal of Biological Psychiatry. 2009; 10 (4 Pt 3):877-883 - 47.
Delgado I, Cussotto S, Anesi A, Dexpert S, Aubert A, Aouizerate B, et al. Association between the indole pathway of tryptophan metabolism and subclinical depressive symptoms in obesity: A preliminary study. International Journal of Obesity. 2022; 46 (4):885-888 - 48.
Cheng Y, Li Y, Benkowitz P, Lamina C, Köttgen A, Sekula P. The relationship between blood metabolites of the tryptophan pathway and kidney function: A bidirectional Mendelian randomization analysis. Scientific Reports. 2020; 10 (1):12675 - 49.
Cansev M, Wurtman R. Aromatic amino acids in the brain. In: Handbook of Neurochemistry and Molecular Neurobiology. New York, NY: Springer Science+Business Media; 2007. pp. 59-97. Available from: https://avesis.uludag.edu.tr/yayin/81facf74-5f8e-4748-9889-8665f7736eac/handbook-of-neurochemistry-and-molecular-neurobiology - 50.
Pietz J, Kreis R, Rupp A, Mayatepek E, Rating D, Boesch C, et al. Large neutral amino acids block phenylalanine transport into brain tissue in patients with phenylketonuria. The Journal of Clinical Investigation. 1999; 103 (8):1169-1178 - 51.
Yanagida O, Kanai Y, Chairoungdua A, Kim DK, Segawa H, Nii T, et al. Human L-type amino acid transporter 1 (LAT1): Characterization of function and expression in tumor cell lines. Biochimica et Biophysica Acta. 2001; 1514 (2):291-302 - 52.
Zepf F. Principles of rapid tryptophan depletion and its use in research on neuropsychiatric disorders. In: Amino Acids in Human Health and Nutrition. Oxfordshire: CABI; 2012. pp. 418-426 - 53.
Moja EA, Stoff DM, Gessa GL, Castoldi D, Assereto R, Tofanetti O. Decrease in plasma tryptophan after tryptophan-free amino acid mixtures in man. Life Sciences. 1988; 42 (16):1551-1556 - 54.
Yuwiler A, Oldendorf WH, Geller E, Braun L. Effect of albumin binding and amino acid competition on tryptophan uptake into brain. Journal of Neurochemistry. 1977; 28 (5):1015-1023 - 55.
Feurté S, Gerozissis K, Regnault A, Paul FM. Plasma Trp/LNAA ratio increases during chronic ingestion of an alpha-lactalbumin diet in rats. Nutritional Neuroscience. 2001; 4 (5):413-418 - 56.
Shen YB, Voilqué G, Odle J, Kim SW. Dietary L-tryptophan supplementation with reduced large neutral amino acids enhances feed efficiency and decreases stress hormone secretion in nursery pigs under social-mixing stress. The Journal of Nutrition. 2012; 142 (8):1540-1546 - 57.
Sainio EL, Närvänen S, Sainio P, Tuohimaa P. Distribution of L-tryptophan in normal and glucose - loaded mice. Amino Acids. 1995; 8 (2):209-216 - 58.
Fernstrom JD, Wurtman RJ. Brain serotonin content: Physiological regulation by plasma neutral amino acids. Science. 1972; 178 (4059):414-416 - 59.
Fernstrom JD, Wurtman RJ. Brain serotonin content: Increase following ingestion of carbohydrate diet. Science. 1971; 174 (4013):1023-1025 - 60.
Spiegelaar N, Martin ID, Tsuji LJS. Indigenous subarctic food Systems in Transition: Amino acid composition (including tryptophan) in wild-harvested and processed meats. International Journal of Food Science. 2019; 27 (2019):1-14 - 61.
United States Department of Agriculture. USDA Nutrient Data Laboratory [Internet]. 2015 [cited Oct 29]. Available from: https://fnic.nal.usda.gov/food-composition/usda-nutrient-data-laboratory - 62.
CFIA (Canadian Food Inspection Agency). Elements within the Nutrition Facts Table [Internet]. [cited 2014 Jul 3]. Available from: http://www.inspection.gc.ca/food/labelling/food-labelling-for-industry/nutrition-labelling/elements-within-the-nutrition-facts-table/eng/1389206763218/1389206811747?chap=8 - 63.
Gibson EL, Vargas K, Hogan E, Holmes A, Rogers PJ, Wittwer J, et al. Effects of acute treatment with a tryptophan-rich protein hydrolysate on plasma amino acids, mood and emotional functioning in older women. Psychopharmacology. 2014; 231 (24):4595-4610 - 64.
Markus CR, Olivier B, de Haan EHF. Whey protein rich in alpha-lactalbumin increases the ratio of plasma tryptophan to the sum of the other large neutral amino acids and improves cognitive performance in stress-vulnerable subjects. The American Journal of Clinical Nutrition. 2002; 75 (6):1051-1056 - 65.
Wurtman RJ, Wurtman JJ, Regan MM, McDermott JM, Tsay RH, Breu JJ. Effects of normal meals rich in carbohydrates or proteins on plasma tryptophan and tyrosine ratios. The American Journal of Clinical Nutrition. 2003; 77 (1):128-132 - 66.
Yokogoshi H, Wurtman RJ. Meal composition and plasma amino acid ratios: Effect of various proteins or carbohydrates, and of various protein concentrations. Metabolism. 1986; 35 (9):837-842 - 67.
Daniel PM, Love ER, Moorhouse SR, Pratt OE. The effect of insulin upon the influx of tryptophan into the brain of the rabbit. The Journal of Physiology. 1981; 312 :551-562 - 68.
Fukagawa NK, Minaker KL, Rowe JW, Young VR. Plasma tryptophan and total neutral amino acid levels in men: Influence of hyperinsulinemia and age. Metabolism. 1987; 36 (7):683-686 - 69.
Lipsett D, Madras BK, Wurtman RJ, Munro HN. Serum tryptophan level after carbohydrate ingestion: Selective decline in non-albumin-bound tryptophan coincident with reduction in serum free fatty acids. Life Sciences. 1973; 12 (Part 2):57-64 - 70.
Pardridge WM. Brain metabolism: A perspective from the blood-brain barrier. Physiological Reviews. 1983; 63 (4):1481-1535 - 71.
Trulson ME, Jacoby JH, MacKenzie RG. Streptozotocin-induced diabetes reduces brain serotonin synthesis in rats. Journal of Neurochemistry. 1986; 46 (4):1068-1072 - 72.
Manjarrez G, Herrera R, Leon M, Hernandez-R J. A low brain serotonergic neurotransmission in children with type 1 diabetes detected through the intensity dependence of auditory-evoked potentials. Diabetes Care. 2006; 29 (1):73-77 - 73.
Jain SK. Can tryptophan oxidation lead to lower tryptophan level in diabetes? A commentary on “Propagation of protein glycation damage involves modification of tryptophan residues via reactive oxygen species: inhibition by pyridoxamine”. Free Radical Biology & Medicine. 2008; 44 (7):1273-1275 - 74.
Herrera-Marquez R, Hernandez- Rodriguez J, Medina-Serrano J, Boyzo-Montes de Oca A, Manjarrez-Gutierrez G. Association of metabolic syndrome with reduced central serotonergic activity. Metabolic Brain Disease. 2011; 26 (1):29-35 - 75.
Chen J, Ning C, Mu J, Li D, Ma Y, Meng X. Role of Wnt signaling pathways in type 2 diabetes mellitus. Molecular and Cellular Biochemistry. 2021; 476 (5):2219-2232 - 76.
Gürcü S, Girgin G, Yorulmaz G, Kılıçarslan B, Efe B, Baydar T. Neopterin and biopterin levels and tryptophan degradation in patients with diabetes. Scientific Reports. 2020; 10 (1):17025 - 77.
Jamnicky B, Slijepcević M, Hadzija M, Juretić D, Borcić O. Tryptophan content in serum and brain of long-term insulin-treated diabetic rats. Acta Diabetologica Latina. 1991; 28 (1):11-18 - 78.
MacKenzie RG, Trulson ME. Does insulin act directly on the brain to increase tryptophan levels? Journal of Neurochemistry. 1978; 30 (5):1205-1208 - 79.
MacKenzie RG, Trulson ME. Regional accumulation of tryptophan and serotonin metabolism following tryptophan loading in diabetic rats. Journal of Neurochemistry. 1978; 31 (1):157-160 - 80.
Curzon G, Fernando JC. Drugs altering insulin secretion: Effects on plasma and brain concentrations of aromatic amino acids and on brain 5-hydroxytryptamine turnover. British Journal of Pharmacology. 1977; 60 (3):401-408 - 81.
Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metabolism. 2012; 15 (5):606-614 - 82.
Würtz P, Soininen P, Kangas AJ, Rönnemaa T, Lehtimäki T, Kähönen M, et al. Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care. 2013; 36 (3):648-655 - 83.
Chen T, Zheng X, Ma X, Bao Y, Ni Y, Hu C, et al. Tryptophan predicts the risk for future type 2 diabetes. PLoS One. 2016; 11 (9):e0162192 - 84.
Vangipurapu J, Stancáková A, Smith U, Kuusisto J, Laakso M. Nine amino acids are associated with decreased insulin secretion and elevated glucose levels in a 7.4-year follow-up study of 5,181 Finnish men. Diabetes. 2019; 68 (6):1353-1358 - 85.
Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circulation Research. 2010; 107 (9):1058-1070 - 86.
Brownlee M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes. 2005; 54 (6):1615-1625 - 87.
Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes. 2003; 52 (1):1-8 - 88.
Chetyrkin SV, Mathis ME, Ham AJL, Hachey DL, Hudson BG, Voziyan PA. Propagation of protein glycation damage involves modification of tryptophan residues via reactive oxygen species: Inhibition by pyridoxamine. Free Radical Biology & Medicine. 2008; 44 (7):1276-1285 - 89.
Herrera R, Manjarrez G, Hernandez J. Inhibition and kinetic changes of brain tryptophan-5-hydroxylase during insulin-dependent diabetes mellitus in the rat. Nutritional Neuroscience. 2005; 8 (1):57-62 - 90.
Kanth VR, Lavanya K, Srinivas J, Raju TN. Elevated expression of indoleamine 2,3-dioxygenase (IDO) and accumulation of kynurenic acid in the pathogenesis of STZ-induced diabetic cataract in Wistar rats. Current Eye Research. 2009; 34 (4):274-281 - 91.
Oxenkrug G. Insulin resistance and dysregulation of tryptophan-kynurenine and kynurenine-nicotinamide adenine dinucleotide metabolic pathways. Molecular Neurobiology. 2013; 48 (2):294-301 - 92.
Friedman M, Cuq JL. Chemistry, analysis, nutritional value, and toxicology of tryptophan in food. A review. Journal of Agricultural and Food Chemistry. 1988; 36 (5):1079-1093 - 93.
Lin HV, Efanov AM, Fang X, Beavers LS, Wang X, Wang J, et al. GPR142 controls tryptophan-induced insulin and Incretin hormone secretion to improve glucose metabolism. PLoS One. 2016; 11 (6):e0157298 - 94.
Ueda Y, Iwakura H, Bando M, Doi A, Ariyasu H, Inaba H, et al. Differential role of GPR142 in tryptophan-mediated enhancement of insulin secretion in obese and lean mice. PLoS One. 2018; 13 (6):e0198762 - 95.
Heikkilä O, Lundbom N, Timonen M, Groop PH, Heikkinen S, Mäkimattila S. Hyperglycaemia is associated with changes in the regional concentrations of glucose and myo-inositol within the brain. Diabetologia. 2009; 52 (3):534-540 - 96.
Muriach M, Flores-Bellver M, Romero FJ, Barcia JM. Diabetes and the brain: Oxidative stress, inflammation, and autophagy. Oxidative Medicine and Cellular Longevity. 2014; 2014 :102158 - 97.
Shah GN, Morofuji Y, Banks WA, Price TO. High glucose-induced mitochondrial respiration and reactive oxygen species in mouse cerebral pericytes is reversed by pharmacological inhibition of mitochondrial carbonic anhydrases: Implications for cerebral microvascular disease in diabetes. Biochemical and Biophysical Research Communications. 2013; 440 (2):354-358 - 98.
Pandey SP, Singh HK, Prasad S. Alterations in hippocampal oxidative stress, expression of AMPA receptor GluR2 subunit and associated spatial memory loss by Bacopa monnieri extract (CDRI-08) in Streptozotocin-induced diabetes mellitus type 2 mice. PLoS One. 2015; 10 (7):e0131862 - 99.
Di Dalmazi G, Pagotto U, Pasquali R, Vicennati V. Glucocorticoids and type 2 diabetes: From physiology to pathology. Journal of Nutrition and Metabolism. 2012; 2012 :525093 - 100.
van Raalte DH, Ouwens DM, Diamant M. Novel insights into glucocorticoid-mediated diabetogenic effects: Towards expansion of therapeutic options? European Journal of Clinical Investigation. 2009; 39 (2):81-93 - 101.
Clark MS, Russo AF. Tissue-specific glucocorticoid regulation of tryptophan hydroxylase mRNA levels. Brain Research. Molecular Brain Research. 1997; 48 (2):346-354 - 102.
Ebou MH, Singh-Estivalet A, Launay JM, Callebert J, Tronche F, Ferré P, et al. Glucocorticoids inhibit basal and hormone-induced serotonin synthesis in pancreatic beta cells. PLoS One. 2016; 11 (2):e0149343 - 103.
Stetler C, Miller GE. Depression and hypothalamic-pituitary-adrenal activation: A quantitative summary of four decades of research. Psychosomatic Medicine. 2011; 73 (2):114-126 - 104.
Gupta D, Kurhe Y, Radhakrishnan M. Antidepressant effects of insulin in streptozotocin induced diabetic mice: Modulation of brain serotonin system. Physiology & Behavior. 2014; 22 (129):73-78 - 105.
Chen CC. Effect of the duration of streptozotocin-induced diabetes on turnover of central biogenic amines in mice. Neuroendocrinology. 1992; 56 (5):629-632 - 106.
Ludwig DS. The glycemic index: Physiological mechanisms relating to obesity, diabetes, and cardiovascular disease. Journal of the American Medical Association. 2002; 287 (18):2414-2423 - 107.
Ighodaro OM, Adeosun AM, Akinloye OA. Alloxan-induced diabetes, a common model for evaluating the glycemic-control potential of therapeutic compounds and plants extracts in experimental studies. Medicina. 2017; 53 (6):365-374 - 108.
Chen X, Yang W. Branched-chain amino acids and the association with type 2 diabetes. Journal of Diabetes Investigation. 2015; 6 (4):369-370 - 109.
Caballero B, Finer N, Wurtman RJ. Plasma amino acids and insulin levels in obesity: Response to carbohydrate intake and tryptophan supplements. Metabolism. 1988; 37 (7):672-676 - 110.
Brandacher G, Hoeller E, Fuchs D, Weiss HG. Chronic immune activation underlies morbid obesity: Is IDO a key player? Current Drug Metabolism. 2007; 8 (3):289-295 - 111.
Breum L, Rasmussen MH, Hilsted J, Fernstrom JD. Twenty-four-hour plasma tryptophan concentrations and ratios are below normal in obese subjects and are not normalized by substantial weight reduction. The American Journal of Clinical Nutrition. 2003; 77 (5):1112-1118 - 112.
Brandacher G, Winkler C, Aigner F, Schwelberger H, Schroecksnadel K, Margreiter R, et al. Bariatric surgery cannot prevent tryptophan depletion due to chronic immune activation in morbidly obese patients. Obesity Surgery. 2006; 16 (5):541-548 - 113.
Cangiano C, Laviano A, Del Ben M, Preziosa I, Angelico F, Cascino A, et al. Effects of oral 5-hydroxy-tryptophan on energy intake and macronutrient selection in non-insulin dependent diabetic patients. International Journal of Obesity and Related Metabolic Disorders. 1998; 22 (7):648-654 - 114.
Wurtman RJ, Wurtman JJ. Brain serotonin, carbohydrate-craving, obesity and depression. Obesity Research. 1995; 3 (Suppl. 4):477S-480S - 115.
Amer A, Breu J, McDermott J, Wurtman RJ, Maher TJ. 5-Hydroxy-L-tryptophan suppresses food intake in food-deprived and stressed rats. Pharmacology, Biochemistry, and Behavior. 2004; 77 (1):137-143 - 116.
Gangwisch JE, Hale L, Garcia L, Malaspina D, Opler MG, Payne ME, et al. High glycemic index diet as a risk factor for depression: Analyses from the Women’s health initiative. The American Journal of Clinical Nutrition. 2015; 102 (2):454-463 - 117.
McKeown NM, Meigs JB, Liu S, Saltzman E, Wilson PWF, Jacques PF. Carbohydrate nutrition, insulin resistance, and the prevalence of the metabolic syndrome in the Framingham offspring cohort. Diabetes Care. 2004; 27 (2):538-546 - 118.
Mwamburi DM, Liebson E, Folstein M, Bungay K, Tucker KL, Qiu WQ. Depression and Glycemic intake in the homebound elderly. Journal of Affective Disorders. 2011; 132 (1-2):94-98 - 119.
Møller SE. Serotonin, carbohydrates, and atypical depression. Pharmacology & Toxicology. 1992; 71 (Suppl. 1):61-71 - 120.
Wallin MS, Rissanen AM. Food and mood: Relationship between food, serotonin and affective disorders. Acta Psychiatrica Scandinavica. Supplementum. 1994; 377 :36-40 - 121.
Andrews PW, Thomson JA, Amstadter A, Neale MC. Primum non Nocere: An evolutionary analysis of whether antidepressants do more harm than good. Frontiers in Psychology. 2012; 24 (3):117 - 122.
Isaac R, Boura-Halfon S, Gurevitch D, Shainskaya A, Levkovitz Y, Zick Y. Selective serotonin reuptake inhibitors (SSRIs) inhibit insulin secretion and action in pancreatic β cells. The Journal of Biological Chemistry. 2013; 288 (8):5682-5693 - 123.
Yao S, Li J, Fan X, Liu Q , Lian J. The effect of selective serotonin re-uptake inhibitors on risk of type II diabetes mellitus and acute pancreatitis: A meta-analysis. Bioscience Reports. 2018; 38 (5):BSR20180967 - 124.
Levkovitz Y, Ben-Shushan G, Hershkovitz A, Isaac R, Gil-Ad I, Shvartsman D, et al. Antidepressants induce cellular insulin resistance by activation of IRS-1 kinases. Molecular and Cellular Neurosciences. 2007; 36 (3):305-312 - 125.
Pan A, Sun Q , Okereke OI, Rexrode KM, Rubin RR, Lucas M, et al. Use of antidepressant medication and risk of type 2 diabetes: Results from three cohorts of US adults. Diabetologia. 2012; 55 (1):63-72 - 126.
Khoza S, Barner JC, Bohman TM, Rascati K, Lawson K, Wilson JP. Use of antidepressant agents and the risk of type 2 diabetes. European Journal of Clinical Pharmacology. 2012; 68 (9):1295-1302 - 127.
Burcu M, Zito JM, Safer DJ, Magder LS, dosReis S, Shaya FT, et al. Association of Antidepressant Medications with Incident Type 2 diabetes among Medicaid-insured youths. JAMA Pediatrics. 2017; 171 (12):1200-1207 - 128.
Miidera H, Enomoto M, Kitamura S, Tachimori H, Mishima K. Association between the use of antidepressants and the risk of type 2 diabetes: A large, population-based cohort study in Japan. Diabetes Care. 2020; 43 (4):885-893 - 129.
Spillmann MK, Van der Does AJ, Rankin MA, Vuolo RD, Alpert JE, Nierenberg AA, et al. Tryptophan depletion in SSRI-recovered depressed outpatients. Psychopharmacology. 2001; 155 (2):123-127 - 130.
Argyropoulos SV, Hood SD, Adrover M, Bell CJ, Rich AS, Nash JR, et al. Tryptophan depletion reverses the therapeutic effect of selective serotonin reuptake inhibitors in social anxiety disorder. Biological Psychiatry. 2004; 56 (7):503-509 - 131.
Hari J. Lost Connections: Uncovering the Real Causes of Depression - and the Unexpected Solutions [Internet]. USA: Bloomsbury Publishing; 2018 [cited 2021 Sep 8]. Available from: https://librarysearch.library.utoronto.ca/discovery/fulldisplay/cdi_proquest_ebookcentral_EBC5235822/01UTORONTO_INST:UTORONTO - 132.
Baker S. Complex Adaptive Systems (CAS) Approach to Biomedicine & Public Health [Internet]. 2021 [cited 2023 Nov 8]. Available from: https://anatomisebiostats.com/biostatistics-blog/complex-adaptive-systems-cas-approach-to-biomedicine-public-health/ - 133.
Wilson T, Holt T. Complexity science: Complexity and clinical care. BMJ : British Medical Journal. 2001; 323 (7314):685 - 134.
Ridder DD, Stöckl T, To WT, Langguth B, Vanneste S. Chapter 7 - Noninvasive transcranial magnetic and electrical stimulation: Working mechanisms. In: Evans JR, Turner RP, editors. Rhythmic Stimulation Procedures in Neuromodulation. London: Academic Press; 2017. pp. 193-223. Available from: https://www.sciencedirect.com/science/article/pii/B9780128037263000079 - 135.
Kay J, Schneider E. Embracing complexity: The challenge of the ecosystem approach. Alternatives. 1994; 20 :32-39 - 136.
Loring PA. Threshold concepts and sustainability: Features of a contested paradigm. Klenk NL, editor. FACETS. 2020; 5 (1):182-199 - 137.
Jayasinghe S. Complexity science to conceptualize health and disease: Is it relevant to clinical medicine? Mayo Clinic Proceedings. 2012; 87 (4):314-319 - 138.
Loring PA. A vernacular for living systems: Alternative framings for the future of food. Futures. 2023; 1 (154):103276 - 139.
Du Plessis C, Brandon P. An ecological worldview as basis for a regenerative sustainability paradigm for the built environment. Journal of Cleaner Production. 2015; 109 :53-61 - 140.
Carey AL, Kingwell BA. Reducing peripheral serotonin turns up the heat in brown fat. Nature Medicine. 2015; 21 (2):114-116 - 141.
Crane JD, Palanivel R, Mottillo EP, Bujak AL, Wang H, Ford RJ, et al. Inhibiting peripheral serotonin synthesis reduces obesity and metabolic dysfunction by promoting brown adipose tissue thermogenesis. Nature Medicine. 2015; 21 (2):166-172 - 142.
Paulmann N, Grohmann M, Voigt JP, Bert B, Vowinckel J, Bader M, et al. Intracellular serotonin modulates insulin secretion from pancreatic beta-cells by protein serotonylation. PLoS Biology. 2009; 7 (10):e1000229 - 143.
Lam DD, Heisler LK. Serotonin and energy balance: Molecular mechanisms and implications for type 2 diabetes. Expert Reviews in Molecular Medicine. 2007; 9 (5):1-24 - 144.
Inubushi T, Kamemura N, Oda M, Sakurai J, Nakaya Y, Harada N, et al. L-tryptophan suppresses rise in blood glucose and preserves insulin secretion in type-2 diabetes mellitus rats. Journal of Nutritional Science and Vitaminology (Tokyo). 2012; 58 (6):415-422 - 145.
Grady CL, Siebner HR, Hornboll B, Macoveanu J, Paulson OB, Knudsen GM. Acute pharmacologically induced shifts in serotonin availability abolish emotion-selective responses to negative face emotions in distinct brain networks. European Neuropsychopharmacology. 2013; 23 (5):368-378 - 146.
Cools R, Robinson OJ, Sahakian B. Acute tryptophan depletion in healthy volunteers enhances punishment prediction but does not affect reward prediction. Neuropsychopharmacology. 2008; 33 (9):2291-2299 - 147.
Robinson OJ, Charney DR, Overstreet C, Vytal K, Grillon C. The adaptive threat bias in anxiety: Amygdala-dorsomedial prefrontal cortex coupling and aversive amplification. NeuroImage. 2012; 60 (1):523-529 - 148.
Bengart P, Gruendler T, Vogt B. Acute tryptophan depletion in healthy subjects increases preferences for negative reciprocity. PLoS One. 2021; 16 (3):e0249339 - 149.
Badcock PB, Davey CG, Whittle S, Allen NB, Friston KJ. The depressed brain: An evolutionary systems theory. Trends in Cognitive Sciences. 2017; 21 (3):182-194 - 150.
Russo SJ, Murrough JW, Han MH, Charney DS, Nestler EJ. Neurobiology of resilience. Nature Neuroscience. 2012; 15 (11):1475-1484 - 151.
Durisko Z, Mulsant BH, Andrews PW. An adaptationist perspective on the etiology of depression. Journal of Affective Disorders. 2015; 172 :315-323 - 152.
Andrews PW, Thomson JA. The bright side of being blue: Depression as an adaptation for analyzing complex problems. Psychological Review. 2009; 116 (3):620-654 - 153.
Booij L, Van der Does W, Benkelfat C, Bremner JD, Cowen PJ, Fava M, et al. Predictors of mood response to acute tryptophan depletion. A reanalysis. Neuropsychopharmacology. 2002; 27 (5):852-861 - 154.
Smith KA, Fairburn CG, Cowen PJ. Relapse of depression after rapid depletion of tryptophan. Lancet. 1997; 349 (9056):915-919 - 155.
Miller HE, Deakin JF, Anderson IM. Effect of acute tryptophan depletion on CO2-induced anxiety in patients with panic disorder and normal volunteers. The British Journal of Psychiatry. 2000; 176 :182-188 - 156.
Maletic V, Robinson M, Oakes T, Iyengar S, Ball SG, Russell J. Neurobiology of depression: An integrated view of key findings. International Journal of Clinical Practice. 2007; 61 (12):2030-2040 - 157.
Slavich GM, Irwin MR. From stress to inflammation and major depressive disorder: A social signal transduction theory of depression. Psychological Bulletin. 2014; 140 (3):774-815 - 158.
Palego L, Betti L, Rossi A, Giannaccini G. Tryptophan biochemistry: Structural, nutritional, metabolic, and medical aspects in humans. Journal of Amino Acids. 2016; 2016 :8952520 - 159.
Zhou J, Li L, Tang S, Cao X, Li Z, Li W, et al. Effects of serotonin depletion on the hippocampal GR/MR and BDNF expression during the stress adaptation. Behavioural Brain Research. 2008; 195 (1):129-138 - 160.
Chung KK, Martinez M, Herbert J. Central serotonin depletion modulates the behavioural, endocrine and physiological responses to repeated social stress and subsequent c-fos expression in the brains of male rats. Neuroscience. 1999; 92 (2):613-625 - 161.
Iacoviello BM, Charney DS. Psychosocial facets of resilience: Implications for preventing posttrauma psychopathology, treating trauma survivors, and enhancing community resilience. European Journal of Psychotraumatology. 2014; 2014 :5 [cited 2019 Mar 17]. Available from:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4185137/ - 162.
Osei K, Rhinesmith S, Gaillard T, Schuster D. Impaired insulin sensitivity, insulin secretion, and glucose effectiveness predict future development of impaired glucose tolerance and type 2 diabetes in pre-diabetic African Americans: Implications for primary diabetes prevention. Diabetes Care. 2004; 27 (6):1439-1446 - 163.
Tabák AG, Herder C, Rathmann W, Brunner EJ, Kivimäki M. Prediabetes: A high-risk state for developing diabetes. Lancet. 2012; 379 (9833):2279-2290
Notes
- Insulin pools throughout the body, excluding pools in the central nervous system.
- Serotonin pools in the central nervous system, separate from serotonin pools in the gut.
- Diagnostic and Statistical Manual of Mental Disorders by the American Psychological Association.
- Except tyrosine.
- Streptozotocin (STZ) impairs normal insulin production by damaging pancreatic cells.