Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
Enhancers are genetically encoded elements that are critical for controlling gene expression. Despite the importance of enhancers in development, normal biological function, and disease, they have been historically overlooked and remain understudied. To understand how enhancers function, appropriate molecular tools are required that can capture the temporal and spatial function of enhancers within appropriate biological contexts. Zebrafish are an excellent in vivo model for the study of enhancer elements and the genetic variants that alter their function. Because zebrafish larvae are transparent, transgenes encoding enhancers tagged by fluorescent reporters can be visualized in the tissues and developmental stages where the enhancers function. Transgenesis of enhancers can be achieved using various plasmid technologies and transgene integration methods. Here, we describe the history and most recent developments in the zebrafish enhancer assay, from vector designs to various transgene integration techniques. We go on to describe how the application of these assays have been integral for our understanding of genetic variants found in humans and within enhancers that can contribute to both human Mendelian and complex polygenic disease.
Department of Clinical Immunology and Rheumatology, University of Alabama at Birmingham (UAB), Birmingham, AL, USA
Caleb Calhoun
Department of Clinical Immunology and Rheumatology, University of Alabama at Birmingham (UAB), Birmingham, AL, USA
Robert Lalonde
Department of Pediatrics, Section of Developmental Biology, University of Colorado School of Medicine, Anschutz Medical Campus, Aurora, CO, USA
Megan Leask*
Department of Clinical Immunology and Rheumatology, University of Alabama at Birmingham (UAB), Birmingham, AL, USA
Department of Physiology, University of Otago, Dunedin, NZ, New Zealand
*Address all correspondence to: megan.leask@otago.ac.nz
1. Introduction
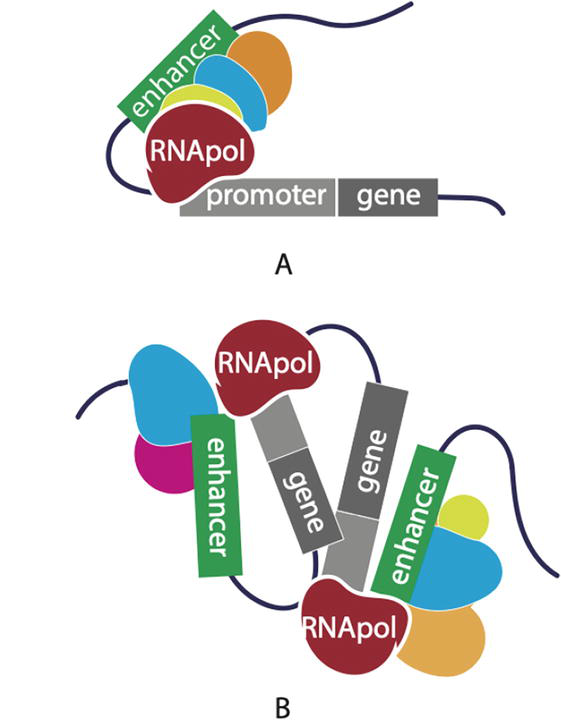
Phenotypic diversity between the cells and tissues of an individual organism is immense, but differences in the protein coding sequences of genes do not fully explain this diversity. Instead, genomic regulatory elements, that reside outside of genes in the non-coding regions of the genome, are largely responsible for this diversity by controlling gene expression [1]. This regulation of gene expression typically involves two types of genomic elements: the gene promoter and the enhancer [2]. The term enhancer reflects its function; the ability to enhance or modify the transcription of a gene from its promoter [3]. Enhancers are defined as pieces of DNA, spanning 100 base pairs in length to a few kb in length, that encode instructions that transcription factors recognize to promote gene transcription (Figure 1) [4]. Importantly, this regulation of gene expression involves the coordination of chromatin looping events that bring the enhancer, bound by transcription factors, and gene promoters into close spatial proximity. The configuration of these physical interactions, such as single enhancer: gene pairs (Figure 1A), single enhancer: multiple genes, or multiple enhancers: multiple genes (Figure 1B) are important for the tissue specificity, developmental timing, and strength of the transcriptional control of genes [5]. This fine tuning of gene expression drives multicellular development, lineage commitment, and ultimately leads to the phenotypic differences that we observe between cells, tissues, individuals and animal species.
Figure 1.
Enhancer mediated regulation of gene expression. A single (A) or multiple enhancer(s) (green) (B) loops to a gene promoter (gray) bound by transcription factors and/or chromatin binding proteins and recruits RNA polymerase to drive gene expression.
The human genome is predicted to have 1 million enhancers, although this is likely an underestimate [6]. Given this, and their role in gene expression, it is not surprising that the disruption of enhancers has been implicated time and time again as the cause of genetic trait differences, Mendelian disease, and predisposition to common diseases. The function of enhancers can be altered in a number of ways; deletions, insertions that separate the target gene from the influence of the enhancer, and single nucleotide changes in the enhancer that either ablate transcription factor binding, or introduce novel or additional binding sites. These alterations in the enhancer code can affect the regulation of gene expression, either by loss of function resulting in diminished gene expression, or gain of function resulting in overexpression of the target gene. A prime example of this, and commonly overlooked as being controlled by the non-coding genome, is the blue eyed human trait. The blue eye trait follows a predictable Mendelian inheritance pattern, which indicates that it is caused by a single genetic change. The causal genetic variant, found in roughly 18% of people with European ancestry, is located in the intron of HERC2 [7]. This non-coding variant disrupts an enhancer that loops to the nearby gene promoter, driving OCA2 gene expression (pigmentation gene), and results in pigments being deposited in a cell-specific manner in the eye. This genetic variant arose from a single shared ancestor, and was likely selected because it conferred an evolutionary advantage [8]. Over evolutionary time, this variant has dramatically increased in frequency in people of European ancestry.
In comparison to the blue eye genetic variant, only a small handful of genetic variants responsible for Mendelian disease that disrupt enhancers have been identified. This is likely due to the historical focus on protein-coding genes, and difficulty in unraveling molecular mechanisms of regulatory genetics. However, genetic variation within non-coding regulatory elements can have profound biological impacts that are equivalent to coding variants. For example, the duffy mutation that impacts susceptibility to malaria [9], and non-coding variants at SLC2A9 that influence serum urate [10, 11].
The recent developments in sequencing technologies, and the reduction in cost, enables us to expand our search to the non-coding genome. The explosion of genome-wide association studies (GWAS), indicates that >90% of the genetic variants underpinning complex human diseases and traits fall in the non-coding genome and overlap significant regions of regulatory activity [10]. Researchers are now interrogating enhancers for discovery of disease and disease susceptibility genetic variants, particularly where variants in the protein-coding genes cannot be identified. Recent examples are emerging of therapeutics that target non-coding elements. For example, an erythroid-specific enhancer that was initially discovered via GWAS has been successfully targeted in gene therapy for sickle cell disease [12]. Importantly because regulatory control by enhancers is often tissue-specific, therapeutic targeting of these non-coding elements raises the possibility that there will be fewer side effects due to systemic off-target effects. Moreover, the sheer number of non-coding elements in the genome offers a wealth of drug targets to exploit. Therefore, identifying and characterizing non-coding disease-causing genetic variants has remarkable therapeutic promise for the treatment of Mendelian and complex disease.
The conserved characteristics of enhancers can be exploited for their discovery. Active enhancers have distinct hallmarks in the chromatin landscape. They are generally depleted of nucleosomes, and have characteristic histone modifications (e.g., acetylation of H3K27 (H3K27ac)). These characteristics can be detected using DNase I hypersensitivity mapping (DHS) and chromatin immunoprecipitation, followed by sequencing (ChIP-seq) using antibodies for specific histone modifications. In an effort to comprehensively annotate the non-coding regions of the genome, international consortia have been developed to consolidate this regulatory information (e.g., ChIP-seq and DHS data) that is being generated by scientists worldwide. Leading the way in this endeavor, is the ENCyclopedia of DNA Elements (ENCODE)) project, which was established in 2003 [6]. As an example of its utility, the publicly available ENCODE data including ChIP-seq, transcription factor binding, and DHS data can be visualized using the UCSC browser, and overlapped with genetic variants of interest to identify putative causal enhancer variants (discussed in more detail in subsequent sections of this chapter). Another extremely valuable dataset that aids in the identification of putative enhancers containing genetic variants, is the Genotype-Tissue and Expression Project (GTEx). This database includes ~1000 individuals who donated their bodies to science and have been genome-wide genotyped, and had RNA-sequencing carried out in >50 tissues. This data allows scientists to connect genetic variants in the non-coding genome to gene expression (expression quantitative trait loci (eQTL)), identifying regions of the genome that control gene expression. This eQTL data can be overlaid with regulatory data from ENCODE to identify putative enhancers.
The wealth of information gleaned from these datasets and repositories indicate that our efforts should now be focussed on the functional validation of these computationally and biochemically predicted enhancer elements, and how the genetic variants within them alter their activity. The gold standard to date for functional validation has utilized reporter assays (e.g., luciferase assays and massively parallel reporter assays). However, because enhancer activity is highly dependent on the availability of the right combination and stoichiometry of specific transcription factors [13], these cell-based assays cannot recapitulate the complexity of the developmental and tissue-specific activity of an enhancer [14]. To truly understand an enhancer’s regulatory mechanisms, a reporter assay must utilize a whole organism (in vivo). In vivo reporter assays of enhancer function, with reporter-tagged non-coding DNA encoding potential enhancers, have been carried out since the late 90s, and were first conducted in Drosophila [15, 16]. These reporter assays, vector construction, and the various transgene insertion techniques have been successfully translated into the zebrafish (the general reporter assay strategy is outlined in Figure 2) over the past 25 years [17, 18, 19, 20, 21, 22, 23], and will be discussed in the following sections of this chapter.
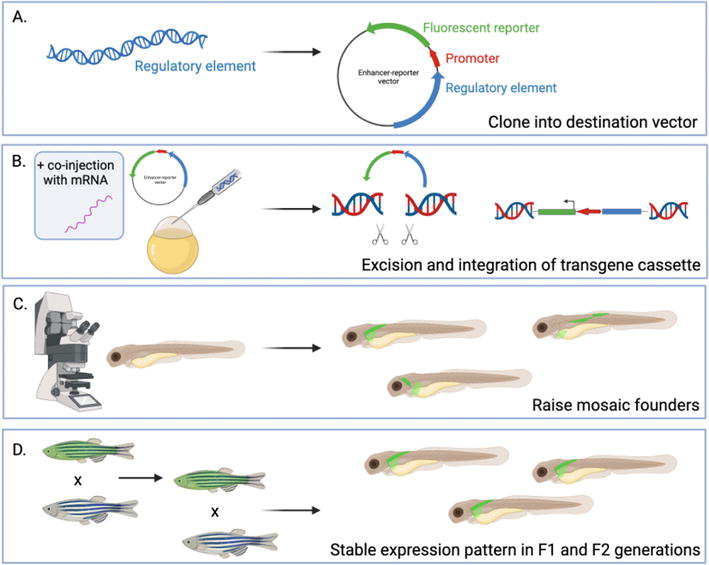
Figure 2.
Reporter assay strategy in zebrafish. (A) a regulatory element is cloned into a destination vector. Vector design varies, but at minimum includes a promoter and a fluorescent reporter. (B) the reporter assay vector is injected into fertilized zebrafish eggs with, or without, the co-injection of additional integration technologies. The transgene is excised from the donor plasmid vector and integrated into the zebrafish genome. (C) Injected fish are screened for reporter fluorescence by confocal microscopy. Individual fish that express the reporter are raised. (D) F0 founders are outcrossed with wild-type fish to establish F1 and F2 transgenic lines, where stable reporter expression patterns emerge. Created using BioRender.com.
3. Zebrafish as an ideal model for enhancer function and application of reporter assays
Zebrafish have been an important in vivo model for the study of enhancers and gene regulation, and studies consistently show that human regulatory elements can be effectively studied using this model [24, 25, 26, 27, 28, 29]. Zebrafish are ideal for the study of enhancers for a number of reasons. For example, humans and zebrafish are only ~420 million years diverged [30]. This equates to >70% of protein-coding genes at the sequence level being conserved between humans and zebrafish. These genes include a large suite of transcriptional machinery, such as transcription factors, which via enhancers function to regulate gene expression [31, 32]. HNF4α is an excellent example of the conservation of transcriptional machinery between humans and zebrafish. HNF4α is a key transcriptional regulator that controls cell-specific gene expression in development, and is conserved between humans and zebrafish [33]. The function of this transcription factor in development is so tightly constrained by evolutionary mechanisms that there are no amino acid differences in the DNA binding domain between zebrafish and humans [34]. In both humans and zebrafish, the conserved DNA binding domain of HNF4α functions to recognize the HNF4α motif that is encoded in the DNA sequence. The ligand binding domain of HNF4α responds to cell-specific cues to drive HNF4α transcriptional regulation [34]. Therefore, zebrafish HNF4α maintains the ability to recognize human HNF4α motifs encoded by human DNA sequence [35, 36].
While a large proportion of the protein-coding genome is conserved, the zebrafish genome lacks many enhancers identifiable by sequence conservation in other species, including humans [37, 38]. However, because the transcriptional machinery involved in gene regulation is conserved, the genetic regulatory mechanisms also persist [39]. For example, enhancer elements within the RET locus exist in both humans and zebrafish, which act analogously to control RET expression. Although the enhancer elements have evolved so that the sequence is no longer orthologous, their function, recruitment of the same transcription factors, is preserved [40].
Additional features of zebrafish, including ease of transgenic manipulation [41] and larval transparency, make it possible to directly observe enhancer activity in vivo [42]. For example, layering imaging of regulatory element expression with fluorescently-labeled cell markers allows for the assessment of tissue-specific enhancer activity over developmental time. Additionally, by comparing tissues where the enhancer drives expression to those where a gene is expressed (e.g., in situ hybridisation), one can approximate the target gene of an enhancer if the enhancer and gene expression colocalizes [36, 43]. Zebrafish are also a cost-effective and relatively low-maintenance model system, particularly when compared with mice (for which reporter assays exist), which makes them suitable for the longer-term rearing and maintenance [18]. This allows for multiple reporter lines to be generated so that qualitative comparisons can be made between different enhancer elements, or variants within the same enhancer.
In zebrafish, a typical in vivo reporter assay (outlined in Figure 2) involves the construction and insertion of a plasmid vector. This vector contains a reporter protein placed downstream of a minimal promoter and a predicted enhancer sequence to be tested (Figure 2A). Insertion of the transgene into the zebrafish genome is mediated via endonuclease, retrovirus, transposon, or recombinase-based integration (Figure 2B) [44, 45]. Expression of the reporter protein in zebrafish larvae is used as a readout of enhancer activity because the reporter gene should only be expressed if the minimal promoter is driven by regulatory activity in the vicinity of the enhancer element.
In the context of human health and disease, alterations to enhancer activity by genetic variants within them can also be assessed in zebrafish reporter assays [19]. Several consequences of variants can be tested. For example, a complete loss of expression in a tissue may indicate the disruption of a key transcription factor binding site within the enhancer, affecting the formation of a transcription factor complex required for regulatory activity [46]. Modification of the strength of expression similarly may indicate a change in transcription factor binding affinity [19]. A novel expression pattern may indicate ‘gain of function’, where the DNA sequence change is such that a transcription factor binds more tightly, or a new transcription factor binding site is introduced [19]. Finally, no change in fluorescence strength or location may indicate that the variant has no functional effect on the enhancer’s activity, or may play some other role that is not captured in the context of this assay, for example in the organization of chromatin in the nucleus [47].
Importantly, there are several factors to consider when interpreting the results of a reporter assay in zebrafish. First, transgenic F0 fish will display mosaic expression of the transgene. This is because not all of the cells of the injected embryos will receive the transgene. However, a proportion of embryos will incorporate the transgene into their gametes, and their F1 progeny (stable lines) will either have the transgene in all cells, or none at all. Additionally, a phenomenon termed ‘position effect’ exists. The position effect is caused by the transgene being integrated into various genomic positions, which can in turn be influenced by the genomic context with which it is inserted [48, 49, 50]. The phenomenon often causes fluorescent expression to be identified that is not driven by the non-coding sequence of interest, but by the random insertion of the transgene into a region where strong regulatory activity exists. This can result in variable ectopic transgene expression, silencing of the transgene, or mutagenic effects if it is inserted within a coding region of the genome. To reduce the potential for bias arising from the over-interpretation of inaccurate fluorescent patterns and overcome mosaicism, multiple independent stable lines (F1 or F2 generations) of zebrafish should be screened for reproducible expression patterns, which may indicate the most likely expression pattern of the putative enhancer’s activity.
Before the advent of superior transgenesis techniques in the early 2000s, the method employed to generate transgenic zebrafish involved the injection of vast quantities of plasmid DNA (~106 plasmid copies) in the hope that some of this DNA would be transcribed and then integrated into the genome to be inherited in subsequent generations [51]. This approach has very low rates of integration and germline transmission, and many different integration techniques have subsequently been developed that have dramatically improved these rates. For example, I-Scel mediated integration, first described in 2002 in medaka fish [45]. This method involves the co-injection of the meganuclease I-SceI, a rare-cutting endonuclease isolated from yeast, alongside a vector containing a transgene flanked by corresponding I-SceI recognition sites. The meganuclease excises the transgene and provides recombinogenic ends to the DNA that facilitate integration into the genome. In comparison to a non-mediated transgenesis approach, this method reduces the amount of mosaicism present in the F0 fish and increases germline transmission rates (Table 1) [45].
Technique
Method
Merits
Limitations
Large plasmid quantity
Inject large quantities of plasmid DNA containing a transgene of interest (~106 plasmid copies) into zebrafish embryos, where some of this DNA is likely to be transcribed and then integrated into the genome.
Low rates of genomic integration Low rates of germline transmission High mosaicism present Position effects Tandem insertions leads to silencing
Endonuclease
Endonuclease mRNA is co-injected with a vector containing a transgene flanked by corresponding endonuclease cutting sites. Once excised from the vector, the transgene is left with recombinogenic ends that facilitate integration into the zebrafish genome.
Vector is injected into zebrafish embryos that contain the transgene and a modified viral sequence that has removed proteins required to replicate the virus but retained those that promote its integration into the genome.
High integration rate High rate of germline transmission
Variable copy number integration Position effects Multicopy insertions
Transposase
Transposase mRNA is co-injected into single cell zebrafish embryo with a vector containing a transgene flanked by non-autonomous transposon sequences recognized by the transposase which catalyzes the transposition of the transgene into the genome.
High integration rate High rate of germline transmission Plenty of published vector technologies for use in reporter assays No tandem insertions
Position effects Multicopy insertions Transgenes can remobilize if you inject with Tol2
Recombinase
Transgenic fish are generated with recombinase ‘landing sites’. Injected into embryos of the progeny of these fish is the recombinase mRNA and a vector containing a recombinase site. The enzyme initiates the recombination of the transgene into the genome at the ‘landing site’.
Very high integration rate Very high rate of germline transmission Single-insertion Targeted integration limits position effects Reduced F0 mosaicism Transgene insertion can be permanent (can inject these with phiC31 + vector)
Application is not widespread in zebrafish Specialized zebrafish transgenic lines required Current lack of understanding of good genomic integration positions and stable lines with mapped landing sites
Table 1.
Integration methods for the insertion of transgenes in zebrafish embryos to study regulatory function of enhancer elements.
The use of a retrovirus-mediated system has also been explored within the zebrafish model for the detection of putative enhancer sequences [52]. A retroviral vector system contains viral sequences that promote reverse transcription from RNA to DNA and integration into the genome, but do not encode the viral proteins required to replicate and produce additional virus. Instead of viral proteins, reporter genes and non-coding sequences can be engineered within the vector [53]. The use of these engineered retroviral vectors in zebrafish reporter assays enables the integration of a transgene into the genome and its inheritance through the germline. However, variation in reporter expression is caused by variable copy number integration and position effects (Table 1).
Over the past two decades, two dominant approaches have emerged for the integration of transgenes into the zebrafish genome that aim to combat the issues described above. The first, transposons, mediate the random integration of a transgene into a genome while the second, recombinase enzymes, mediate site-directed integration. These two methods will be discussed further in sections 3.1 and 3.2 and are outlined in Table 1.
3.1 Transposon-based integration
Transposons are genetic elements that can move from one area in the genome to another. Some elements are autonomous and encode a functional transposase protein that can catalyze the transposition of DNA [54], and other elements are non-autonomous, and do not encode a functional transposase but maintain essential sequences that can be transposed in the presence of transposase [55, 56]. An autonomous Tol2 mobile element, belonging to the hAT transposon family [57], was discovered in the medaka fish [58]. In zebrafish, where this element is naturally absent, a two-component transposition system utilizing the Tol2 transposon to integrate a transgene of interest has been developed [59]. This system requires both a plasmid containing a nonautonomous Tol2 element lacking part of the transposase gene, and the mRNA of the Tol2 transposon. These components are co-injected into a single-cell zebrafish embryo, where the Tol2 transposon catalyzes the transposition of the nonautonomous element from the plasmid into the zebrafish genome (Figure 2) [60, 61, 62]. This technology has been adapted to study predicted enhancer elements by designing a vector with an enhancer-promoter-reporter cassette in-between two nonautonomous Tol2 elements [63]. Fish injected using this system are analyzed for their fluorescent expression patterns indicative of enhancer activity, and these fish are then raised to adulthood and used to create transgenic lines. The rate of transgene integration is very high, particularly when compared to simply injecting just plasmid DNA, and this technology has been successfully applied to study a range of human enhancers. However, a large caveat to transposon-mediated transgenesis is that the transgene is inserted randomly and predominantly into active genomic regions, which causes position effects [63, 64]. Many groups have utilized this Tol2 transposon-based system to functionally investigate enhancers in zebrafish through the use of specifically designed vectors.
Some vector designs containing Tol2 sequences are of a relatively simple design; a green fluorescent protein (GFP) reporter, a minimal promoter, and a Gateway cassette for easy recombination of DNA of interest [17]. While these designs are successfully used to generate transgenic fish, there are additional components that can be added for ease of use, and that have improved our ability to assess enhancer activity in zebrafish. For example, the more simple vector designs lack an internal positive control, which makes it difficult to determine the success of a genome integration event or the passage of this integration down the germline. This is particularly challenging when the spatiotemporal nature of enhancer activity is considered, whereby an enhancer may only drive expression of the reporter at a specific developmental stage and/or a tissue that may not be visually obvious.
The Zebrafish Enhancer Detector (ZED) vector was designed to address the need for a positive control for transgenesis and additionally, inconsistent fluorescent expression caused by the position effect phenomenon (Figure 3) [65]. The ZED vector contains a positive control cassette which includes a Cardiac Actin promoter and red fluorescent protein (RFP). This cassette drives RFP expression in the somites of the fish and therefore, can be used as an obvious indicator of the success of vector integration into the genome. In comparison, the enhancer detection cassette contains the green fluorescent protein (GFP) as a reporter for activity so that visualization of the success of integration and potential enhancer activity can be visualized on different light channels. The ZED vector decreases position effects by flanking the reporter cassette with insulator sequences so that the cassette is shielded from position effects. Furthermore, this vector contains an improved minimal promoter to drive GFP which was selected for optimal enhancer activity detection, and two excision cassettes. The excision cassettes allow for the genetic deletion of the transgene, should a researcher want to confirm that the fluorescent activity is dependent specifically on the enhancer element.
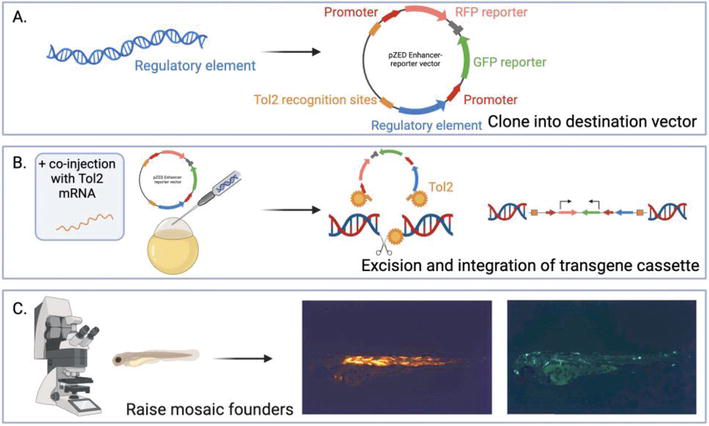
Figure 3.
Zebrafish enhancer detection (ZED) vector system. (A) a regulatory element is cloned into the ZED vector which contains a positive control cassette, consisting of a cardiac actin promoter and RFP, and a reporter cassette, consisting of a gateway entry site upstream of a minimal promoter and GFP. (B) the vector is co-injected into single-cell fertilized zebrafish eggs alongside Tol2 mRNA. The Tol2 transposase is transcribed into an enzyme that binds to the recognition sites on the vector and excises the transgene, and then binds to a sequence in the genome where it mediates the transgene’s integration. (C) Confocal microscopy is used to screen injected embryos for RFP (integration control), and GFP (regulatory element activity) expression. Zebrafish photos are original work. Created using BioRender.com.
The ZED vector’s key improvements allow for the efficient and rapid generation of transgenic zebrafish that contain an internal transgenesis control and reduced position effects. However, limitations to this system remain. For example, while position effects have been reduced, they unfortunately still remain which can interfere with the interpretation of fluorescent signals. Another issue with this system, is while RFP expression in somites makes for an obvious and effective control, the expression of this protein can be so strong and so abundant throughout the fish that the RFP can bleed through other fluorescent light channels (as seen in Figure 3C) and it is hard to analyze the location of specific GFP expression in the fish. Despite the short-comings of this particular vector, various improved transgenesis reporter fluorescent markers continue to be designed and incorporated into reporter assays for example, cryaa: venus is a cerulean fluorescent marker of transgenesis expressed only in the lens on the fish [66].
Another example of an assay that applies the Tol2-mediated transgenesis is the dual-color reporter-based assay, used for the analysis of variants within enhancer elements in parallel (Figure 4) [19]. In this assay, two Tol2 constructs bearing the alternate alleles of a genetic variant within an enhancer are recombined with different fluorescent reporter cassettes in a multi-gateway reaction. These two plasmids are then co-injected with Tol2 mRNA into zebrafish embryos. Developing fish are then screened for reporter fluorescence and the positive fish are outcrossed to generate stable dual-reporter-expressing lines. Using confocal microscopy, tissues where the activity of two enhancers overlap are observed as yellow, while independent activities are green or red. This system allows for the direct comparison of the spatial and temporal activities of different enhancers, or different variants within the same enhancer, in a single zebrafish [19]. This assay significantly reduces the number of zebrafish normally required in a reporter assay. A limitation is that this system cannot control for the position of insertion sites, and the zebrafish tend to display variation in expression patterns due to positional effects. Additionally, the two enhancer sequences will integrate into the genome in variable numbers because they are within different plasmids, limiting the system for quantitative assessment of altered enhancer activity.
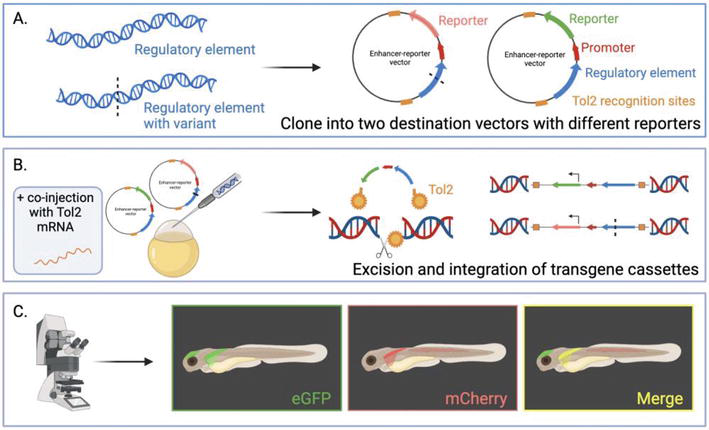
Figure 4.
Dual-color zebrafish transgenesis strategy. (A) Two regulatory elements are cloned into vectors containing a minimal promoter and two different fluorescent reporters. (B) both vectors are co-injected into single-cell fertilized zebrafish eggs alongside Tol2 mRNA. The Tol2 transposase is transcribed into an enzyme that binds to the recognition sites on the vector and excises both transgenes, and then binds to a sequence in the genome where it mediates the transgene’s integration. (C) Confocal microscopy is used to screen injected embryos for fluorescence where independent regulatory activity is observed as green or red, and overlapping activities are yellow. These mosaic F0 founders are raised to generate transgenic lines. Created using BioRender.com.
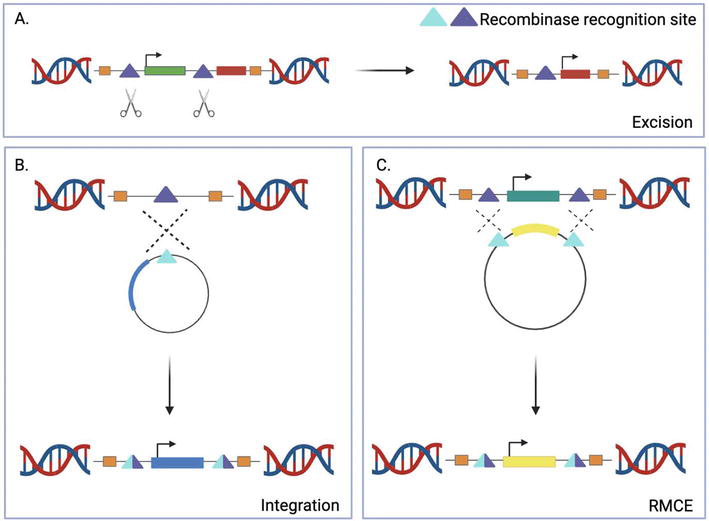
3.2 Recombinase-based integration
In comparison to the random integration methodology of transposons such as Tol2, site-specific recombinases are enzymes that catalyze recombination events at highly specific target sequences. This method has the potential to mitigate the variability caused by the random integration of transgenes and resulting positional effects by creating an efficient and reproducible site-directed integration system, where enhancer elements and their variation can be quantitatively compared. Owing to their specificity, pre-defined recombinase sites can be established within a model organism’s genome, permitting the integration (Figure 5B) or excision (Figure 5A/6B) of transgenes at a defined position by recombinases [67, 68, 69, 70]. Notable recombinases that have been employed within the zebrafish model system include Cre, derived from bacteriophage P1, Flp, from yeast, and phiC31 recombinase, from phage phiC31. In comparison to the recombinases Cre and Flp who target loxP and FRT sites respectively, phiC31 targets two heterotypic sites termed attB and attP, resulting in the directional and irreversible integration of DNA (Figure 6D) [71, 72, 73]. PhiC31’s canonical role is the integration of a phage genome into a bacterial genome via recombination between attP (phage) and attB (bacteria) sequences. Utilizing the specificity of phiC31 recombinase to these sites, an exogenous plasmid containing a transgene with an attP (or attB) site can be injected alongside phiC31 encoding mRNA into a zebrafish embryo harboring a previously integrated attB (or attP) landing site in their genome (Figure 6). The phiC31 initiates the recombination of this transgene into the genome at these attB/attP sites [74, 75, 76, 77, 78]. The irreversible nature of this system caused by the completed attP/attB recombination, produces hybrid attL (left) and attR (right) sites which cannot be used by phiC31 as substrates. This lends itself to heightened efficiency because even in the presence of large quantities of plasmid, the recombination between the attP/attB sites cannot be reversed [71].
Figure 5.
Recombinase-based transgene integration strategies. (A) Excision of a previously inserted cassette, typically employed to induce regulatory activity at a specific tissue or point in development. (B) Site-directed integration of an introduced transgene in the genome. (C) Recombination-mediated cassette exchange (RMCE) between a new and previously introduced transgene. The phiC31 integrase system is effective at controlling cassette excision, integration, as well as RMCE. Created using BioRender.com.
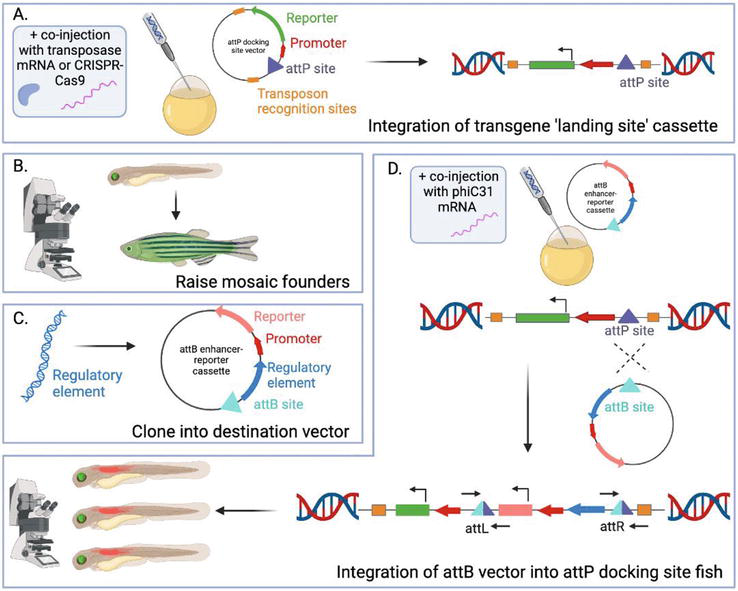
Figure 6.
Generation at attP/attB-recombinant zebrafish transgenic lines using phiC31 recombinase. (A) a vector containing GFP-tagged attP sites is co-injected with integration technologies, such as transposase mRNA, into fertilized single-cell zebrafish embryos, where the transgene is integrated into the genome. (B) Using confocal microscopy, larvae are screened for fluorescence, reflecting the integration of the landing site cassette into the zebrafish genome, and these fish are raised to generate transgenic lines. (C) a regulatory element is cloned into a vector containing an attB site and a fluorescence reporter. (D) the vector is co-injected with phiC31 mRNA into transgenic embryos containing an attP integration site. A recombination event occurs, mediated by phiC31, between the attB and attP sites, and the transgene is integrated at this landing site. Using confocal microscopy, larvae are screened for fluorescence expression. Created using BioRender.com.
The use of phiC31 attP/attB recombination for the insertion of transgenes such as putative enhancer sequences, is routine in Drosophila work [68, 79] however, its translation into the zebrafish model has been largely limited to proof-of-principle studies. Two such studies describe a technique by which phiC31 catalyzes a recombination event in a part of a previously inserted transgene, causing the excision of a control cassette and the activation of a second coding sequence (Figure 5A) [75, 76]. In comparison to the single phiC31-mediated recombination event, recombinase-mediated cassette exchange (RMCE) involves the phiC31 integrase excising and replacing a reporter-tagged integrated transgene with another reporter labeled transgene (Figure 5C) [80]. During RMCE, a transgene containing a fluorescent reporter protein flanked by attP (or attB) sites is integrated into the zebrafish genome via Tol2-mediated transgenesis. These zebrafish are screened for germline transmission, and the fluorescent-positive embryos co-injected with phiC31 mRNA and a vector containing an enhancer sequence tagged with a different fluorescent reporter, flanked by attB (or attP) sites. PhiC31 facilitates recombination between the attP/attB sites which excise the fluorescently labeled reporter cassette and integrates the other fluorescently labeled cassette. A limitation of this approach comes down to the initial random insertion of the transgene by Tol2, as the number and position of these insertions are not controlled. Therefore, zebrafish can have multiple independent insertions and many transmit both fluorescent markers following injection, reflecting that recombination did not occur at every attP/attB site [80]. To improve upon this system, stable transgenic zebrafish lines with well characterized attP/attB sequences that support efficient phiC31-mediated recombination are required. These lines would eliminate the time and labor required to generate new attP/attB-containing transgenic acceptor lines for every experiment and additionally, would eliminate the need to screen and map each transgene insertion site.
A large amount of time and labor is dedicated to the generation of stable single-insertion attP/attB transgenic zebrafish lines. A research group in 2013 reported to have generated two such lines [78]. Tol2-mediated transgenesis was employed to generate several independent attP landing site zebrafish lines containing a full-length attP acceptor site alongside a GFP-tagged promoter. These lines were assessed for a 50% Mendelian inheritance pattern in the F2 generation, reflective of a single transgene insertion. These lines were co-injected with phiC31 mRNA and a vector containing a GFP-tagged transgenesis marker flanked by full-length attB sites, and screened for fluorescence reflecting a successful integration of the transgene. Two landing site lines consistently demonstrated reproducible attP/attB recombination, and the integration position was mapped and characterized. Reproducible attP/attB recombination was determined through a variety of experiments. One such screen tested integration in mutant backgrounds, where attP landing-site lines were crossed with fish lacking a transcription factor essential for melanocyte development. The progeny were injected with phiC31 mRNA and a vector containing the missing transcription factor, and skin pigmentation was successfully rescued [78]. Ultimately, this group was able to successfully establish, characterize, and maintain functional attP landing site fish for the site-directed generation of transgenic lines. Multiple other teams of scientists have also been generating single landing-site zebrafish [42, 77], and have demonstrated that this technique successfully limits variability of transgene activity caused by positional effects, particularly when compared to lines generated with random Tol2-mediated transgene insertions [77].
While this technique has proven successful for the integration of transgenes at a single genomic position, our understanding of the genomic positions where the rate of this integration is most successful is lacking. The use of gene editing technologies such as CRISPR-Cas9, would enable a specific locus to be targeted for attP/attB-site insertion, removing the unpredictability caused by the initial Tol2 transposon-mediated insertion. Validated integration loci have been identified in mammalian systems at the Hipp11 (h11) locus [81, 82, 83], and work has been undertaken to identify similar sites in zebrafish [84]. There is some evidence that zebrafish lines with stable transgene expression have integrated transgenes adjacent to regions marked with native histone marks, reflecting open and active chromatin [84]. In the future, we strongly encourage researchers to map the chromatin dynamics and the genomic coordinates of transgene integration sites produced by random integration. These insights will help identify features that support successful, reproducible, and inherited integration which can then be applied to generate landing site lines with targeted technologies, such as CRISPR-Cas9.
4. Application of reporter assays to understand human disease
Historically, the search for genetic variants contributing to human disease has been restricted to exons, representing only 1–2% of the human genome. In comparison, variants within non-coding regions and their contribution to disease remain poorly investigated. This is because variants within coding regions are much easier to interpret, as mutations that truncate the mRNA transcript or alter the structure of the protein intuitively result in aberrant protein function and disease phenotypes. Less clear are the potential effects of variants within non-coding enhancer elements. However, the impact of non-coding variants have become well recognized in the etiology of Mendelian and polygenic diseases, and the utilization of the zebrafish reporter assay has been an invaluable tool for our understanding of their contribution.
4.1 Mendelian disease
Complex phenotypes and diseases are influenced by thousands of genes and genetic variants that are common. Comparatively, Mendelian disorders are caused by rare and typically singular genetic variants that have predictable Mendelian inheritance patterns. For example, Cystic Fibrosis is an autosomal recessive disorder that is caused by disruptions, most commonly the p.Phe508del variant, in the cystic fibrosis transmembrane conductance regulator (CFTR) gene [85]. The search for genetic variants contributing to Mendelian disorders has largely been limited to exons. Historically, it was understood that there are enough redundancies between enhancers that regulate the same gene, such that any pathogenic impact of an enhancer mutation would be mitigated by this redundancy [86, 87, 88]. However, a large percentage of people with a disease that follows a Mendelian inheritance pattern do not have a genetic variant in the coding sequence [89], indicating that the causal genetic variant may instead reside in functional non-coding sequences, such as enhancers. The reduction in the costs of DNA sequencing has led to the more routine use of targeted or whole genome sequencing in the clinic, which in turn has led to singular pathogenic enhancer variants being uncovered. For example, several variants have been identified in an enhancer 25 kb upstream of PTF1A in patients with pancreatic agenesis [90]. These genetic variants display Mendelian inheritance patterns in affected families. The enhancer activates a developmental enhancer cluster, and loss-of-function variants prevent the hierarchical activation of other enhancer domains therefore, circumventing functional redundancy, and causing the disease phenotype [90]. This finding highlights the necessity of investigating the non-coding genome for regions such as enhancers that are disrupted by Mendelian genetic variants.
The zebrafish reporter assay can be employed to validate Mendelian genetic variants discovered in enhancers, furthering our understanding of their contribution to disease. For example, a regulatory variant was identified in a person with the cardiac features of Holt Oram disease (congenital condition of the heart and limbs) that disrupts the expression of TBX5 (a transcription factor with defined roles in cardiogenesis and forelimb development [91, 92]). In this study, the region surrounding TBX5 was sequenced in an individual with the most common heart defect observed in Holt-Oram Syndrome, but who did not have limb defects. A reporter assay was employed in zebrafish (see Figure 2) to interrogate potential enhancer activity and the variant’s effect. The enhancer sequence was PCR amplified from the individual with the variant and an individual without. These regions were Gateway cloned into an entry vector containing a GFP reporter and two Tol2 transposon recognition sites. Each construct was co-injected alongside Tol2 mRNA into one-cell zebrafish embryos, and these F0s were screened for GFP expression after 24 hrs. In fish injected without the variant-containing construct, 34% of fish displayed reproducible GFP expression in the heart. Comparatively, only 1.8% of fish injected with the variant-containing construct displayed weak GFP expression in the heart [26]. These data indicate that the enhancer sequence has regulatory activity that recapitulates TBX5 expression in the heart but no other tissues associated with Holt-Oram syndrome (e.g., limb progenitors). The data also indicate that there is a functional consequence of the variant, virtually eliminating the enhancer’s ability to drive gene expression in a cardiac-specific manner. Importantly, this information decoupled the molecular mechanisms underpinning the limb and cardiac malformations that are seen in Holt-Oram syndrome, broadening understanding of the disease. A caveat to this study is that they did not make transgenic stable zebrafish lines which would have reduced any position effect bias caused by the random transposon-mediated integration. However, the study provided a convincing example of the contribution of non-coding variants to Mendelian disease.
Another example of an enhancer that is causal of Mendelian disease, that was characterized in zebrafish, is found upstream of the SOX9 gene and linked to the Mendelian disease Pierre Robin sequence (PRS) [93, 94]. SOX9 is a transcription factor essential for chondrocyte differentiation, neural stem state determination, and male sexual development [95]. Pathogenic PRS variants have been identified within an enhancer ~1.5 Mb upstream of SOX9 [93]. It was hypothesized that the enhancer has specific spatiotemporal activity, and that the disruption of this activity leads to misexpression of SOX9, and consequently the craniofacial abnormalities seen in people with PRS. A dual-color reporter-based assay in zebrafish (see Figure 4) [19] was employed to test the enhancer region, termed HCNE-F2 [93], for enhancer activity, and the ability of a variant identified in people with PRS to alter this activity [96]. The HCNE-F2 region of interest was PCR amplified from the DNA of a person with PRS carrying the variant of interest, and a control. These were cloned into a vector containing either a GFP or mCherry reporter respectively, flanked by Tol2 recombination sites. The vectors were co-injected alongside Tol2 mRNA into one-cell stage embryos and screened for fluorescence, and positive fish raised to adulthood. A number of stable lines were generated by in-crossing fish with stable germline transmission and their progeny were screened for dual-fluorescence from both transgenes. The HCNE-F2 region that did not contain the variant drove fluorescence in the developing pharyngeal arches and craniofacial cartilage, and this expression was lost in the HCNE-F2 region containing the variant [96]. The application of this dual-reporter assay enabled the direct comparison between control and variant-containing sequences of a putative enhancer region. The results indicate that the enhancer region has craniofacial regulatory activity, and the variant ablates this activity, potentially leading to the misexpression of SOX9 and the craniofacial defects seen in people with PRS.
4.2 Complex polygenic disease
Complex polygenic conditions, such as type 2 diabetes and cardiovascular disease, are those where individual common genetic variants contribute only very small effects. To capture important regions of the genome that contribute to the development of polygenic disease, genome-wide association studies (GWAS) that consist of many hundreds of thousands of participants are the gold standard approach [97]. Crucially, GWAS find that <10% of associated regions of the genome have causal mechanisms implicating a protein-coding variant. Instead, the majority of the polygenic associations are outside of the coding regions, suggesting that altered gene regulation is a major contributor to complex disease. Integrated meta-analyses of genomic and epigenomic annotations have demonstrated that GWAS variants are significantly enriched in functional non-coding regions [98, 99], particularly in close proximity to enhancers [100, 101]. For example, ~60% of likely causal variants map to enhancer elements in an autoimmune disease GWAS [102]. These data suggest that a substantial proportion of genetic variants identified using GWAS contribute to disease by disrupting enhancer activity. However, studying the biological consequences of genetic variation at GWAS loci is challenging for a number of reasons. Identifying the causal gene(s) can be difficult, as candidate enhancers do not always affect nearby genes, but can have long-range interactions with promoters [103]. In addition, in most cases, the causal variant at a GWAS locus will also be associated with many other genetic variants due to linkage disequilibrium. Unfortunately, there has been very little progress in translating these loci into biology since the advent of GWAS in the early 2000s, and identifying causal variants and determining how and where non-coding variants affect target genes remains poorly understood [104].
For example, it took almost 10 years for the well-known obesity associated locus in intron 1 of FTO, first identified by GWAS in 2007 [105, 106], to be biologically linked to an enhancer that functions to regulate IRX3 and IRX5 [107, 108]. This example at FTO highlights the challenge of pinpointing both the causal gene(s) and causal genetic variant underpinning a genetic association with disease. It was initially hypothesized that FTO was the causal gene at this locus, however numerous studies using human cell lines, primary human adipocytes, and mouse models eventually showed that IRX3 and IRX5 were the causal genes underpinning the genetic association [108]. Additionally, more than 100 linked variants (LD R2 > 0.8) associated with obesity were identified at FTO, over a 47 kb block of linkage disequilibrium [109], such that the causal variant(s) have been hard to define.
In an attempt to assign function to the FTO region of association, without having to systemically test the individual genetic variants within this LD block, a research group took a sledgehammer approach by applying Bacterial Artificial Chromosome (BAC) vectors [110, 111, 112] in an enhancer-reporter zebrafish assay. Importantly, this type of vector enables testing of very large portions of DNA for enhancer function using GFP reporter cassettes. Fluorescent-positive zebrafish, injected with modified BAC vectors and Tol2 mRNA, were identified, raised, and used to create stable transgenic lines. In this study they were able to show that 183 kb of human genomic DNA spanning ~30 kb upstream of the FTO promoter, and 150 kb into the FTO gene including exon 4, were able to drive expression of GFP in the brain of the developing zebrafish larvae. In addition, immunohistochemistry for GFP showed GFP expression indicative of enhancer activity within the hypothalamus of the adult zebrafish, recapitulating the brain expression of IRX3. Intriguingly, this zebrafish study is not well cited and largely ignored by researchers working on untangling the mechanisms underpinning this genetic association at FTO. Crucially, the exact molecular mechanisms of the FTO genetic association are still unclear, complicated by multiple signals of association and population-specific effects [113]. A putative causal variant was identified in 2015, and was found to disrupt a conserved ARID5B motif [108], but it is not clear if the ARID5B disrupting variant is the only causal variant at this locus. This ambiguity highlights the potential for next generation zebrafish assays to substantially contribute to the scientific debate, and our understanding of the mechanisms surrounding this locus.
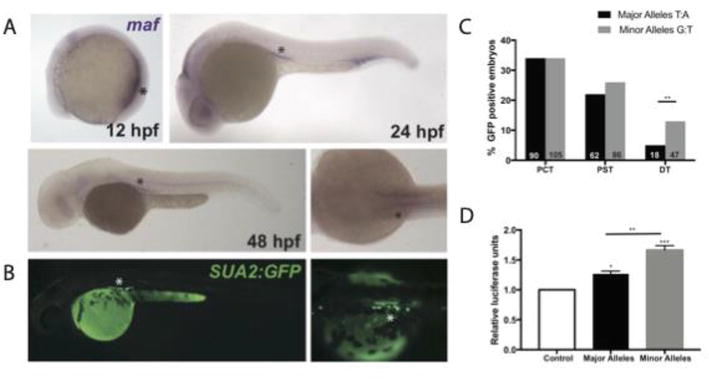
The BAC approach described above, where very large pieces of DNA are inserted, was particularly useful before we had better prediction methods for regulatory function for genetic variants. Now, with data made available by the ENCODE consortium [6] and GTEx, it is possible to overlay genetic variants with regulatory data and gene expression data (eQTL) to prioritize putative causal variants before testing in zebrafish assays. The success of this approach is highlighted by two studies [35, 36] that investigated loci with genetic associations of serum urate, a key metabolite involved in the development of the inflammatory arthritis, gout [114]. In these studies, transcription factor binding sites were overlaid with urate-associated genetic variants at the PDZK1 and MAFTRR genes. At both loci, PAINTOR (a bioinformatic tool that statistically tests the likelihood a genetic variant is causal) identified that genetic variants that were within, or close to, HNF4A core consensus motifs had the highest PAINTOR probability scores. Using the ZED-vector based assay described in Section 3.1 (see Figure 3) [65], the regions encompassing these HNF4A binding sites, and the genetic variants associated with serum urate, were introduced into zebrafish embryos. GFP expression was observed in the kidney tubule of developing zebrafish and additionally, in the liver and intestine for the PDZK1 enhancer [35]. Importantly, enhancer activity overlapped expression of zebrafish HNF4A, PDZK1, and MAF(the regulatory target of the lncRNA MAFTRR), causally linking the enhancers to target gene expression (Figure 7A–B).
Figure 7.
Zebrafish maf coincides with enhancer activity. A. in situ hybridization for zebrafish maf indicates expression in the pronephros at 12 hpf and the proximal tubules at 24 and 48 hpf. B. Enhancer expression (GFP) in the proximal tubules at 48 hpf. C. Percentage of transgene positive embryos (RFP) that present with GFP-positive cells in the PCT, PST, and DT. D. Luciferase reporter assay in HEK293 cells. A one-way ANOVA test resulted in a significant difference between the means of the control and the enhancer containing each variant. This figure has been adapted from Leask et al. 2019 with permission from the authors.
For the enhancer at MAFTRR, differences in GFP expression between genetic variant alleles was semi-quantified in F0 fish (Figure 7C), showing that the urate raising alleles had a greater ability to drive expression in the proximal convoluted tubule [36]. The allelic differences in expression were confirmed by luciferase assay in kidney cell lines (Figure 7D). Importantly, this is another example that highlights the need for a zebrafish assay that can robustly quantify allelic differences at enhancers. Such an assay would dispense with the cell-based assays, such as luciferase assays and CRISPR activation screens, for allelic quantification, which require prior knowledge about the types of cells and tissues where the enhancer functions. The value of an in vivo assay was highlighted in the PDZK1 example, as although GFP expression was observed in the kidney, liver and intestine of the zebrafish, only the liver cell lines selected for the luciferase assay have HNF4A expression [35]. Therefore, it would not have been possible to robustly test the allelic effects of the enhancer variants in kidney cell lines. In this context, the zebrafish assay circumvents this challenge because the transcriptional machinery required to drive gene expression is within the appropriate biological context.
This chapter has highlighted the role of the zebrafish report assay to test enhancer activity in vivo and the various tools available to researchers using this model. We describe how in zebrafish, reporter assays have been applied and improved over the years to test for regulatory activity of predicted enhancer elements. In the future, recombinase landing site zebrafish lines will reduce the intensive labor required to screen transgenic fish generated using random integration technologies. These lines will also be an essential tool for the accurate in vivo quantification that both Mendelian and complex-disease variants have on enhancer function. The advent of cost-effective whole genome sequencing and large-scale GWAS has enabled us to comprehensively discover non-coding Mendelian disease variants and common variants that contribute to complex disease. It is therefore more important than ever to develop tools that are scalable and can accurately recapitulate enhancer activity in the relevant biological context, to comprehensively understand the underlying molecular and disease mechanisms.
The authors would like to thank the University of Alabama at Birmingham for funding (Underrepresented in Medicine Early Career Investigator Award award to Dr. Leask).
1.Mattick JS. Non-coding RNAs: The architects of eukaryotic complexity. EMBO Reports. 2001;2(11):986-991
2.Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annual Review of Genomics and Human Genetics. 2006;7:29-59
3.Banerji J, Rusconi S, Schaffner W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981;27(2 Pt 1):299-308
4.Spitz F, Furlong EEM. Transcription factors: From enhancer binding to developmental control. Nature Reviews. Genetics. 2012;13(9):613-626
5.He B, Chen C, Teng L, Tan K. Global view of enhancer–promoter interactome in human cells. National Academy of Sciences of the United States of America. 2014;111(21):E2191-E2199
6.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57-74
7.Visser M, Kayser M, Palstra RJ. HERC2 rs12913832 modulates human pigmentation by attenuating chromatin-loop formation between a long-range enhancer and the OCA2 promoter. Genome Research. 2012;22(3):446-455
8.Eiberg H, Troelsen J, Nielsen M, Mikkelsen A, Mengel-From J, Kjaer KW, et al. Blue eye color in humans may be caused by a perfectly associated founder mutation in a regulatory element located within the HERC2 gene inhibiting OCA2 expression. Human Genetics. 2008;123(2):177-187
9.McManus KF, Taravella AM, Henn BM, Bustamante CD, Sikora M, Cornejo OE. Population genetic analysis of the DARC locus (Duffy) reveals adaptation from standing variation associated with malaria resistance in humans. PLoS Genetics. 2017;13(3):e1006560
10.Boocock J, Leask M, Okada Y, Matsuo H, Kawamura Y, Asian Genetic Epidemiology Network (AGEN) Consortium, et al. Genomic dissection of 43 serum urate-associated loci provides multiple insights into molecular mechanisms of urate control. Human Molecular Genetics. 2020;29(6):923-943
11.Tin A, Marten J, Halperin Kuhns VL, Li Y, Wuttke M, Kirsten H, et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nature Genetics. 2019;51(10):1459-1474
12.Frangoul H, Altshuler D, Cappellini MD, Chen YS, Domm J, Eustace BK, et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. The New England Journal of Medicine. 2021;384(3):252-260
13.Jindal GA, Bantle AT, Solvason JJ, Grudzien JL, D’Antonio-Chronowska A, Lim F, et al. Single-nucleotide variants within heart enhancers increase binding affinity and disrupt heart development. Developmental Cell. 2023;58(21):2206-16.e5
14.Ryan GE, Farley EK. Functional genomic approaches to elucidate the role of enhancers during development. Wiley Interdisciplinary Reviews. Systems Biology and Medicine. 2020;12(2):e1467
15.Barolo S, Carver LA, Posakony JW. GFP and beta-galactosidase transformation vectors for promoter/enhancer analysis in drosophila. BioTechniques. 2000;29(4):726, 728, 730, 732
17.Ishibashi M, Mechaly AS, Becker TS, Rinkwitz S. Using zebrafish transgenesis to test human genomic sequences for specific enhancer activity. Methods. 2013;62(3):216-225
18.Bhatia S, Kleinjan DA. Disruption of long-range gene regulation in human genetic disease: A kaleidoscope of general principles, diverse mechanisms and unique phenotypic consequences. Human Genetics. 2014;133(7):815-845
19.Bhatia S, Gordon CT, Foster RG, Melin L, Abadie V, Baujat G, et al. Functional assessment of disease-associated regulatory variants in vivo using a versatile dual colour transgenesis strategy in zebrafish. PLoS Genetics. 2015;11(6):e1005193
20.Chahal G, Tyagi S, Ramialison M. Navigating the non-coding genome in heart development and congenital heart disease. Differentiation. 2019;107:11-23
21.Goode DK, Elgar G. Capturing the regulatory interactions of eukaryote genomes. Briefings in Functional Genomics. 2013;12(2):142-160
22.Rainger JK, Bhatia S, Bengani H, Gautier P, Rainger J, Pearson M, et al. Disruption of SATB2 or its long-range cis-regulation by SOX9 causes a syndromic form of Pierre Robin sequence. Human Molecular Genetics. 2014;23(10):2569-2579
23.Yuan X, Song M, Devine P, Bruneau BG, Scott IC, Wilson MD. Heart enhancers with deeply conserved regulatory activity are established early in zebrafish development. Nature Communications. 2018;9(1):4977
24.Ghiasvand NM, Rudolph DD, Mashayekhi M, Brzezinski JA 4th, Goldman D, Glaser T. Deletion of a remote enhancer near ATOH7 disrupts retinal neurogenesis, causing NCRNA disease. Nature Neuroscience. 2011;14(5):578-586
25.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470(7333):279-283
26.Smemo S, Campos LC, Moskowitz IP, Krieger JE, Pereira AC, Nobrega MA. Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Human Molecular Genetics. 2012;21(14):3255-3263
27.Pasquali L, Gaulton KJ, Rodríguez-Seguí SA, Mularoni L, Miguel-Escalada I, Akerman İ, et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nature Genetics. 2014;46(2):136-143
28.Kramer ET, Godoy PM, Kaufman CK. Transcriptional profile and chromatin accessibility in zebrafish melanocytes and melanoma tumors. G3 [Internet]. 2022;12(1):jkab379. DOI: 10.1093/g3journal/jkab379
29.Ferre-Fernández JJ, Muheisen S, Thompson S, Semina EV. CRISPR-Cas9-mediated functional dissection of the foxc1 genomic region in zebrafish identifies critical conserved cis-regulatory elements. Human Genomics. 2022;16(1):49
30.Webb AE, Kimelman D. Analysis of early epidermal development in zebrafish. Methods in Molecular Biology. 2005;289:137-146
31.Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496(7446):498-503
32.Kettleborough RNW, Busch- Nentwich EM, Harvey SA, Dooley CM, de Bruijn E, van Eeden F, et al. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature. 2013;496(7446):494-497
33.Bertrand S, Brunet FG, Escriva H, Parmentier G, Laudet V, Robinson-Rechavi M. Evolutionary genomics of nuclear receptors: From twenty-five ancestral genes to derived endocrine systems. Molecular Biology and Evolution. 2004;21(10):1923-1937
34.Taniguchi H, Fujimoto A, Kono H, Furuta M, Fujita M, Nakagawa H. Loss-of-function mutations in Zn-finger DNA-binding domain of HNF4A cause aberrant transcriptional regulation in liver cancer. Oncotarget. 2018;9(40):26144-26156
35.Ketharnathan S, Leask M, Boocock J, Phipps-Green AJ, Antony J, O’Sullivan JM, et al. A non-coding genetic variant maximally associated with serum urate levels is functionally linked to HNF4A-dependent PDZK1 expression. Human Molecular Genetics. 2018;27(22):3964-3973
36.Leask M, Dowdle A, Salvesen H, Topless R, Fadason T, Wei W, et al. Functional urate-associated genetic variants influence expression of lincRNAs LINC01229 and MAFTRR. Frontiers in Genetics. 2018;9:733
37.Lee AP, Kerk SY, Tan YY, Brenner S, Venkatesh B. Ancient vertebrate conserved noncoding elements have been evolving rapidly in teleost fishes. Molecular Biology and Evolution. 2011;28(3):1205-1215
38.Ravi V, Venkatesh B. Rapidly evolving fish genomes and teleost diversity. Current Opinion in Genetics & Development. 2008;18(6):544-550
39.Taher L, McGaughey DM, Maragh S, Aneas I, Bessling SL, Miller W, et al. Genome-wide identification of conserved regulatory function in diverged sequences. Genome Research. 2011;21(7):1139-1149
40.Fisher S, Grice EA, Vinton RM, Bessling SL, McCallion AS. Conservation of RET regulatory function from human to zebrafish without sequence similarity. Science. 2006;312(5771):276-279
41.Howe DG, Bradford YM, Eagle A, Fashena D, Frazer K, Kalita P, et al. The zebrafish model organism database: New support for human disease models, mutation details, gene expression phenotypes and searching. Nucleic Acids Research. 2017;45(D1):D758-D768
42.Bhatia S, Kleinjan DJ, Uttley K, Mann A, Dellepiane N, Bickmore WA. Quantitative spatial and temporal assessment of regulatory element activity in zebrafish. Elife [Internet]. 2021;10:e65601. DOI: 10.7554/eLife.65601
43.Taminato T, Yokota D, Araki S, Ovara H, Yamasu K, Kawamura A. Enhancer activity-based identification of functional enhancers using zebrafish embryos. Genomics. 2016;108(2):102-107
44.Rogers WA, Williams TM. Quantitative comparison of cis-regulatory element (CRE) activities in transgenic Drosophila melanogaster. Journal of Visualized Experiments [Internet]. 2011;58:3395. DOI: 10.3791/3395
45.Thermes V, Grabher C, Ristoratore F, Bourrat F, Choulika A, Wittbrodt J, et al. I-SceI meganuclease mediates highly efficient transgenesis in fish. Mechanisms of Development. 2002;118(1-2):91-98
46.Kleinjan DJ, Coutinho P. Cis-ruption mechanisms: Disruption of cis-regulatory control as a cause of human genetic disease. Briefings in Functional Genomics & Proteomics. 2009;8(4):317-332
47.Helmsauer K, Valieva ME, Ali S, Chamorro González R, Schöpflin R, Röefzaad C, et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nature Communications. 2020;11(1):5823
48.Jaenisch R, Jähner D, Nobis P, Simon I, Löhler J, Harbers K, et al. Chromosomal position and activation of retroviral genomes inserted into the germ line of mice. Cell. 1981;24(2):519-529
49.Wilson C, Bellen HJ, Gehring WJ. Position effects on eukaryotic gene expression. Annual Review of Cell Biology. 1990;6:679-714
50.Rossant J, Nutter LMJ, Gertsenstein M. Engineering the embryo. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(19):7659-7660
51.Westerfield M, Wegner J, Jegalian BG, DeRobertis EM, Püschel AW. Specific activation of mammalian Hox promoters in mosaic transgenic zebrafish. Genes & Development. 1992;6(4):591-598
52.Laplante M, Kikuta H, König M, Becker TS. Enhancer detection in the zebrafish using pseudotyped murine retroviruses. Methods. 2006;39(3):189-198
53.Amsterdam A, Becker TS. Transgenes as screening tools to probe and manipulate the zebrafish genome. Developmental Dynamics. 2005;234(2):255-268
54.Fedoroff N, Wessler S, Shure M. Isolation of the transposable maize controlling elements Ac and Ds. Cell. 1983;35(1):235-242
55.Döring HP, Tillmann E, Starlinger P. DNA sequence of the maize transposable element dissociation. Nature. 1984;307(5947):127-130
56.Sutton WD, Gerlach WL, Peacock WJ, Schwartz D. Molecular analysis of ds controlling element mutations at the adh1 locus of maize. Science. 1984;223(4642):1265-1268
57.Koga A, Suzuki M, Inagaki H, Bessho Y, Hori H. Transposable element in fish. Nature. 1996;383(6595):30-30
58.Kawakami K, Koga A, Hori H, Shima A. Excision of the tol2 transposable element of the medaka fish, oryzias latipes, in zebrafish, danio rerio. Gene. 1998;225(1-2):17-22
59.Parinov S, Kondrichin I, Korzh V, Emelyanov A. Tol2 transposon-mediated enhancer trap to identify developmentally regulated zebrafish genes in vivo. Developmental Dynamics. 2004;231(2):449-459
60.Kawakami K, Shima A. Identification of the Tol2 transposase of the medaka fish Oryzias latipes that catalyzes excision of a nonautonomous Tol2 element in zebrafish Danio rerio. Gene. 1999;240(1):239-244
61.Kawakami K, Shima A, Kawakami N. Identification of a functional transposase of the Tol2 element, an Ac-like element from the Japanese medaka fish, and its transposition in the zebrafish germ lineage. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(21):11403-11408
62.Kawakami K. Transgenesis and gene trap methods in zebrafish by using the Tol2 transposable element. Methods in Cell Biology. 2004;77:201-222
63.Urasaki A, Morvan G, Kawakami K. Functional dissection of the Tol2 transposable element identified the minimal cis-sequence and a highly repetitive sequence in the subterminal region essential for transposition. Genetics. 2006;174(2):639-649
64.Vrljicak P, Tao S, Varshney GK, Quach HNB, Joshi A, LaFave MC, et al. Genome-wide analysis of transposon and retroviral insertions reveals preferential integrations in regions of DNA flexibility. G3. 2016;6(4):805-817
65.Bessa J, Tena JJ, de la Calle-Mustienes E, Fernández-Miñán A, Naranjo S, Fernández A, et al. Zebrafish enhancer detection (ZED) vector: A new tool to facilitate transgenesis and the functional analysis of cis-regulatory regions in zebrafish. Developmental Dynamics. 2009;238(9):2409-2417
66.Zhong Y, Huang W, Du J, Wang Z, He J, Luo L. Improved Tol2-mediated enhancer trap identifies weakly expressed genes during liver and β cell development and regeneration in zebrafish. The Journal of Biological Chemistry. 2019;294(3):932-940
67.Branda CS, Dymecki SM. Talking about a revolution: The impact of site-specific recombinases on genetic analyses in mice. Developmental Cell. 2004;6(1):7-28
68.Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for drosophila using germ-line-specific φC31 integrases. National Academy of Sciences of the United States of America. 2007;104(9):3312-3317
69.Allen BG, Weeks DL. Transgenic Xenopus laevis embryos can be generated using phiC31 integrase. Nature Methods. 2005;2(12):975-979
70.Bateman JR, Lee AM, Wu CT. Site-specific transformation of drosophila via ϕC31 integrase-mediated cassette exchange. Genetics. 2006;173(2):769-777
71.Groth AC, Olivares EC, Thyagarajan B, Calos MP. A phage integrase directs efficient site-specific integration in human cells. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(11):5995-6000
72.Groth AC, Fish M, Nusse R, Calos MP. Construction of transgenic drosophila by using the site-specific integrase from phage φC31. Genetics. 2004;166(4):1775-1782
73.Smith MCA, Till R, Brady K, Soultanas P, Thorpe H, Smith MCM. Synapsis and DNA cleavage in ϕC31 integrase-mediated site-specific recombination. Nucleic Acids Research. 2004;32(8):2607-2617
74.Lister JA. Use of phage φC31 integrase as a tool for zebrafish genome manipulation. Methods in Cell Biology. 2011;104:195-208
75.Lister JA. Transgene excision in zebrafish using the phiC31 integrase. Genesis. 2010;48(2):137-143
76.Lu J, Maddison LA, Chen W. PhiC31 integrase induces efficient site-specific excision in zebrafish. Transgenic Research. 2011;20(1):183-189
77.Roberts JA, Miguel-Escalada I, Slovik KJ, Walsh KT, Hadzhiev Y, Sanges R, et al. Targeted transgene integration overcomes variability of position effects in zebrafish. Development. 2014;141(3):715-724
78.Mosimann C, Puller AC, Lawson KL, Tschopp P, Amsterdam A, Zon LI. Site-directed zebrafish transgenesis into single landing sites with the phiC31 integrase system. Developmental Dynamics. 2013;242(8):949-963
79.Fogg PCM, Colloms S, Rosser S, Stark M, Smith MCM. New applications for phage integrases. Journal of Molecular Biology. 2014;426(15):2703-2716
80.Hu G, Goll MG, Fisher S. ΦC31 integrase mediates efficient cassette exchange in the zebrafish germline. Developmental Dynamics. 2011;240(9):2101-2107
81.Hippenmeyer S, Youn YH, Moon HM, Miyamichi K, Zong H, Wynshaw-Boris A, et al. Genetic mosaic dissection of Lis1 and Ndel1 in neuronal migration. Neuron. 2010;68(4):695-709
82.Tasic B, Hippenmeyer S, Wang C, Gamboa M, Zong H, Chen-Tsai Y, et al. Site-specific integrase-mediated transgenesis in mice via pronuclear injection. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(19):7902-7907
83.Zhu F, Gamboa M, Farruggio AP, Hippenmeyer S, Tasic B, Schüle B, et al. DICE, an efficient system for iterative genomic editing in human pluripotent stem cells. Nucleic Acids Research. 2014;42(5):e34
84.Lalonde RL, Kemmler CL, Riemslagh FW, Aman AJ, Kresoja-Rakic J, Moran HR, et al. Heterogeneity and genomic loci of ubiquitous transgenic Cre reporter lines in zebrafish. Developmental Dynamics. 2022;251(10):1754-1773
85.Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science. 1989;245(4922):1059-1065
86.Cannavò E, Khoueiry P, Garfield DA, Geeleher P, Zichner T, Gustafson EH, et al. Shadow enhancers are pervasive features of developmental regulatory networks. Current Biology. 2016;26(1):38-51
87.Hay D, Hughes JR, Babbs C, Davies JOJ, Graham BJ, Hanssen L, et al. Genetic dissection of the α-globin super-enhancer in vivo. Nature Genetics. 2016;48(8):895-903
88.Osterwalder M, Barozzi I, Tissières V, Fukuda-Yuzawa Y, Mannion BJ, Afzal SY, et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature. 2018;554(7691):239-243
89.Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, et al. The genetic basis of Mendelian phenotypes: Discoveries, challenges, and opportunities. American Journal of Human Genetics. 2015;97(2):199-215
90.Weedon MN, Cebola I, Patch AM, Flanagan SE, De Franco E, Caswell R, et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nature Genetics. 2014;46(1):61-64
91.Liberatore CM, Searcy-Schrick RD, Yutzey KE. Ventricular expression of tbx5 inhibits normal heart chamber development. Developmental Biology. 2000;223(1):169-180
92.Takeuchi JK, Koshiba-Takeuchi K, Suzuki T, Kamimura M, Ogura K, Ogura T. Tbx5 and Tbx4 trigger limb initiation through activation of the Wnt/Fgf signaling cascade. Development. 2003;130(12):2729-2739
93.Benko S, Fantes JA, Amiel J, Kleinjan DJ, Thomas S, Ramsay J, et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nature Genetics. 2009;41(3):359-364
94.Jakobsen LP, Ullmann R, Christensen SB, Jensen KE, Mølsted K, Henriksen KF, et al. Pierre Robin sequence may be caused by dysregulation of SOX9 and KCNJ2. Journal of Medical Genetics. 2007;44(6):381-386
95.Pritchett J, Athwal V, Roberts N, Hanley NA, Hanley KP. Understanding the role of SOX9 in acquired diseases: Lessons from development. Trends in Molecular Medicine. 2011;17(3):166-174
96.Gordon CT, Attanasio C, Bhatia S, Benko S, Ansari M, Tan TY, et al. Identification of novel craniofacial regulatory domains located far upstream of SOX9 and disrupted in Pierre Robin sequence. Human Mutation. 2014;35(8):1011-1020
97.Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: Illuminating the dark road from association to function. American Journal of Human Genetics. 2013;93(5):779-797
98.Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482(7385):390-394
99.Trynka G, Sandor C, Han B, Xu H, Stranger BE, Liu XS, et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nature Genetics. 2013;45(2):124-130
100.Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome Research. 2012;22(9):1748-1759
101.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43-49
102.Farh KKH, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518(7539):337-343
103.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Research. 2019;47(D1):D1005-D1012
104.Ritchie GR, Flicek P. Computational approaches to interpreting genomic sequence variation. Genome Medicine. 2014;6(10):87
105.Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316(5826):889-894
106.Scuteri A, Sanna S, Chen WM, Uda M, Albai G, Strait J, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genetics. 2007;3(7):e115
107.Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gómez-Marín C, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507(7492):371-375
108.Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, Haugen C, et al. FTO obesity variant circuitry and adipocyte browning in humans. The New England Journal of Medicine. 2015;373(10):895-907
109.Rinkwitz S, Geng FS, Manning E, Suster M, Kawakami K, Becker TS. BAC transgenic zebrafish reveal hypothalamic enhancer activity around obesity associated SNP rs9939609 within the human FTO gene. Genesis. 2015;53(10):640-651
110.Kraus P, Winata CL, Lufkin T. BAC transgenic zebrafish for transcriptional promoter and enhancer studies. Methods in Molecular Biology. 2015;1227:245-258
111.Zhang Y, Muyrers JP, Testa G, Stewart AF. DNA cloning by homologous recombination in Escherichia coli. Nature Biotechnology. 2000;18(12):1314-1317
112.Zhang Y, Buchholz F, Muyrers JP, Stewart AF. A new logic for DNA engineering using recombination in Escherichia coli. Nature Genetics. 1998;20(2):123-128
113.Pahl MC, Grant SFA, Leibel RL, Stratigopoulos G. Technologies, strategies, and cautions when deconvoluting genome-wide association signals: FTO in focus. Obesity Reviews. 2023;24(5):e13558
114.Lin KC, Lin HY, Chou P. The interaction between uric acid level and other risk factors on the development of gout among asymptomatic hyperuricemic men in a prospective study. The Journal of Rheumatology. 2000;27(6):1501-1505
Written By
Emily Morice, Caleb Calhoun, Robert Lalonde and Megan Leask
Submitted: 23 November 2023Reviewed: 26 November 2023Published: 19 December 2023