
Abstract
This chapter digs into the complexities of diabetic foot ulcer (DFU) wound healing, encompassing cellular responses from fibroblasts, keratinocytes, and macrophages, as well as humoral responses involving the release of growth factors and cytokines. The normal wound healing process is hindered in diabetics by factors like infections, venous insufficiency, impaired oxygenation, age-related changes, immune dysfunction, and a dry environment, contributing to delayed and challenging wound healing. The discussion then focuses on the intricate interplay of signaling cascades, including PI3K/Akt, MAPK/ERK, and Wnt/β-catenin, in the pathology of DFUs. Diabetes induces disruptions in the PI3K/Akt pathway, impeding cell migration and angiogenesis due to compromised insulin signaling and increased oxidative stress. Abnormalities in the MAPK pathway, essential for inflammation and tissue remodeling, further impact wound closure in DFUs. Additionally, downregulation of the Wnt/β-catenin pathway, crucial for tissue regeneration, contributes to immune dysfunction, delaying healing in diabetic wounds. Finally, the chapter explores multifaceted factors contributing to the pathogenesis of DFUs, including epigenetic modifications, oxidative stress, advanced glycation end products (AGEs), the polyol pathway, diacylglycerol-protein kinase C (DAG-PKC) activation, and the nitric oxide (NO) pathway. Persistent hyperglycemia in diabetes hinders wound healing, causing chronic ulcers and complications. Addressing these mechanisms is crucial for revolutionizing management.
Keywords
- wound healing
- tissue regeneration
- signaling pathways
- diabetes
- foot ulcer
1. Introduction
This chapter delves into the intricate world of diabetic foot ulcer wound healing and tissue regeneration. Diabetic foot ulcers pose significant challenges due to impaired healing linked to diabetes-related complications. The chapter explores the inflammatory phase of wound healing, growth factors’ crucial roles, and the impact of signaling pathways like PI3K/Akt and MAPK/ERK in diabetic wound healing. It also touches on epigenetic modifications, oxidative stress, and AGEs’ effects on cellular function. Emerging therapeutic approaches, including growth factor therapies and stem cells, offer hope for enhancing diabetic foot ulcer healing. Ultimately, this understanding of signaling pathways and mechanisms could revolutionize the management of this challenging condition. We will start our chapter by talking about how the healing process of a wound happens and how it is affected in DFU.
2. Wound healing in DFU
Wound healing is a normal physiological reaction to tissue injury that usually involves cellular and humeral responses. The cellular response from fibroblasts, keratinocytes, endothelial cells, macrophages, lymphocytes, and platelets [1]. The humeral response involves the release of many growth factors and pro-inflammatory cytokines, such as transforming growth factor (TGF)-β, platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), and epidermal growth factor (EGF).
2.1 Phases of normal wound healing
The wound healing process mainly consists of four overlapping and dynamic phases, which are: hemostasis, inflammation, proliferation, and remodeling [2]. In acute wounds, the healing process goes through four phases, and they heal within a specific time as they have a specific start point and endpoint. However, the existence of some local or systemic factors can delay the acute wound-healing process and cause chronic wounds [1]. The immediate initial response after tissue injury is vasoconstriction to achieve adequate hemostasis for 5–10 minutes [1]. Simultaneously, platelets adhere to the vascular wall
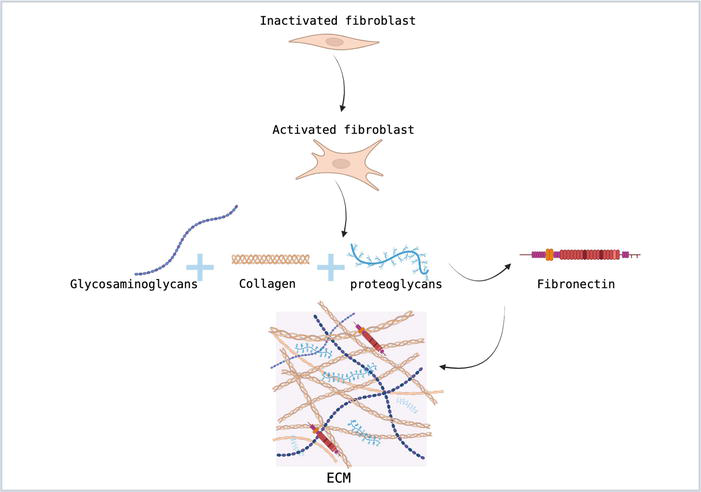
Figure 1.
Fibroblast activation as part of the proliferation phase.
2.2 Factors affecting wound healing process in diabetics
The wound healing process can be impaired and affected by various factors, as in Figure 2, that can be either local or systemic factors. Local factors directly affect the wound itself, while the systemic are those that are related to the diseases and overall health.
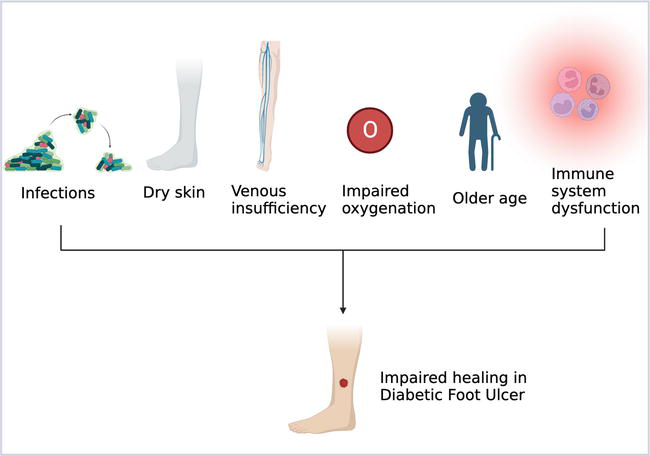
Figure 2.
Factors affecting wound healing process in DFU.
Some of the local factors that affect the wound-healing process are:
Infections: If the wound got infected, this would prolong the inflammatory phase as the clearance of the microbes would not be completed. In addition, the presence of the microbes and endotoxins can lead to excessive and prolonged production of pro-inflammatory mediators such as IL-1 which in turn delay the healing process and can lead to its failure. Furthermore, some bacteria, such as
Venous insufficiency: venous insufficiency leads to venous hypertension. The increase in hydrostatic pressure causes fibrin deposition around the blood vessels affecting oxygen transportation and permeability, which can lead to tissue hypoxia [6].
Impaired oxygenation: oxygen is an important element in inducing angiogenesis, fibroblast proliferation, collagen synthesis, and preventing infections. The early wound microenvironment is usually deprived of oxygen due to vascular disruption and increased oxygen demand by cells that are metabolically active. Systemic diseases, such as diabetes, can compromise vascular flow, laying the stage for inadequate tissue oxygenation [2, 5].
Dry environment: usually wet environment enhances the wound healing process. A hydrated and moist environment enhances the migration of epithelial cells thus improving the proliferation stage [7]. However, in the case of hyperglycemia, the body pulls fluid from its cells to make enough urine to eliminate the excess sugar. This might cause diabetic patients’ skin to become dry and affect wound healing process in these patients [8].
On the other side, some of the systemic factors that affect the wound healing process are:
Age: diabetes is a common disease among elderly, as around 40% of adult diabetics are over the age of 65 [9]. Elderly people usually have delayed wound healing compared to the youth. In addition, wound healing in the elderly has some age-related changes such as, intensified platelet aggregation, increase in inflammatory mediators, delayed infiltration of macrophages and lymphocytes, decrease in growth factors production, delayed re-epithelialization, and delayed angiogenesis and collagen deposition that causes decreased wound strength [2].
Immune system dysfunction: suppression or dysfunction of the immune system causes impairment and delay of wound healing process. Diabetic patients may suffer from immune cell dysfunction in the wound healing process. One of the immune cellular dysfunctions in diabetic wound healing is the failure of macrophage transition from M1 to M2. The hyperglycemic state can increase pro-inflammatory cytokines which in turn leads to decreased phagocytosis of M1 type and block its transition to M2 phenotype. The persistence of M1 phenotype results in a sustained inflammatory phase delaying the wound healing process [10].
2.3 Wound healing in diabetic foot ulcers (DFU)
Specifically, wound healing in diabetic foot ulcers (DFU) is a serious problem that faces many diabetic patients, as around 60–80% of ulcers heal, 10–15% remain active, and 5–24% lead to amputation of the limb after the first evaluation within 6–18 months [11]. Wound healing times in diabetic people were found to be higher than in non-diabetic people, and even they differed between diabetic people, showing a direct correlation with the level of hyperglycemia. As noted, diabetic patients who experience higher levels of hyperglycemia and high HBA1c showed longer healing times for the wound compared to diabetic patients who have a lower level of HBA1c [12]. Furthermore, wound healing phases in DFU are affected by many factors. One of the factors that affect wound healing in DFU is repeated trauma to the foot due to loss of sensation after ischemic neuropathy because of micro- and macrovascular diseases that compromise blood flow to the nerves. Capillary basement membrane thickening in diabetic patients delays the healing process by affecting the infiltration of cells to the wound site [13]. In addition, another study on animals showed that the total number of infiltrated leucocytes in the wound site is lower in diabetic animals compared to non-diabetic animals [14]. Capillary basement membrane thickening has been studied well especially in diabetic patients’ retina. As, it was found that hyperglycemia can induce biosynthetic increase of basement membrane (BM) components like fibronectin, type IV collagen, and laminin increase. The overexpression of BM components can be explained by different mechanisms, such as increased polyol pathway flux and inflammation. Also, increased growth factor activity, protein kinase C activation, and advanced glycation end-product (AGE) accumulation are some factors that contribute to thickening of BM [15]. Prolongation of the inflammatory phase in DFU can also be attributed to excessive production of pro-inflammatory cytokines by the macrophages. Additionally, neutrophils upregulate the production of extracellular traps, which target microorganisms, causing the perpetuation of the inflammation phase [16]. Also, the phagocytic function of polymorphonuclear cells is impaired compared to non-diabetic patients due to high levels of glycosylated protein in the wound site [12]. The concentration of glycosylated wound proteins directly correlates with the activity of proteases and collagenases. The increase in the activity of proteases and collagenases inversely correlates with the amount and concentration of collagen in the wound site, affecting the proliferation phase of the ulcer healing process [17]. Insufficient angiogenesis is one of the major contributors to delayed wound healing in DFU. Deficiency in proangiogenic factors such as VEGF and upregulation of antiangiogenic factors such as Ang-1 affect maturation, regression, and stabilization of newly formed blood vessels. Angiogenesis deficiency affects oxygen transportation to the wound site, leads to impairment of leucocyte migration into the wound, and thus increases the risk of infection [16]. Analysis of non-healing DFU in one of the studies showed high levels of inflammatory cytokines, fewer active growth factors, and higher levels of proteases. The low number of active growth factors can be attributed to the high levels of proteases, as it has been shown that the concentration of MMP is 65-fold higher in the biopsies of DFU [18]. In addition, to have a normal wound healing and remodeling phase, there should be a balance between collagenous and non-collagenous extracellular matrix components. Usually, MMP and the tissue inhibitor of metalloproteinase TIMP are responsible for the remodeling phase. In chronic wounds, as in DFU, high levels of MMP and low levels of TIMP were found and contributed to their chronicity [19]. Now after diving into how a wound is affected in a DFU patient, we can proceed to learn about the tissue repair process and what are the signaling pathways involved.
3. Signaling pathways in diabetic foot ulcers: insights into PI3K/Akt, MAPK, and Wnt/β-catenin signaling and implications for tissue repair
This section aims to provide a detailed understanding of the PI3K/Akt, MAPK, and Wnt/B-catenin signaling pathways. In addition to examining how aberrant activation or inhibition of these pathways in diabetes can disrupt wound healing and impair tissue regeneration.
3.1 PI3K/Akt signaling pathway
Starting with, the serine/threonine kinase Akt (also known as protein kinase B or PKB) which was initially identified as a proto-oncogene is an integral intracellular signaling cascade that contributes to the process of wound healing. This signaling cascade has received widespread attention due to its critical role in regulating a wide range of cellular activities, including cell cycle, apoptosis, angiogenesis, and glucose metabolism [20]. When this pathway is activated, Akt is phosphorylated and activated, which regulates several downstream targets involved in numerous cellular functions [21, 22]. The PI3K/Akt pathway is dysregulated in DFUs as a result of poor insulin signaling and increased oxidative stress [23]. Furthermore, the PI3K/Akt pathway regulates cell migration by promoting keratinocyte relocation for re-epithelialization and enhancing fibroblast proliferation and extracellular matrix formation [24]. It additionally executes an important role in angiogenesis by influencing endothelial cell activity [24]. This part provides an overview of the PI3K/Akt pathway and its relevance in wound healing, discusses the dysregulation of this pathway in diabetes, and explores its impact on cell migration, proliferation, and differentiation.
3.1.1 Overview of the PI3K/Akt pathway and its relevance in wound healing
Activation of the pathway is initiated by the binding of extracellular ligands to receptor tyrosine kinases (RTKs), leading to the recruitment and activation of phosphatidylinositol 3-kinase (PI3K), which phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3) as illustrated in Figure 3. Some of the components involved in the PI3K/Akt signaling pathway, such as glycogen syntheses kinase-3 (GSK-3) and mammalian target of rapamycin (mTOR) have been found to contribute to glucose metabolism and wound healing processes in diabetes mellitus [21].
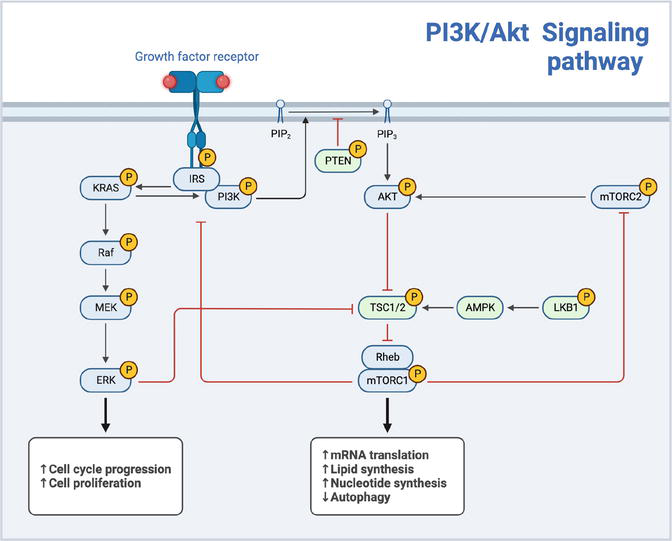
Figure 3.
PI3K/Akt signaling pathway.
3.1.2 Impact of PI3K/Akt pathway on cell migration, proliferation, and differentiation
The PI3K/Akt pathway exerts a profound influence on wound healing. Through its regulation of cytoskeletal dynamics and focal adhesion turnover, the pathway facilitates efficient cell migration into the wound bed, promoting re-epithelialization and tissue repair [24]. Akt activation stimulates the formation of lamellipodia and filopodia, facilitating cell motility and wound closure [21]. Moreover, the pathway influences cell proliferation by promoting cell cycle progression and inhibiting apoptosis, ensuring an adequate number of cells for tissue repair [24]. Additionally, the PI3K/Akt pathway regulates cell differentiation, promoting the differentiation of progenitor cells into specialized cell types such as keratinocytes, fibroblasts, and endothelial cells, required for tissue reconstruction [21].
3.1.3 Dysregulation of PI3K/Akt pathway in diabetic foot ulcers
In diabetes, the PI3K/Akt pathway is frequently dysregulated, primarily due to impaired insulin signaling and increased oxidative stress. Insulin resistance, a hallmark of type 2 diabetes, leads to decreased activation of the pathway [25]. This disruption hinders the phosphorylation and activation of Akt, impairing its downstream signaling. Additionally, chronic hyperglycemia in diabetes induces oxidative stress, resulting in the generation of reactive oxygen species (ROS) that can directly inhibit Akt activity [26, 27, 28]. The dysregulated PI3K/Akt pathway in diabetes contributes to impaired angiogenesis, cell survival, and proliferation, ultimately delaying wound healing, as observed in diabetic foot ulcers and other diabetes-related complications.
3.2 MAPK signaling pathway
Another key highly conserved intracellular signaling cascade involved in wound healing is the MAPK (Mitogen-Activated Protein Kinase) signaling pathway. It has been established that MAPK genes can be divided into three major subfamilies: extracellular signal-regulated kinases (ERKs), Jun N-terminal kinases (JNKs), and p38 MAPKs, which are involved in signal transduction and cellular response regulation [29, 30]. By regulating cell survival, proliferation, immune response, inflammation, and differentiation during tissue repair, the MAPK pathway plays a crucial role in wound healing [31]. A dysfunction of MAPK signaling has been observed in DFUs, resulting in impaired wound healing. Hyperglycemia-induced oxidative stress and chronic inflammation are two contributors to the dysregulation of the MAPK pathway [31]. This dysregulation negatively impacts inflammatory responses, immune cell activation, collagen synthesis, remodeling, and re-epithelialization [31]. It is of great interest to understand how the MAPK pathway influences these cellular functions to identify potential therapeutic targets for restoring normal pathway function in wound healing and enhancing wound healing outcomes.
3.2.1 Influence of the MAPK pathway on cellular functions during wound healing
The MAPK pathway exerts a significant influence on various cellular functions critical for wound healing. Activation of the ERK pathway promotes keratinocyte proliferation, migration, granulation tissue formation, and wound closure [31]. The JNK pathway regulates the production of pro-inflammatory cytokines such as TNF-α and IL-1β, chemokines, and matrix metalloproteinases, influencing the inflammatory response and immune cell recruitment [32]. Fibroblast activation, myofibroblast differentiation, and collagen synthesis, crucial for wound contraction and remodeling are regulated through a non-canonical pathway mediated by mitogen-activated protein kinase (MAPK) effectors such as MAPK kinases 6 (MKK6) and its direct downstream target kinase, p38 [33]. Collectively, the MAPK pathway ensures coordinated cellular responses during different phases of wound healing.
3.2.2 Aberrations in MAPK signaling in diabetic foot ulcers
In diabetic foot ulcers, aberrations in MAPK signaling have been observed, contributing to impaired wound healing. Chronic hyperglycemia, a hallmark of diabetes that impairs keratinocyte migration, is one of the major mechanisms contributing to diabetic wound healing delays [34]. It has been proposed that the P38/MAPK pathway affects keratinocyte migration and controls autophagy during wound healing [34]. A study discovered that the P38/MAPK pathway was down-regulated, which was accompanied by autophagy inactivation, inhibiting keratinocyte migration in high-glucose conditions. Additionally, enhanced activation of p38 MAPK and JNK pathways in response to pro-inflammatory cytokines, such as TNF-α and IL-1β has been observed, leading to excessive inflammation, and hindering the progression of the wound healing process [35]. It has been demonstrated that negative-pressure wound therapy can limit inflammation and promote wound healing in diabetic foot patients by down-regulating the MAPK/JNK signaling pathway [35], demonstrating the importance of the MAPK pathway in diabetic wound healing. It is thought that persistent and prolonged inflammation is a significant factor that hinders the healing process of diabetic wounds. In recent times, exosomes have emerged as novel mediators of intercellular communication and play crucial roles in regulating the inflammatory immune micro-environments of diabetic wounds. According to a recent study, exosomes derived from mesenchymal stem cells (MSC-exos) possess the ability to protect β cells from apoptosis caused by hypoxia. This protective effect is attributed to their carrying of miRNA-21, which helps alleviate endoplasmic reticulum (ER) stress and inhibit P38 MAPK signaling [36]. These findings suggest that MSC-exos hold great promise in promoting the healing of diabetic wounds.
3.3 The Wnt/β-catenin signaling pathway
Finally, The Wnt/B-catenin pathway is a highly conserved signaling pathway that regulates cellular processes, including cell proliferation, differentiation, embryonic development, and adult tissue homeostasis. However, in diabetic foot ulcers, the Wnt/B-catenin pathway is often dysregulated, leading to impaired wound healing [37].
3.3.1 Wnt/β-catenin pathway and its significance
The Wnt signaling pathway is closely linked to diabetic wound healing, as it plays a crucial role in regulating processes like skin development, angiogenesis, and epithelial remodeling [38]. This pathway is a complex network comprising three branches: the canonical pathway also known as the classical Wnt signaling pathway and the noncanonical Wnt pathways are independent of B-catenin-T-cell factor/lymphoid enhancer-binding factor (TCF/LEF), such as the Wnt/planar cell polarity (PCP) pathway, and the Wnt/Ca2+ pathway [39]. The primary function of the canonical Wnt pathway is to regulate cell proliferation, while the noncanonical Wnt pathways control cell polarity and migration. These two main pathways work together in a network of reciprocal regulation. The classical Wnt signaling pathway is particularly relevant to diabetic wound healing. Various studies have confirmed its involvement in promoting angiogenesis, epithelial remodeling, and regulating the proliferation, and differentiation of skin cells [40]. The Wnt/B-catenin pathway consists of four segments: the extracellular signal, membrane segment, cytoplasmic segment, and nuclear segment. The signaling process is initiated by Wnt proteins, including Wnt3a, Wnt1, and Wnt5a, which serve as extracellular signals. The cell membrane segment contains specific receptors, Frizzled (a sevenfold transmembrane receptor protein), and LRP5/6. The cytoplasmic segment includes key proteins like B-catenin, DVL, GSK-3β, AXIN, APC, and CK1. In the nuclear segment, B-catenin translocates to the nucleus, where it interacts with TCF/LEF family members and regulates downstream target genes, such as MMPs and c-Myc [41]. Activation of the canonical Wnt pathway typically occurs when extracellular Wnt ligands bind to membrane receptors through autocrine/paracrine mechanisms. This leads to the stabilization of B-catenin, allowing its translocation to the nucleus, where it interacts with transcription factors to regulate gene expression involved in various cellular processes critical for wound healing [42]. In the absence of Wnts, the transmembrane receptors FZD and LRP5/6 remain separate on the plasma membrane. In the cytoplasm, a “destruction complex,” comprising APC, AXIN, CK1, and GSK3 protein, captures B-catenin and initiates its degradation. This prevents B-catenin from entering the nucleus, and target gene transcription is inhibited by the association of GROUCHOU with TCF/LEF [43]. Upon recognition of Wnts by FZD and LRP5/6, the “destruction complex” is recruited to the cell membrane through interactions with FZD, which prevents the degradation of B-catenin. Subsequently, β-catenin translocates to the nucleus and activates target gene transcription by interacting with TCF/LEF. Figure 4 shows that the movement of B-catenin between the cytoplasm and nucleus is a crucial characteristic of Wnt/B-catenin pathway activation [44].
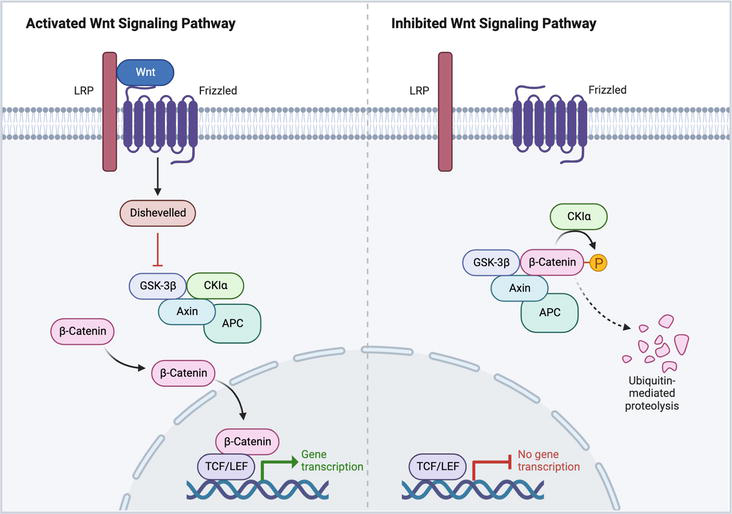
Figure 4.
Wnt signaling pathway activation and inhibition.
3.3.2 Effects of Wnt/B-catenin pathway on cell behavior, tissue repair, and wound healing
The Wnt/B-catenin pathway enhances the proliferation of progenitor cells, promoting their self-renewal and differentiation into tissue-specific cell types Through these mechanisms, the Wnt/B-catenin pathway is instrumental in orchestrating tissue regeneration processes [45]. Furthermore, during wound healing, this pathway interacts with a variety of different signaling cascades. For example, it cross-talks with growth factors such as TGF-B and VEGF, regulating angiogenesis and promoting the formation of new blood vessels necessary for nutrient supply and oxygenation during tissue repair [45]. In addition, interactions with the Notch and Hedgehog signaling pathways influence cell fate decisions during tissue repair [46]. This complex interaction between multiple pathways influences the cellular responses that eventually dictate the wound-healing outcome in DFUs.
3.3.3 Altered Wnt/B-catenin signaling in diabetic foot ulcers
In diabetic foot ulcers, the Wnt/B-catenin signaling pathway is often downregulated, leading to the inhibition of skin cells’ biological activity and reduced expression of cytokines [47]. This results in immune dysfunction in the wound, abnormal granulation tissue formation, and impaired reepithelialization, ultimately causing delays in the healing process [47]. Moreover, in diabetic wounds, pigment epithelium-derived factor inhibits Wnt/β-catenin signaling, leading to the mobilization and dysfunction of endothelial progenitor cells (EPCs). This inhibition of angiogenesis hinders the process of wound healing and causes delays in the overall healing response [48]. During the wound-healing stage of ulcers, macrophages play a significant role in the regulation of the Wnt/B-catenin pathway. Wnt5a influences the inflammatory response by modulating the phenotype of macrophages. Additionally, Wnt7b is a crucial protein that facilitates wound angiogenesis [49]. Some studies suggest that downregulation of the Wnt/B-catenin pathway might be linked to a decrease in Rspo protein caused by diabetes [50]. Multiple factors contribute to the altered signaling, including chronic hyperglycemia, oxidative stress, and inflammation associated with diabetes. Hyperglycemia has been shown to impact fibroblasts’ proliferation, migration, and collagen secretion through the degradation of B-catenin [51]. In diabetic mice, impaired wound healing was associated with altered expression of WNT ligands, receptors, and downstream components, such as B-catenin and T-cell factor (TCF). These disruptions in Wnt/B-catenin signaling contribute to an imbalance in the B-catenin pool, compromising the activation of target genes involved in angiogenesis, cell proliferation, and tissue regeneration [40]. After getting to know in detail about the signaling pathways involved in the tissue repair process, it is time to comprehend how variable mechanisms play a role in the pathogenesis of a diabetic foot ulcer.
4. The role of epigenetic modifications, oxidative stress, and advanced glycation end products (AGEs) in diabetic foot ulcer pathogenesis and healing
Diabetic foot ulcers (DFUs) are a common and severe complication of diabetes mellitus [52]. Several risk factors, like arterial insufficiency, peripheral neuropathy, peripheral vascular disease, impaired resistance to infection, trauma, and foot deformities, contribute to diabetic foot ulcers [53]. It is characterized by impaired wound healing leading to chronic, non-healing ulcers. Several factors contribute to the altered gene expression, impaired cellular function, and compromised wound healing observed in diabetic individuals. Among these factors, epigenetic modifications, oxidative stress, and advanced glycation end products (AGEs) play crucial roles in the pathogenesis of DFUs.
Diabetes is characterized by a condition called stress, which is related to high levels of glucose in the blood (hyperglycemia). Among the cells affected, microvascular endothelial cells are particularly vulnerable to damage caused by hyperglycemia. These cells struggle to adjust their glucose intake when faced with high glucose levels. It is believed that this damage occurs through four pathways: (1) the polyol pathway flux, (2) increased production of advanced glycation end products (AGEs) and activation of their receptor (RAGE), (3) activation of different forms of the enzyme protein kinase C (PKC) and (4) overactivation of the hexosamine pathway. They all lead to increased production of mitochondrial superoxide by the mitochondrial electron transport chain, which then transforms into other reactive oxygen species (ROS) leading to oxidative stress within the cell. This process also leads to the inhibition of an enzyme called glyceraldehyde 3 phosphate dehydrogenase (GAPDH). The activation of these pathways plays a role in microvascular disease and tissue damage seen in diabetic patients significantly contributing to the development of foot ulcers. Moreover, in the case of foot ulcers, there is an increase in the levels of nitric oxide (NO). This can react with superoxide to create oxygen species (ROS). At the time two reducing agents, glutathione and cysteine, were found to be downregulated. As a consequence, not only is the production of ROS increased in the diabetic foot, but the mechanisms for neutralizing them are also impaired, ultimately leading to poor wound healing outcomes [54]. This chapter provides an in-depth analysis of the influence of these factors on diabetic foot ulcer healing.
4.1 Advanced glycation end products (AGEs)
Uncontrolled hyperglycemia is the main cause of diabetic complications, as it leads to the production of advanced glycation end products (AGEs) through a complex series of reactions involving the non-enzymatic interaction of reducing sugars with proteins, lipids, and nucleic acids. Under normal physiological conditions, there is moderate AGE production, but it becomes significantly accelerated in the presence of persistent hyperglycemia due to increased glucose availability.
The pathophysiology of diabetes mellitus involves the detrimental effects of advanced glycation end products (AGEs), which can lead to insulin resistance through two primary mechanisms. AGEs can directly harm the body by trapping and cross-linking proteins, or indirectly by binding to cell surface receptors. While AGEs can interact with various receptors, the most important one is the receptor for advanced glycation end products (RAGE), a member of the immunoglobulin superfamily that was originally identified for its ability to bind AGEs.
RAGE is a multiligand receptor capable of recognizing three-dimensional structures, making it a pattern-recognition receptor. Its gene is located near the major histocompatibility complex III (MHC class III), indicating its involvement in immune responses. The full-length RAGE (fl-RAGE) is composed of extracellular domains (N-terminal V-type domain and two C-type immunoglobulin domains), a transmembrane domain, and a cytosolic domain. Recently, several naturally occurring RAGE protein isoforms have been described due to alternative splicing and proteolytic cleavage. These isoforms include truncated RAGE variants, such as soluble RAGE (sRAGE) and dominant-negative RAGE (dnRAGE), which can interfere with the signaling of fl-RAGE.
In diabetes, persistent hyperglycemia leads to elevated levels of AGEs in the bloodstream, which engage RAGE, triggering a cascade of signaling events. This includes the activation of various kinases and transcription factors, such as MAPK, p38, SAPK/JNK, ERK1/2, and JAK/STAT, ultimately leading to sustained activation of transcription factors like NF-κB, STAT3, HIF-1α, and AP-1. These signaling pathways contribute to insulin resistance through negative regulation of insulin signal transduction and altered insulin receptor signaling. Abnormal activation of the ERK1/2 pathway also influences diabetogenic factors and promotes adipogenesis.
Inflammation plays a critical role in insulin resistance as well. AGEs/RAGE signaling and increased inflammation activate additional mediators and transcription factors, including STAT3, which leads to degradation of IRS-1, exacerbating insulin resistance. The positive feedback loop of RAGE activation further upregulates RAGE receptor expression, perpetuating the process. NF-κB activation is a central player in inflammation and insulin resistance, regulating the expression of inflammatory cytokines like IL-1β, IL-6, and TNFα.
Furthermore, RAGE/NF-κB signaling is linked to the activation of the NLRP3 inflammasome, a key component of the innate immune system that mediates the maturation and secretion of inflammatory cytokines, contributing to insulin resistance. Increased NLRP3 expression has been correlated with insulin resistance in various human studies.
In response to AGEs/RAGE signaling, increased levels of reactive oxygen species (ROS) are generated through NADPH oxidase activation, leading to oxidative stress. Mitochondrial dysfunction and ER stress also contribute to oxidative stress propagation. Mitochondrial ROS production activates kinases involved in stress responses, creating a vicious cycle of inflammation and ROS generation.
The interplay between RAGE-induced cellular dysfunction, protein kinases, inflammation, and oxidative stress attenuates insulin sensitivity in target cells. Persistent NF-κB activation positively regulates RAGE expression, further amplifying the process. Additionally, NF-κB p65 directly induces insulin resistance by repressing the transcription of the glucose transporter GLUT4 protein in skeletal muscles.
The AGEs/RAGE interaction activates several signaling cascades such as ERK/MAPK, IKK/NF-κB, PKC, and JAK/STAT and transcription factors such as STAT3, CREB, NFκB, and AP-1 which intensifies the inflammatory responses and causes oxidative stress. The AGEs/RAGE axis stimulates the infiltration of macrophages and causes an increase in the gene expression of inflammatory cytokines, extracellular matrix proteins and adhesion molecules. TGF-β, fibronectin, and collagen are overexpressed. The AGEs/RAGE axis-induced NF-B activation will result in an upregulation of RAGE expression as a positive feedback mechanism. The ongoing inflammation and oxidative stress of the signaling cascade driven by AGEs/RAGE/NFκB play are crucial in the pathogenesis of diabetic complications, such as diabetic foot ulcers [55].
4.2 Polyol pathway
In addition to advanced glycation end products (AGEs), the polyol pathway plays a role in the impaired healing of diabetic foot ulcers (DFUs). The polyol pathway is a metabolic process that converts glucose into sorbitol and fructose with the help of an enzyme called aldose reductase.
In diabetes due to increased glucose levels, the polyol pathway becomes active. This conversion of glucose to sorbitol consumes NADPH, which is a coenzyme involved in antioxidant defense mechanisms. Consequently, this decreases the cellular antioxidant capacity leading to the accumulation of oxygen species (ROS) and oxidative stress in DFUs.
The buildup of sorbitol and fructose within cells creates stress disrupting balance and hindering various essential functions required for effective wound healing. Osmotic stress causes swelling and damages cell membranes. Impairs the supply of nutrients and oxygen to the cells involved in the wound-healing process.
Moreover, converting sorbitol into fructose generates NADH that stimulates ROS production through dysfunction and activation of oxidative stress pathways. The increased production of ROS intensifies stress in DFUs causing harm to proteins, lipids, and DNA.
Activation of the polyol pathway also impacts cellular redox balance by altering the ratio between reduced glutathione (GSH) and oxidized glutathione (GSSG) affecting overall intracellular health.
The reduced GSH plays a role, as an antioxidant molecule while the oxidized GSSG serves as an indicator of stress. When there is an imbalance in the ratio of GSH to GSSG it disrupts the redox signaling system that’s essential for wound healing processes as illustrated in Figure 5. This disruption can impair functions and compromise tissue repair.
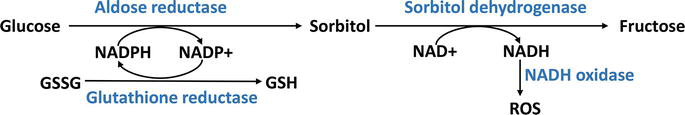
Figure 5.
This diagram illustrates how diabetes leads to the activation of the polyol pathway, converting glucose to sorbitol and fructose, which consumes NADPH and generates oxidative stress. NADPH: nicotinamide adenine dinucleotide phosphate, NADP+: oxidized Nicotinamide adenine dinucleotide phosphate, GSSG: glutathione disulfide, GSH: glutathione, NAD+: oxidized nicotinamide adenine dinucleotide, NADH: nicotinamide adenine dinucleotide, ROS: reactive oxygen species.
In diabetes, the activation of the polyol pathway has been found to contribute to stress, cellular damage, and hindered wound healing. Researchers have explored targeting this pathway as an approach to enhance wound healing in diabetic foot ulcers (DFUs). Studies using models of ulcers inhibiting aldose reductase—the key enzyme, in the polyol pathway—have shown promise in reducing oxidative stress restoring cellular redox balance, and improving wound healing outcomes [56].
4.3 Diacylglycerol-protein kinase C (DAG-PKC) activation
Numerous mechanisms have been proposed to elucidate the detrimental impacts of elevated glucose levels, encompassing the polyol pathway flux, oxidative stress, non-enzymatic glycation, and the activation of diacylglycerol-protein kinase C (DAG-PKC) pathway. The DAG-PKC pathway plays a vital role in vascular function, influencing endothelial permeability, vasoconstriction, extracellular matrix (ECM) synthesis/turnover, cell growth, angiogenesis, cytokine activation, and leucocyte adhesion. Dysfunction in these systems has been associated with the diabetic state.
Studies have consistently reported heightened levels of total DAG in vascular tissues during diabetes. This increase in DAG levels can occur through multiple pathways, involving hydrolysis of phosphatidylinositides, metabolism of phosphatidylcholine by phospholipase C (PLC), or de novo synthesis from glycolytic intermediates like dihydroxyacetone phosphate and glycerol-3-phosphate. Metabolic labeling studies have demonstrated increased glucose incorporation into the glycerol backbone of DAG. The de novo synthesis of DAG involves stepwise acylation catalyzed by glycerol-3-phosphate acyltransferase and monoacylglycerol-3-phosphate acyltransferase. PKC activation is stimulated by DAG, which contains both saturated and unsaturated fatty acids, with 1-palmitoyl-2-oleoyl-sn-glycerol being significant, although 1-stearoyl-2-arachidonyl-sn-glycerol may be the most active. Moreover, the DAG-PKC pathway can be activated by hyperglycemia-induced increases in oxidants such as H2O2, which directly activate PKC or enhance DAG production. In hyperglycemic conditions, elevated DAG levels predominantly consist of palmitate or oleate fatty acids, supporting the notion that increased DAG levels primarily result from de novo synthesis or PLD pathways, rather than rapid formation from the actions of phospholipase C. Enhanced PKC activation has been associated with multiple diabetic complications, including alterations in blood flow, basement membrane thickening, ECM expansion, vascular permeability, angiogenesis, cell growth, and changes in enzymatic activity, such as Na+-K+-ATPase, cPLA2, and MAP kinase [57].
4.4 The nitric oxide (NO) pathway
The nitric oxide (NO) pathway plays a crucial role in wound healing, including diabetic foot ulcers (DFUs). NO is a signaling molecule synthesized by the enzyme nitric oxide synthase (NOS) from the amino acid L-arginine. In normal physiological conditions, NO exerts beneficial effects on wound healing by promoting angiogenesis, regulating inflammation, and stimulating collagen synthesis. It has many protective roles in the treatment and prevention of diabetes complications.
However, in diabetes, the NO pathway is impaired, contributing to the delayed wound healing observed in DFUs. Several mechanisms contribute to the dysregulation of the NO pathway in diabetes:
Reduced NO bioavailability: diabetes is associated with decreased bioavailability of NO due to decreased NOS activity and increased oxidative stress. This imbalance disrupts the signaling cascade mediated by NO, leading to impaired vasodilation, angiogenesis, and endothelial function, which are critical for proper wound healing.
Advanced glycation end products (AGEs): AGEs, discussed earlier, also play a role in impairing the NO pathway. AGEs can react with NO, leading to the formation of reactive nitrogen species (RNS), such as peroxynitrite. RNS can further exacerbate oxidative stress, impair NO-mediated signaling, and induce cellular damage.
Diminished endothelial nitric oxide synthase (eNOS) activity: in diabetes, eNOS activity is reduced, leading to decreased NO production. This reduction is linked to alterations in intracellular signaling pathways, including decreased activation of the Akt pathway and increased protein kinase C (PKC) activity, both of which negatively impact eNOS function.
The impaired NO pathway in DFUs has detrimental effects on wound healing. Reduced NO bioavailability compromises angiogenesis, impairing the formation of new blood vessels necessary for delivering oxygen and nutrients to the wound bed. Moreover, impaired NO signaling disrupts the inflammatory response and compromises immune cell recruitment, leading to prolonged inflammation and increased susceptibility to infection.
Restoring the NO pathway represents a potential therapeutic strategy for improving wound healing in DFUs. Approaches such as the use of NO donors, NOS cofactors, and modulators of eNOS activity have been explored to enhance NO bioavailability and promote wound healing in diabetes. These interventions aim to restore proper NO signaling, improve angiogenesis, reduce inflammation, and enhance tissue repair processes.
In conclusion, the dysregulation of the NO pathway in diabetic foot ulcers contributes to impaired wound healing. Restoring NO bioavailability and improving NO-mediated signaling hold promise for developing novel therapeutic interventions to address the impaired wound healing observed in DFUs. Further research is needed to elucidate the specific mechanisms underlying NO pathway dysfunction in diabetes and to explore targeted strategies for therapeutic intervention [58].
4.5 Epigenetic modifications
To add to what has been said, hyperglycemia was highlighted in the literature to be involved in mediating epigenetic changes that contribute to forming diabetic complications, including the cell types involved in impaired wound healing in diabetics [54].
Epigenetic modifications are heritable changes in gene expression that do not involve alterations in the DNA sequence. Various epigenetic mechanisms exist, including changes in DNA methylation, microRNA expression, and histone post-translational modifications. In the context of DFUs, epigenetic modifications have been implicated in the dysregulation of various cellular processes crucial for wound healing, such as cell proliferation, migration, angiogenesis, and inflammation.
Studies have shown that hyperglycemia-induced epigenetic changes occur in key cell types involved in wound healing, including keratinocytes, fibroblasts, and immune cells. DNA methylation, a prominent epigenetic mechanism, plays a significant role in gene silencing. Hypermethylation of specific gene promoter regions in DFUs can suppress the expression of critical genes involved in wound healing, impairing the regeneration of new tissue. Conversely, hypomethylation of genes associated with inflammation and oxidative stress exacerbates the inflammatory response and oxidative damage, further impeding the healing process.
The endothelium, which plays a critical role in maintaining vascular balance, closely interacts with various factors present in the diabetic environment. It is highly sensitive to metabolic and inflammatory signals, making it susceptible to damage and contributing to the development of atherogenesis. The harmful impact of hyperglycemia on endothelial function is widely recognized, and as a result, both activated endothelial cells and endothelial progenitor cells have become promising targets for therapeutic interventions in diabetes mellitus [59, 60].
Temporary elevation of glucose levels leads to different changes in histone lysine modifications, including the addition of a methyl group to the H3 histones at lysine 4 (H3K4m1). This modification remains present at the promoter region of the RELA gene, responsible for encoding the nuclear factor (NF)-κB-p65 subunit, for up to 6 days after vascular endothelial cells return to normal glucose levels (normoglycemia). The NF-κB-p65 subunit is a crucial proinflammatory transcription factor that plays a central role in regulating genes involved in vascular inflammation and atherosclerosis, including those responsible for adhesion molecules, cytokines, and chemokines [61].
Furthermore, there is a phenomenon, termed metabolic memory or the legacy effect, says that hyperglycemia induces vasculature damage which stays long after there is a state of normoglycemia. The mechanism is unknown yet, but it has been proposed that epigenetic changes, which happen independently of changes in the genotype, may be responsible. This is because these changes, especially histone modifications and DNA methylation, have been shown to be heritable after several rounds of cell division [54, 62].
4.6 Oxidative stress
Oxidative stress refers to an imbalance between the production of oxygen species (ROS) and the cellular defense mechanisms that protect against them. In diabetes, prolonged high blood sugar levels lead to increased ROS production through processes, such as glucose oxidation problems with mitochondria functioning and activation of enzymes.
ROS can have effects on cellular processes that are crucial for wound healing. These effects include hindering collagen synthesis reducing the response to growth factors impeding blood vessel formation (angiogenesis) and disrupting the remodeling of the matrix. Additionally, ROS interferes with signaling pathways that are important, for cell movement, reproduction, and specialization. This disruption affects the coordinated efforts of cell types involved in repair. The persistent oxidative stress seen in foot ulcers contributes to inflammation and delays the healing process [63].
5. Conclusion
Diabetes foot ulcers (DFUs) are a common and devastating consequence of diabetes mellitus. Despite advancements in wound care, DFUs often exhibit delayed wound healing and tissue regeneration, offering substantial problems to patients and healthcare practitioners, emphasizing the need for a deeper understanding of the underlying molecular mechanisms involved in wound healing. This chapter explored the inflammatory phase of wound healing, highlighted the crucial roles of growth factors, and examined the impact of signaling pathways like PI3K/Akt and MAPK/ERK in diabetic wound healing. It also touched on epigenetic modifications, oxidative stress, and the effects of advanced glycation end products (AGEs) on cellular function. Ultimately, this understanding of signaling pathways and mechanisms has the potential to revolutionize the management.
References
- 1.
UpToDate. n.d.-a. Available from: https://www.uptodate.cn/contents/basic-principles-of-wound-management?source=related_link - 2.
Guo S, DiPietro LA. Factors affecting wound healing. Journal of Dental Research. 2010; 89 (3):219-229. DOI: 10.1177/0022034509359125 - 3.
Locatelli L, Colciago A, Castiglioni S, Maier JA. Platelets in wound healing: What happens in space? Frontiers in Bioengineering and Biotechnology. 25 Oct 2021; 9 :2-2. DOI: 10.3389/fbioe.2021.716184 - 4.
Vaidyanathan L. Growth factors in wound healing – A review. Biomedical and Pharmacology Journal. 2021; 14 (3):1469-1480. DOI: 10.13005/bpj/2249 - 5.
Wallace HA, Zito PM, Basehore BM. Wound Healing Phases - Statpearls - NCBI Bookshelf. 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470443/ - 6.
Lalieu RC, Akkerman I, van Hulst RA. Hyperbaric oxygen therapy for venous leg ulcers: A 6 year retrospective study of results of a single center. Frontiers in Medicine. 2021; 8 :671678. DOI: 10.3389/fmed.2021.671678 - 7.
Thomas Hess C. Checklist for factors affecting wound healing. Advances in Skin and Wound Care. 2011; 24 (4):192. DOI: 10.1097/01.asw.0000396300.04173.ec - 8.
Diabetes and your skin [Internet]. Centers for Disease Control and Prevention; 2022. Available from: https://www.cdc.gov/diabetes/library/features/diabetes-and-your-skin.html - 9.
Laiteerapong N, Huang ES. Diabetes in older adults [Internet]. U.S. National Library of Medicine; 2018. Available from: https://pubmed.ncbi.nlm.nih.gov/33651542/ - 10.
Lin S, Wang Q , Huang X, Feng J, Wang Y, Shao T, et al. Wounds under diabetic milieu: The role of immune cellar components and signaling pathways. Biomedicine and Pharmacotherapy. 2022; 157 :114052. DOI: 10.1016/j.biopha.2022.114052 - 11.
Alexiadou K, Doupis J. Management of diabetic foot ulcers. Diabetes Therapy. 20 Apr 2012; 3 (1):1-1. DOI: 10.1007/s13300-012-0004-9 - 12.
Markuson M, Hanson D, Anderson J, Langemo D, Hunter S, Thompson P, et al. The relationship between hemoglobin A1C values and healing time for lower extremity ulcers in individuals with diabetes. Advances in Skin and Wound Care. 2009; 22 (8):365-372. DOI: 10.1097/01.asw.0000358639.45784.cd - 13.
Tsourdi E, Barthel A, Rietzsch H, Reichel A, Bornstein SR. Current aspects in the pathophysiology and treatment of chronic wounds in diabetes mellitus. BioMed Research International. 2013; 2013 :1-6. DOI: 10.1155/2013/385641 - 14.
Fahey TJ, Sadaty A, Jones WG, Barber A, Smoller B, Shires GT. Diabetes impairs the late inflammatory response to wound healing. Journal of Surgical Research. 1991; 50 (4):308-313. DOI: 10.1016/0022-4804(91)90196-s - 15.
Roy S, Ha J, Trudeau K, Beglova E. Vascular basement membrane thickening in diabetic retinopathy. Current Eye Research. 2010; 35 (12):1045-1056. DOI: 10.3109/02713683.2010.514659 - 16.
Dasari N, Jiang A, Skochdopole A, Chung J, Reece EM, Vorstenbosch J, et al. Updates in diabetic wound healing, inflammation, and scarring. Seminars in Plastic Surgery. 2021; 35 (03):153-158. DOI: 10.1055/s-0041-1731460 - 17.
Hennessey PJ, Ford EG, Black CT, Andrassy RJ. Wound collagenase activity correlates directly with collagen glycosylation in diabetic rats. Journal of Pediatric Surgery. 1990; 25 (1):75-78. DOI: 10.1016/s0022-3468(05)80167-8 - 18.
Duckworth WC, Fawcett J, Reddy S, Page JC. Insulin-degrading activity in wound fluid. The Journal of Clinical Endocrinology and Metabolism. 2004; 89 (2):847-851. DOI: 10.1210/jc.2003-031371 - 19.
Lobmann R, Ambrosch A, Schultz G, Waldmann K, Schiweck S, Lehnert H. Expression of matrix-metalloproteinases and their inhibitors in the wounds of diabetic and non-diabetic patients. Diabetologia. 2002; 45 (7):1011-1016. DOI: 10.1007/s00125-002-0868-8 - 20.
Xie Y, Shi X, Sheng K, Han G, Li W, Zhao Q , et al. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (review). Molecular Medicine Reports. 2019; 19 (2):783-791. DOI: 10.3892/mmr.2018.9713. Epub 2018 Dec 3 - 21.
Jere SW, Houreld NN, Abrahamse H. Role of the PI3K/AKT (mtor and gsk3β) signalling pathway and photobiomodulation in diabetic wound healing. Cytokine and Growth Factor Reviews. 2019; 50 :52-59. DOI: 10.1016/j.cytogfr.2019.03.001 - 22.
Manning BD, Toker A. AKT/PKB signaling: Navigating the network. Cell. 2017; 169 (3):381-405. DOI: 10.1016/j.cell.2017.04.001 - 23.
Burgess JL, Wyant WA, Abdo Abujamra B, Kirsner RS, Jozic I. Diabetic wound-healing science. Medicina. 2021; 57 (10):1072. DOI: 10.3390/medicina57101072 - 24.
Xu X, Ma S, Feng J, Zhu Y, Luo D. Dysregulation of the PI3K/AKT signaling pathway and the implications for diabetic neuropathy. Molecular Biology Reports. 2018; 45 (1):203-210. DOI: 10.1007/s11033-017-4150-7 - 25.
Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001; 414 (6865):799-806. DOI: 10.1038/414799a - 26.
Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: A unifying hypothesis of type 2 diabetes. Endocrine Reviews. 2002; 23 (5):599-622. DOI: 10.1210/er.2001-0039 - 27.
Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000; 404 (6779):787-790. DOI: 10.1038/35008121 - 28.
Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001; 414 (6865):813-820. DOI: 10.1038/414813a - 29.
Morrison DK. MAP kinase pathways. Cold Spring Harbor Perspectives in Biology. 2012; 4 (11):2-4. DOI: 10.1101/cshperspect.a011254 - 30.
Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochemical Journal. 2010; 429 (3):403-417. DOI: 10.1042/bj20100323 - 31.
Tamama K, Sen CK, Wells A. Differentiation of bone marrow mesenchymal stem cells into the smooth muscle lineage by blocking Erk/MAPK signaling pathway. Stem Cells and Development. 2008; 17 (5):897-908. DOI: 10.1089/scd.2007.0155 - 32.
Nikoloudaki G, Brooks S, Peidl AP, Tinney D, Hamilton DW. JNK signaling as a key modulator of soft connective tissue physiology, pathology, and healing. International Journal of Molecular Sciences. 2020; 21 (3):1015. DOI: 10.3390/ijms21031015 - 33.
Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, et al. Fibroblast-specific genetic manipulation of p38 mitogen-activated protein kinase in vivo reveals its central regulatory role in fibrosis. Circulation. 2017; 136 (6):549-561. DOI: 10.1161/circulationaha.116.026238 - 34.
Li L, Zhang J, Zhang Q , Zhang D, Xiang F, Jia J, et al. High glucose suppresses keratinocyte migration through the inhibition of p38 MAPK/autophagy pathway. Frontiers in Physiology. 2019; 10 (24):1-3. DOI: 10.3389/fphys.2019.00024 - 35.
Wang T, Li X, Fan L, Chen B, Liu J, Tao Y, et al. Negative pressure wound therapy promoted wound healing by suppressing inflammation via down-regulating MAPK-jnk signaling pathway in diabetic foot patients. Diabetes Research and Clinical Practice. 2019; 150 :81-89. DOI: 10.1016/j.diabres.2019.02.024 - 36.
Jin C, Chen J, Cheng Y, Fu Y, Zhao H, Tang M, et al. Mesenchymal stem cell-derived exosomes protect beta cells against hypoxia-induced apoptosis via miR-21 by alleviating ER stress and inhibiting p38 MAPK phosphorylation. Stem Cell Research & Therapy. 2020; 11 (1):7-10. DOI: 10.1186/s13287-020-01610-0 - 37.
Deng H, Li B, Shen Q , Zhang C, Kuang L, Chen R, et al. Mechanisms of diabetic foot ulceration: A review. Journal of Diabetes. 2023; 15 (4):299-312. DOI: 10.1111/1753-0407.13372 - 38.
Nie XQ , Zhang H, Shi XJ, Zhao JF, Chen Y, Wu FM, et al. Asiaticoside nitric oxide gel accelerates diabetic cutaneous ulcers healing by activating Wnt/beta-catenin signaling pathway. International Immunopharmacology. 2020; 79 :106109. DOI: 10.1016/j.intimp.2019.106109 - 39.
De A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochimica et Biophysica Sinica. 2011; 43 (10):745-756. DOI: 10.1093/abbs/gmr079 - 40.
Zhang H, Nie X, Shi X, Zhao J, Chen Y, Yao Q , et al. Regulatory mechanisms of the Wnt/β-catenin pathway in diabetic cutaneous ulcers. Frontiers in Pharmacology. 2018; 9 :1114. DOI: 10.3389/fphar.2018.01114 - 41.
Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017; 169 (6):985-999. DOI: 10.1016/j.cell.2017.05.016 - 42.
Cruciat CM, Niehrs C. Secreted and transmembrane Wnt inhibitors and activators. Cold Spring Harbor Perspectives in Biology. 2013; 5 (3):a015081. DOI: 10.1101/cshperspect.a015081 - 43.
Reyes M, Flores T, Betancur D, Peña-Oyarzún D, Torres V. Wnt/β-catenin signaling in oral carcinogenesis. International Journal of Molecular Science. 2020; 21 (5):4682. DOI: 10.3390/ijms21134682 - 44.
Muñoz-Castañeda JR et al. Klotho/FGF23 and Wnt signaling as important players in the comorbidities associated with chronic kidney disease. Toxins. 2020; 12 :185. DOI: 10.3390/toxins12030185 - 45.
Liu J, Xiao Q , Xiao J, et al. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduction and Targeted Therapy. 2022; 7 (1):3. DOI: 10.1038/s41392-021-00762-6 - 46.
Shi Y, Shu B, Yang R, et al. Wnt and notch signaling pathway involved in wound healing by targeting c-Myc and Hes1 separately. Stem Cell Research and Therapy. 2015; 6 (1):120. DOI: 10.1186/s13287-015-0103 - 47.
Yamaguchi Y, Passeron T, Hoashi T, et al. Dickkopf 1 (DKK1) regulates skin pigmentation and thickness by affecting Wnt/beta, catenin signaling in keratinocytes. FASEB Journal. 2008; 22 (4):1009-1020. DOI: 10.1096/fj.07-9475com - 48.
Qi W, Yang C, Dai Z, et al. High levels of pigment epithelium-derived factor in diabetes impair wound healing through suppression of Wnt signaling. Diabetes. 2015; 64 (4):1407-1419. DOI: 10.2337/db14-1111 - 49.
Newman AC, Hughes CC. Macrophages and angiogenesis: A role for Wnt signaling. Vascular Cell. 2012; 4 (1):13. DOI: 10.1186/2045-824X-4-13 - 50.
Zhao Y, Ming L, Wei Z, et al. Changes in the expression of Wnt/β-catenin signaling pathway in diabetic ulcers. Chinese Journal of Pathophysiology. 2015; 17 :2033-2038 - 51.
Mullin NK, Mallipeddi NV, Hamburg-Shields E, Ibarra B, Khalil AM, Atit RP. Wnt/β-catenin signaling pathway regulates specific lncRNAs that impact dermal fibroblasts and skin fibrosis. Frontiers in Genetics. 2017; 8 :183. DOI: 10.3389/fgene.2017.00183 - 52.
Mariadoss AV, Sivakumar AS, Lee C-H, Kim SJ. Diabetes mellitus and diabetic foot ulcer: Etiology, biochemical and molecular based treatment strategies via gene and nanotherapy. Biomedicine and Pharmacotherapy. 2022; 151 :113134. DOI: 10.1016/j.biopha.2022.113134 - 53.
Noor S, Zubair M, Ahmad J. Diabetic foot ulcer—a review on pathophysiology, classification and microbial etiology. Diabetes and Metabolic Syndrome: Clinical Research and Reviews. 2015; 9 (3):192-199. DOI: 10.1016/j.dsx.2015.04.007 - 54.
Rafehi H, El-Osta A, Karagiannis TC. Epigenetic mechanisms in the pathogenesis of diabetic foot ulcers. Journal of Diabetes and its Complications. 2012; 26 (6):554-561. DOI: 10.1016/j.jdiacomp.2012.05.015 - 55.
Khalid M, Petroianu G, Adem A. Advanced glycation end products and diabetes mellitus: Mechanisms and perspectives. Biomolecules. 2022; 12 (4):542. DOI: 10.3390/biom12040542 - 56.
Garg SS, Gupta J. Polyol pathway and redox balance in diabetes. Pharmacological Research. 2022; 182 :106326. DOI: 10.1016/j.phrs.2022.106326 - 57.
Dasevcimen N, King G. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacological Research. 2007; 55 (6):498-510. DOI: 10.1016/j.phrs.2007.04.016 - 58.
Gheibi S, Samsonov AP, Gheibi S, Vazquez AB, Kashfi K. Regulation of carbohydrate metabolism by nitric oxide and hydrogen sulfide: Implications in diabetes. Biochemical Pharmacology. 2020; 176 :113819. DOI: 10.1016/j.bcp.2020.113819 - 59.
Nomoto H, Miyoshi H, Furumoto T, Oba K, Tsutsui H, Miyoshi A, et al. A comparison of the effects of the GLP-1 analogue liraglutide and insulin glargine on endothelial function and metabolic parameters: A randomized, controlled trial Sapporo Athero-Incretin Study 2 (SAIS2). PLOS ONE. 18 Aug 2015; 10 (8):2-2. DOI: 10.1371/journal.pone.0135854 - 60.
Sukmawati D, Tanaka R. Introduction to next generation of endothelial progenitor cell therapy: A promise in vascular medicine. American Journal of Translational Research. 2015; 7 (3):411-421 - 61.
Keating ST, Plutzky J, El-Osta A. Epigenetic changes in diabetes and cardiovascular risk. Circulation Research. 2016; 118 (11):1706-1722. DOI: 10.1161/circresaha.116.306819 - 62.
El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. Journal of Experimental Medicine. 2008; 205 (10):2409-2417. DOI: 10.1084/jem.20081188 - 63.
Darenskaya MA, Kolesnikova LI, Kolesnikov SI. Oxidative stress: Pathogenetic role in diabetes mellitus and its complications and therapeutic approaches to correction. Bulletin of Experimental Biology and Medicine. 2021; 171 (2):179-189. DOI: 10.1007/s10517-021-05191-7