
Abstract
Biomarker study on dementia has developed and the most reliable fluid markers are amyloid peptide (Aβ), TAU, and phosphorylated TAU detected in cerebrospinal fluid (CSF). We have focused on novel Alzheimer’s disease (AD) biomarker candidates (annexin A5 and Milk fat globule-EGF factor 8 protein [MFG-E8]), Ca2+ and phospholipid binding properties, which were elevated in the neuronal cell culture medium by Aβ42 treatment. We have previously reported annexin A5 as an AD biomarker. In this chapter, we focused on MFG-E8. An immunohistochemical study using AD mouse model (APP/PS1) brains revealed characteristic distributions of the staining with anti-MFG-E8 antibody. Anti-MFG-E8 antibody staining was detected in the core regions of the anti-Aβ-antibody stained plaques in 20 weeks old and older APP/PS1 mice, while no staining was observed in control (wild mouse) and anti-Aβ-antibody staining was detected outside of it. The volume of the staining was augmented with advancing age. It was further revealed that the MFG-E8 protein changed to amyloidotic features over time from the Congo red spectral peak shift and electron microscopic study in vitro. As the emergence of senile plaque takes a long time, MFG-E8 present in the plaque might be in an amyloidotic form. From these results, MFG-E8 is a novel biomarker candidate for AD.
Keywords
- Alzheimer’s disease
- biomarker
- MFG-E8
- amyloid
- immunohistochemistry
- Congo red
- electron microscopy
1. Introduction
Accumulating data of biomarker study for Alzheimer’s disease (AD) show that the cerebrospinal fluid (CSF) biomarkers [amyloid β peptides, total tau (T-tau), and phosphorylated tau (P-tau)] are significant elements of AD pathophysiology, which has been confirmed from the clinical research. On the other hand, although many blood biomarkers have been suggested, reliable biomarker is scarce at present compared with CSF biomarkers.
We have been engaged in Alzheimer biomarker studies, especially blood plasma biomarkers. To identify biomarker candidates for AD, we prepared primary culture neuronal cells from mice embryos and isolated the proteins increased in culture media by Aβ42-treatment, possibly secreted proteins, using a proteomic approach [1]. Since plasma biomarkers are expected to be blood–brain barrier permeable, we focused on lipid-binding proteins. For the isolation of biomarker candidates, we prepared phosphatidylserine (PS) binding proteins using a unique method with our original synthetic thermoresponsive magnetic nanoparticles (Therma-max, Magnabeat, Chiba, Japan) coated with PS, denoted as nanoliposome [2]. As PS is exposed to the outer membrane of cells during the apoptotic process, PS-binding proteins may be involved in neuronal damage and might become plasma biomarkers. Lipid-coated nanoliposomes, a mimic of liposomes, disperse well at low temperatures such as at 4°C and aggregate at room temperature, and stick to a magnet, which is a simple method [3]. Using this method, we isolated the Ca2+ fraction which was released with EGTA. As Aβ42-dependent cytotoxicity involves disruption of Ca2+ homeostasis, we focused on the proteins involved in Ca2+ signaling, Ca2+ binding proteins. Among the proteins identified, we have previously focused on annexin A5 as a biomarker candidate [1]. We have shown that secretion of annexin A5 was augmented in the neuronal culture model and that it was significantly increased in plasma from both mouse model and AD patients, compared with control and published several papers [1, 4, 5]. In addition to annexin A5, we also have been interested in another candidate, Milk fat globule EGF factor 8 (MFG-E8) having PS and Ca2+-binding properties [2]. MFG-E8, known as lactadherin, is a secreted glycoprotein originally identified as a component of milk fat globules [6]. MFG-E8 is expressed and secreted by a variety of cells and tissues such as macrophages and dendritic cells. It was shown that MFG-E8 plays a role in the activation of engulfment by phagocytic cells with its specific binding sites for both apoptotic cell membrane and phagocytic cell [6, 7]. It was reported that MFG-E8 interacted with Aβ peptides, and also had a role in the activation of Aβ42 phagocytosis by brain cells in vitro [8, 9, 10]. In our previous study using the AD mouse model (TG2576), we identified the distinctive localization of MFG-E8, in the center of amyloid plaque [2]. In this chapter, using another mouse model (APP/PS1) we report the time-dependent pattern of immuno-staining of MFG-E8 and the unique physical property of MFG-E8 (amyloidotic) in vitro, with Congo red (CR) spectrophotometry and electron microgram. Formation of amyloidogenic proteins characteristic to neurodegenerative disorders, such as AD, and dementia with Lewy body [11, 12]. Thus, MFG-E8 might be a novel marker having amyloidotic properties.
2. Materials and methods
2.1 Reagents
Ultrapure grade CR was purchased from Merck KGaA (Darmstadt, German). Human recombinant MFG-E8 (rMFG-E8) was purchased from R&D Systems, Inc. (Minneapolis, MN, USA). 3, 3′-diaminobenzizine (DAB) was purchased from Tokyo Chemical Industry Co. Ltd. (Tokyo, Japan). Collodion film on the TEM grids (200 mesh) was purchased from Nisshin EM Co., Ltd. (Tokyo, Japan).
2.2 Antibody
Armenian hamster monoclonal antibody against mouse MFG-E8 and goat polyclonal antibody against Armenian hamster IgG were purchased from Medical & Biological Laboratories Co. (Nagoya, Japan). Mouse monoclonal antibody (IgG) against human amyloid β, mouse monoclonal antibody against human MFG-E8, and HRP-conjugated rabbit anti-mouse IgG (H+L) were purchased from Immuno-Biological Laboratories Co. Ltd. (Gunma, Japan). Alexa Fluoro™594-conjugated donkey anti-mouse IgG and DyLight™488-conjugated goat anti-Armenian hamster IgG were from BioLegend (Tokyo, Japan).
2.3 Animals
Alzheimer model mice (APP/PS1), double transgenic mice expressing a chimeric mouse/human amyloid precursor protein (Mo/HuAPP695swe), and a mutant human presenilin 1 (PS1-dE9) were purchased from The Jackson Laboratories (Bar Harbor, ME, USA). This project was approved by the Sapporo Medical University Animal Experimental Ethics Committee (the authorization numbers: 11-011 and 14-014).
2.4 Immunohistochemistry
For immunohistochemistry (IHC) staining, formalin-fixed paraffin-embedded mouse brain sections (either 4 or 10 μm thick) were deparaffinized with xylene and treated with 0.3% H2O2, and then incubated in Histofine (Nichirei Biosciences Inc., Tokyo Japan) with microwave (500 W) for 10 minutes for antigen activation. The sections were then incubated in 70% formic acid for 5 minutes and Bloc Ace (KAC Co., Ltd., Tokyo, Japan) for 1 hour when HRP-labeled secondary antibody was used for visualization. The sections were incubated with 5% FBS in 4% Bloc Ace for blocking, and then with primary antibodies diluted with 4% Bloc Ace in PBS (1:100 for both anti-Aβ and anti-MFG-E8 antibodies) overnight at 4°C. The primary antibody binding was then detected by incubating the sections with HRP-labeled secondary antibody [anti-mouse IgG (1:400 dilution) or anti-Armenian hamster antibody (1:6000 dilution)], or, DyLight 488-conjugated anti-mouse antibody (1:30 dilution) and Alexa 594-conjugated anti-Armenian hamster antibody (1:30 dilution) for 45 minutes at room temperature. Antibody visualization was done with DAB for HRP-labeled secondary antibodies. For observing fluorescently labeled sections after final washing in PBS, they were mounted with VECTASIELD containing 4′, 6-diamidino-2 phenylindole (DAPI) (VECTOR, Burlingame, CA, USA) and used confocal microscopy (LSM520 META, Zeiss, Tokyo, Japan). Fluorescent images were acquired with laser lines 405, 488, and 543 nm using bandpass filters 420–480, 505–530, and 560–605 nm, respectively.
2.5 Congo red spectral shift assay for amyloid (spectrophotometry)
CR is a well-known for histological stain to verify the presence of amyloidal deposits in tissue. The binding of CR to amyloid has since been characterized and shown to depend on the secondary conformation of the amyloid, the β-pleated sheet conformation. In the present study, about 200 μM CR stock solution was prepared by dissolving in a solution of 90% phosphate-buffered saline (PBS) and 10% ethanol and filtered twice through a cellulose acetate membrane (pore size 0.45 μm) and stored at room temperature. Determination of the accurate concentration of CR stock solution was done by measuring the absorbance of a diluted aliquot in a solution of 1 mM sodium phosphate (pH 7.0) and 40% ethanol with
2.6 Electron microscopy
Aliquots of rMFG-E8 solution dissolved in PBS and 0.01% NaN3 were placed on a Collodion film on the TEM grids, and let sit for 10 minutes at room temperature. The solution that remained on the film was adsorbed with filter paper and the film was air-dried. Staining was done with a 4% uranyl acetate for 1 minute. After washing with H2O and air-died, samples were viewed under a transmission electron microscope (JEM-1400, JEO Tokyo, Japan).
3. Results
3.1 Immunohistochemical studies on mouse brain
In this study, to explore the localization of MFG-E8 in the brain we used double transgenic mice (APP/PS1) expressing a chimeric mutant mouse/human amyloid precursor protein and a mutant human presenilin 1, both directed to CNS neurons. As these mutations are associated with early-onset AD, APP/PS1 mice have been widely used in studying neurological disorders of the brain, specifically AD and amyloid plaque formation. From the IHC study anti-Aβ-antibody stained spots, (amyloid plaques) were observed at 20 weeks old and older mice (Figure 1). Characteristically, the anti-Aβ antibody stained plaques with larger radiuses developed in older mice (Figure 1). On the other hand, no staining was observed in control (wild type) mice even at 40 months old (data not shown). It is noticeable that the staining with anti-MFG-E8 antibody was also observed at 20 weeks old and older mice (Figure 2) whereas no staining was observed in control (wild type) mice (data not shown), which seems similar to the pattern of the anti-Aβ antibody stained plaques although the intensity of staining was weak.
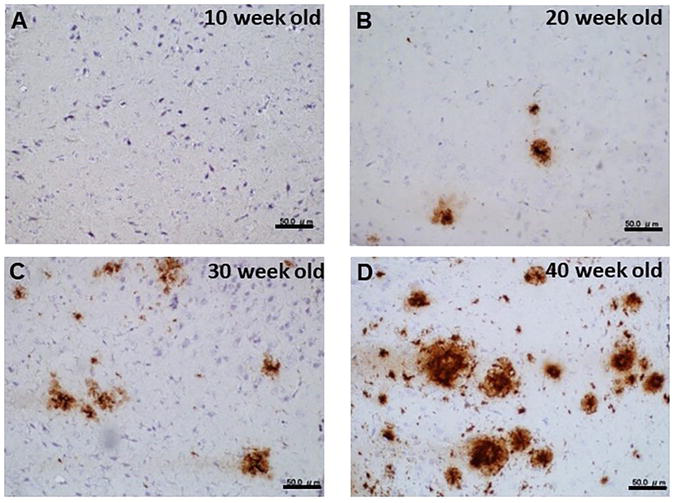
Figure 1.
Immunohistochemical staining of APP/PS1 mouse brain (Aβ). Antibody visualization was done with DAB. A. 10 weeks old; B. 20 weeks old; C. 30 weeks old; D. 40 weeks old. Bars in the insets show 50 μm.
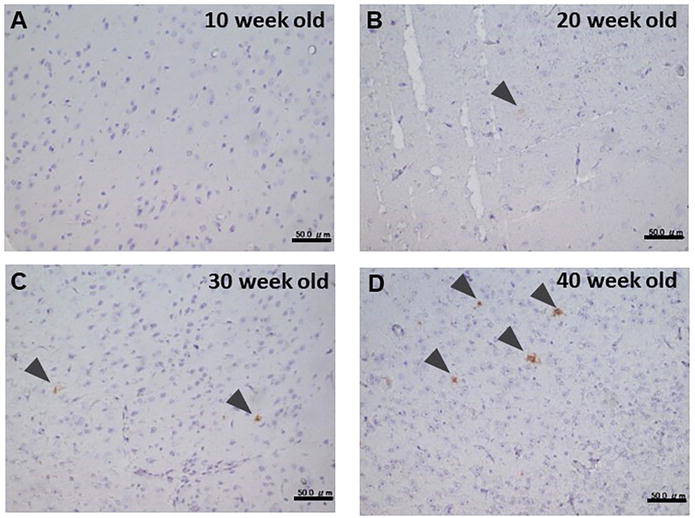
Figure 2.
Immunohistochemical staining of APP/PS1 mouse brain (MFG-E8). Antibody visualization was done with DAB. A. 10 weeks old; B. 20 weeks old; C. 30 weeks old; D. 40 weeks old. Bars in the insets show 50 μm.
To further explore the localization of both Aβ and MFG-E8, we next performed immunofluorescent staining (double staining) and observed specimens with confocal laser microscopy. The immuno-stained pattern with both anti-Aβ and anti-MFG-E8 antibodies shows a characteristic pattern. Anti-MFG-E8 antibody was detected in the core region and anti-Aβ antibody was detected in the outer side of MFG-E8 in all plaques identified (Figure 3), and partly overlapping (yellow) sites were observed. Relatively, the magnitude of the anti-MFG-E8 antibody staining core region was larger in older mice compared with younger ones. On the other hand, no anti-MFG-E8 antibody staining was observed in wild-type mice (control) (data not shown). The characteristic localization of both Aβ and MFG-E8 was well shown in the 3D reconstruction image (Figure 3).
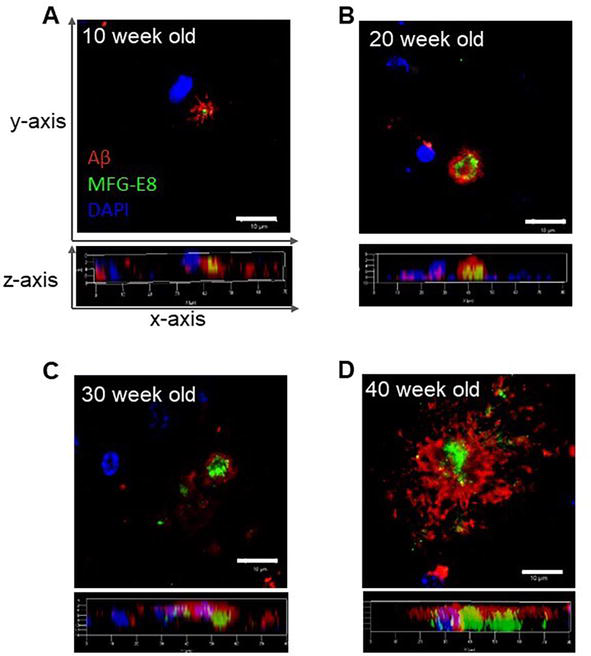
Figure 3.
Fluorescent immunohistochemical staining of APP/PS1 mouse brain (MFG-E8). Fluorescently labeled antibodies against Aβ and MFG-E8 are shown in red and green, respectively. For nuclear staining, DAPI is included in an aqueous anti-fade mounting medium, which shows in blue. A. 10 weeks old; B. 20 weeks old; C. 30 weeks old; D. 40 weeks old. Bars in the insets show 10 μm.
3.2 Congo red spectrum shift by MFG-E8
Particular amyloidogenic proteins have been reported in neurodegenerative diseases such as Aβ for AD and α-synuclein for Dementia with Lewy bodies [11]. It is well known that Aβ peptide is a major component in senile plaque and it forms amyloid, whose physicochemical and biological properties have been elucidated and published in many papers [13, 14]. In vitro study demonstrated that when Aβ peptide with 40 or 42 residues is solubilized in H2O, it gradually forms an amyloid filament [15]. CR, a well-known histological stain, is extensively used to reveal the presence of amyloidal deposits in tissue. It is also demonstrated that binding of CR to the amyloid form of Aβ peptide caused specific spectral peak shifts of CR in vitro (redshift) [13, 14]. It is plausible that the secondary conformation of the amyloid specifically contributes to the spectral change. CR peak shift was also used for the quantitative assay of the amyloid form of Aβ peptide [13]. Based on these findings, we next analyzed the CR spectrum with rMFG-E8. As amyloid formation takes place taking time, we compared the spectra of rMFG-E8 with different pre-incubation times: short-term (2 hours) and long-term (14 days) incubation at 37°C after lyophilized rMFG-E8 was dissolved. Interestingly, the maximum peak around 500 nm of the spectrum of rMFG-E8 after 14-day incubation shifted to a longer wavelength (redshift) compared with that after 2 hours pre-incubated MFG-E8 (Figure 4). The difference spectra show the peak shift more clearly. These results suggest that MFG-E8 is an amyloidotic protein.
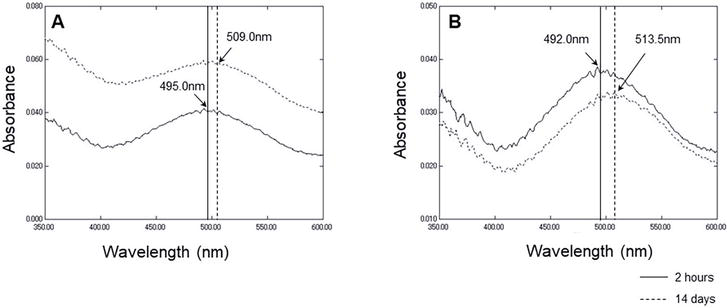
Figure 4.
CR spectrum shift assay of MFG-E8. A. Absorbance spectrum of CR with MFG-E8 after pre-incubation for 2 hours or 14 days. B. Different absorbance spectrum between CR with MFG-E8 after pre-incubation for 2 hours or 14 days, and CR alone. Vertical lines in the insets show the maximum wavelengths of each spectrum.
3.3 Electron microscopic analysis of MFG-E8
Negative staining transmission electron micrograms of rMFG-E8 solution pre-incubated for 2 hours, 14 days, and 21 days after dissolving were compared. It was shown that fibrous aggregates appeared in the rMFG-E8 solutions pre-incubated for 14 and 21 days, whereas much less fibrous aggregates in the rMFG-E8 solution after 2 hours incubation (Figure 5). Thus, electron microscopy also reveals that rMFG-E8 is likely to form amyloidotic form over time.

Figure 5.
Electron microscopic analysis of rMFG-E8 (negative staining). Electron microgram of rMFG-E8 after different times pre-incubation. Pre-incubation times are as follows: A. 2 hours; B. 14 weeks; C. 21 weeks.
Taking the results of CR spectral peak shift in 14-day preincubated rMFG-E8 into account, it is conceivable that the rMFG-E8 whole molecule is an amyloidotic protein.
4. Discussion
In our previous study, we explored brain MFG-E8 using an AD model mice (tg2576) [2]. IHC study with tg2576 mice at 12 months old revealed that the staining pattern with anti-MFG-E8 antibody was similar to that with APP/PS1 mice ([2] and Figure 1). Immuno-staining with the antibody was observed in the core region of the senile plaques in both tg2576 and APP/PS1. In the present study, we further demonstrated with APP/PS1 mice that localization of the anti-MFG-E8 antibody staining was observed in the core region of the plaques that emerged from the early stage (20 weeks old) (Figures 1 and 2). All areas stained with anti-MFG-E8 antibodies have always anti-Aβ antibodies staining areas (Figure 3). The characteristic localization of MFG-E8 might suggest that MFG-E8 plays a role in the formation of plaque. In this study, as the plaque was identified by anti-Aβ-antibody staining, it is uncertain whether or not the emergence of MFG-E8 in the plaque was a preceding event to the assembling of Aβ.
The radius of plaque in older mice relatively becomes more extensive over time. The anti-MFG-E8 antibody-stained regions, the cores of the plaques, also become more extensive, suggesting that accumulation of MFG-E8 took place in the core regions during the growth of plaque. As amyloid forms with homogeneous proteins, there might exist an unknown mechanism for growing MFG-E8 only in the core region when MFG-E8 is secreted. In the present study, MFG-E8 was shown to have an amyloidotic property (Figures 4 and 5). As rMFG-E8 used in this study was prepared from secreted mammalian cells, freshly dissolved rMFG-E8 is not likely to be amyloidotic. It was also shown amyloidotic aggregates of MFG-E8 were observed after 14 days in this study. The formation of amyloid plaques requires a long time in the brain. Thus, MFG-E8 in plaque is in amyloidotic conformation. The amyloidotic feature of MFG-E8 might be favorable in part for the specific location in the plaque. Many constituents in the plaque have been identified [16, 17], but MFG-E8 has not been reported before.
Medin is an integral fragment derived from MFG-E8, which was first found in aortic smooth muscle cells over 50 years of age [18, 19]. Medin, a 5.5 kDa component located in the MFG-E8 FF5/8-C2 domain, contains the amino acid sequence favorable for amyloid formation [18, 20]. Recently medin amyloid in blood vessels was shown to cause cerebrovascular dysfunction in aged mice [21]. It was further shown the medin co-localized with vascular Aβ in the AD mouse model and the vascular Aβ deposition was reduced in the medin-deficient mice [22]. In the present study, MFG-E8 was shown to localize in the senile plaque (Figure 3). Moreover, rMFG-E8 was suggested to make an amyloidotic form in time (Figures 4 and 5). Even after 14 days of incubation, no fragment was observed on SDS PAGE (data not shown). Thus, both CR peak shift and electron microscopic image suggest an amyloidotic conformation of a whole molecule of rMFG-E8. Although it is not certain if the immunostained MFG-E8 in the plaque contained a fragment of MFG-E8 or not, either form of MFG-E8 possibly exists in the plaque.
On the other hand, MFG-E8 was initially identified as a component of milk fat globules and indicated to have a role in pathological and physiological processes, including the clearance of apoptotic cells and angiogenesis [23]. It was also suggested that MFG-E8 has a protective role against Aβ-dependent toxicity in the cell cultural assay [8, 9]. These results indicate that secreted MFG-E8, not amyloidotic form, is likely to have a protective role. It has been demonstrated that medin enhances various reactive oxygen species and reactive nitrogen species produced by both endothelial cells and vascular smooth muscle cells, which promotes vascular endothelial dysfunction and arterial stiffening [24]. On the other hand, it is obscure if the amyloidotic MFG-E8 has a role pathologically or physiologically.
It has been reported that MFG-E8 in the brain was identified in the vasculature but not in plaque-rich areas [10]. In contrast, the present study suggested the localization of MFG-E8 in the core region in plaques by anti-MFG-E8 antibody staining. The reason for the discrepancy between our results and other reports is uncertain.
A significant decrease in MFG-E8 mRNA was reported in AD patients [8]. On the other hand, our previous in vitro study showed an increase in MFG-8 by Aβ42 treatment. In the present study, the magnitude of the area of anti-MFG-E8 antibody staining was increased in older mice, while no staining by the anti-MFG-E8 antibody was detected in the control. These findings may indicate that the production and secretion of MFG-E8 increased or that the clearance of MFG-E8 was attenuated in older APP/PS1 mice. It has been elucidated that the disruption of Ca2+ homeostasis can lead to widespread impairment of cellular and synaptic signaling, subsequently contributing to dementia including AD [25]. It might be possible that MFG-E8 detected in plaques (outer of cells) was relevant to cell damage.
Previously, we demonstrated that annexin A5 is a potential plasma biomarker. Annexin A5 was reported to be able to cross the blood–brain barrier [26], in which annexin’s lipid binding properties may be favorable for crossing. It is conceivable that MFG-E8 may also cross the blood–brain barrier due to its phospholipid-binding properties. Our previous results suggest that the blood level of MFG-E8 in AD patients was higher than in control, although the number of samples was limited [2]. In spite of that MFG-E8 is an amyloidotic protein and it requires time for amyloidotic fiber (a high molecular mass aggregate) formation, it is plausible that a fraction of MFG-E8 might cross the blood–brain barrier after secretion from brain cells. In the next step, we would like to investigate further the verification of MFG-E8 as a blood biomarker.
References
- 1.
Yamaguchi M, Kokai Y, Imai S, Utsumi K, Matsumoto K, Honda H, et al. Investigation of annexin A5 as a biomarker for Alzheimer's disease using neuronal cell culture and mouse model. Journal of Neuroscience Research. 2010; 88 :2682-2692 - 2.
Kimura M, Sohma H, Matsuki K, Yamaguchi M, Imai S, Kokai Y. Milk fat globule-EGF-factor 8 is induced from neuronal cells upon stimulation of Aβ oligomer and specifically localizes in amyloid plaques in the brain of mouse model for Alzheimer's disease. Sapporo Medical Journal. 2017; 85 (supplement):23-34 - 3.
Ohnishi N, Furukawa H, Hata H, Wang J-M, An C-I, Fukusaki E, et al. High-efficiency bioaffinity of cells and proteins using novel thermoresponsive biotinylated magnetic nanoparticles. NanoBiotechnology. 2006; 2 :43-49 - 4.
Sohma H, Imai S, Takei N, Honda H, Matsumoto K, Utsumi K, et al. Evaluation of annexin A5 as a biomarker for Alzheimer's disease and dementia with Lewy bodies. Frontiers in Aging Neuroscience. 2013; 5 :1-7 - 5.
Sohma H, Kokai Y. Plasma Biomarkers in Alzheimer’s Disease. London, UK: InTechOpen; 2016 - 6.
Aziz M, Jacob A, Matsuda A, Wang P. Review: Milk fat globule-EGF factor 8 expression, function and plausible signal transduction in resolving inflammation. Apoptosis. 2011; 16 :1077-1086 - 7.
Yi Y-S. Functional role of milk fat globule-epidermal growth factor VIII in macrophage-mediated inflammatory responses and inflammatory/ autoimmune diseases. Mediators of Inflammation (Hindawi Publishing Corporation). 2016; 2016 :12. Article ID 5628486. DOI: 10.1155/2016/5628486 - 8.
Boddaert J, Kinugawa K, Lambert J-C, Boukhtouche F, Zoll J, Merval R, et al. Evidence of a role for lactadherin in Alzheimer's disease. The American Journal of Pathology. 2007; 170 :921-929 - 9.
Li E, Noda M, Doi Y, Parajuli B, Kawanokuchi J, Sonobe Y, et al. The neuroprotective effects of milk fat globule-EGF factor 8 against oligomeric amyloid β toxicity. Journal of Neuroinflammation. 2012; 9 :148. Available from:http://www.jneuroinflammation.com/content/9/1/148 - 10.
Marazuela P, Solé M, Bonaterra-Pastra A, Pizarro J, Camacho J, Martínez-Sáez E, et al. MFG-E8 (LACTADHERIN): A novel marker associated with cerebral amyloid angiopathy. Acta Neuropathologica Communications. 2021; 9 (145):1-17 - 11.
Irwin DJ, Hurtig HI. The contribution of Tau, amyloid-beta and alpha-synuclein pathology to dementia in Lewy body disorders. Journal of Alzheimers Disease and Parkinsonism. 2018; 8 :1-13 - 12.
Zerovnik E, Stoka V, Mirtic A, Guncar G, Grdadolnik J, Staniforth RA, et al. Mechanisms of amyloid fibril formation – focus on domain-swapping. The FEBS Journal. 2011; 278 :2263-2282 - 13.
Klunk WE, Jacob RF, Mason RP. Quantitating amyloid by Congo red spectral shift assay. Methods in Enzymology. 1999; 309 :285-305 - 14.
Yokoyama K, Fisher AD, Amori AR, Welchons DR, McKnight RE. Spectroscopic and calorimetric studies of Congo red dye-amyloid β peptide complexes. Journal of Biophysical Chemistry. 2010; 1 (3):153-163 - 15.
Finder VH, Glockshuber R. Amyloid-β aggregation. Neurodegenerative Diseases. 2007; 4 :13-27 - 16.
Liao L, Cheng D, Wang J, Duong DM, Losik TG, Gearing M, et al. Proteomic characterization of postmortem Amyloid plaques isolated by laser capture microdissection. The Journal of Biological Chemistry. 2004; 279 :37061-37068 - 17.
Rahman MM, Lendel C. Extracellular protein components of amyloid plaques and their roles in Alzheimer’s disease pathology. Neurodegeneration. 2021; 16 :59. DOI: 10.1186/s13024-021-00465-0 - 18.
Haeggqvist B, Naeslund J, Sletten K, Westermark GT, Mucchiano G, Tjernberg LO, et al. Medin: An integral fragment of aortic smooth muscle cell-produced lactadherin forms the most common human amyloid. Proceedings of the National Academy of Sciences of the United States of America. 1999; 96 :8669-8674 - 19.
Peng S, Westermark GT, Naeslund J, Haeggqvist B, Glennert J, Westermark P. Medin and medin-amyloid in aging inflamed and non-inflamed temporal arteries. Journal of Pathology. 2002; 196 :91-96 - 20.
Larsson A, Söderberg L, Westermark GT, Sletten K, Engström U, Tjernberg LO, et al. Unwinding fibril formation of medin, the peptide of the most common form of human amyloid. Biochemical and Biophysical Research Communications. 2007; 361 :822-828 - 21.
Degenhardt K, Wagner J, Skodras A, Candlish M, Koppelmann AJ, Wild K, et al. Medin aggregation causes cerebrovascular dysfunction in aging wild-type mice. Proceedings of the National Academy of Sciences. 2020; 117 :23925-23931 - 22.
Wagner J, Degenhardt K, Veit M, Louros N, Konstantoulea K, Skodras A, et al. Medin co-aggregates with vascular amyloid-β in Alzheimer’s disease. Nature. 2022; 612 :123-131 - 23.
Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002; 417 :182-187 - 24.
Wang M, McGrawand KR, Monticone RE. Milk fat globule epidermal growth factor VIII fragment medin in age-associated arterial adverse remodeling and arterial disease. Cells. 2023; 12 :253. DOI: 10.3390/cells12020253 - 25.
Cascella R, Cecch C. Calcium dyshomeostasis in Alzheimer’s disease pathogenesis. International Journal of Molecular Sciences. 2021; 22 :4914. DOI: 10.3390/ijms22094914 - 26.
Caserta MT, Caccioppo D, Lapin GD, Ragin A, Groothuis DR. Blood-brain barrier integrity in Alzheimer's disease patients and elderly control subjects. Journal of Neuropsychiatry and Clinical Neurosciences. 1998; 10 :78-84