
Abstract
The 21st century ushers us into an information explosive era in modern medicine development. At the cusp of biology and technology convergence, significant advances are being made in the strategic approach toward preventing and treating retinal degeneration and neovascular diseases, in particular of a restoration of macular vision, from new surgical interventions to emerging pharmacotherapies through minimally invasive ocular drug delivery and genetic bio factory, and rapidly evolving retinal gene therapies and cell transplant, which are changing the landscape of retinal therapeutic horizon. Understanding disease physio pathological processes is becoming ever more important for developing and delivering these therapeutic modalities to individual patients with precision; in the light of cell & gene therapy, host-donor interplay is of strategic importance.
Keywords
- macular degeneration
- stem cell transplant
- gene therapy
- choroidal ischemia
- Bruch’s membrane
- tissue restoration
- stargardts macular degeneration
- age-related macular degeneration
1. Introduction
The first retinal gene therapy product, Luxturna for Leber’s congenital amaurosis −2 (LCA2) approved in 2017, was built upon the historical success of the human genome project (1990–2003) [1, 2]. Stem cell therapy is a younger cousin; the first creation of human embryonic stem cell (hESC) line in 1998 by James Thompson et al. revolutionized stem cell therapy with the promise of generating unlimited tissue lineage specific cells (donor) for retinal transplant [3, 4]. The eye is an excellent proof of concept (POC) model, for which subretinal space presents an immune privilege site for testing novel regenerative cell & genetic medicine in human conditions. Unlike pharmacotherapy (singular) that targets living cells by inhibiting or activating cell receptor signaling pathways to prevent neuronal cell loss, cell transplant or gene therapies are means to replace the cell loss (RPE or photoreceptor) or correct genetic defect in metabolically compromised cells in a complex diseased environment. In order to achieve clinical success, understanding the donor-host interplays is vital. This chapter will focus on current therapeutic advances in age-related macular degeneration (AMD) and Stargardts macular degeneration and how the disease state impacts drug discovery and the future development of regenerative medicine.
2. Age-related macular degeneration (AMD)
2.1 Therapeutic advances
AMD is the leading cause of irreversible blindness in western countries in ages over 65-years. It is a multifactorial, slow progressive, and neural degenerative disease, clinically featured with drusen formation in the early stage; as the disease progresses, about 85% of patients progress toward geographic atrophy (GA), an advanced form of AMD (dry type), and 15% end up with choroidal neovascularization (CNV), leading to subretinal fluid or leakage (wet type), which accounts for 90% of vision loss in AMD [5]. In the past 15–20 years, the success of anti-VEGF therapies, from Macugen, Avastin (off-label) to Lucentis, Eylea, Beovu, and the recent approval of Vabysmo (bispecific Faricimab) [6], has revolutionized retinal specialty care for treating retinal angiogenesis pathologies by removing VEGF to prevent such devastating vision loss, caused by wet AMD, diabetic macular edema, retinal vein occlusion, and CNV secondary to other retinal diseases such as myopia and polypoidal choroidal vasculopathy (Figure 1). However, 1/3 to 2/3 of CNV lesions gradually evolve (switch) to less VEGF-dependent pathological entities with subretinal inflammatory or fibrotic scarring, surrounded by macular ischemia associated with the underneath choroidal microcapillary dropout or atrophy, which is similar to a GA lesion. In this sense, Wet AMD and GA represent a spectrum of AMD disease progression at a different time. The physio–pathological root causes associated with AMD can be categorized as RPE aging, Bruch’s membrane thickening or atrophy, choroidal ischemia, photoreceptor loss by intricate metabolic abnormalities, and neuroinflammation or para-inflammation such as complement imbalance (Figure 2A) [7]. Recent development of the C3 inhibitor (e.g., Syfovre by Apellis Pharma) and C5 inhibitor (e.g., Izervay by Iveric Bio) has demonstrated that complement inhibition by Syfovre or Izervay can slow down GA progression by about 20% and reduce vison loss at some degrees [9]. This is an exciting and first step that we have a viable therapeutic drug to offer to GA patients other than vitamin supplement. Other pathway-based therapies such as removing N-retinylidene-N-retinylethanolamine (A2E, Gildeuretinol @Ph3 by Alkeus Pharma) and integrin modulation (Risuteganib@ Ph2b/3 by Allegro Pharma) are also being actively explored for treating early and intermediate AMD.
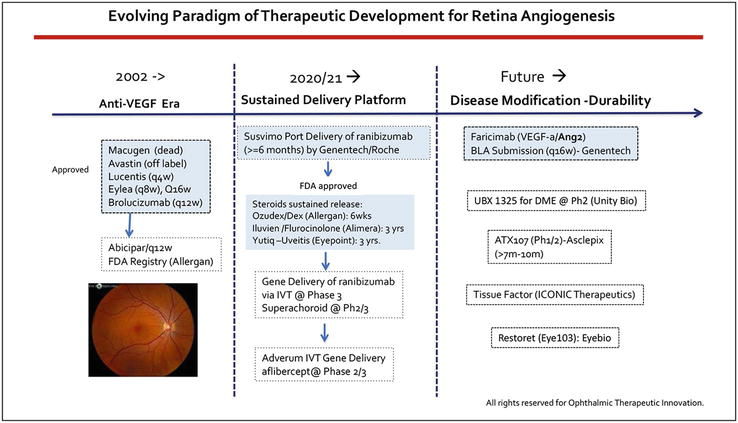
Figure 1.
Highlights anti-VEGF therapeutic development for treating retinal angiogenesis, from the first generation Macugen, Avastin (off-label) and Lucentis to current standard care of Eylea and Faricimab, which was recently approved by FDA for treating wAMD, DME and RVO. Brolucizumab is the smallest anti-VEGF drug approved by FDA in 2019, due to sight threatening vasculitis and endophthalmitis (rare cases), it did not become the market dominance as anticipated. The centra column represents sustained release drug delivery system, including Suvismo, ranibizumab in port delivery system, and Ozudex, Iluvien, and Yutiq which delivers steroid from 6 months to 3 years. Tyrosine kinase inhibitors (TKI) are being actively studied in sustained release delivery formula for treating wAMD and retinal angiogenesis. Due to the ceiling effect of anti-VEGF therapy, there is an unmet need for new pathway based therapies to address subretinal inflammatory scarring and prevent photoreceptor loss, such as UBX1325 (Unity bio), ATX 107 (Asclepix), Restoret (Eyebio) etc. FDA approved products are showed in blue box. All rights reserved @ 2023 Dr. Tina Guanting Qiu.
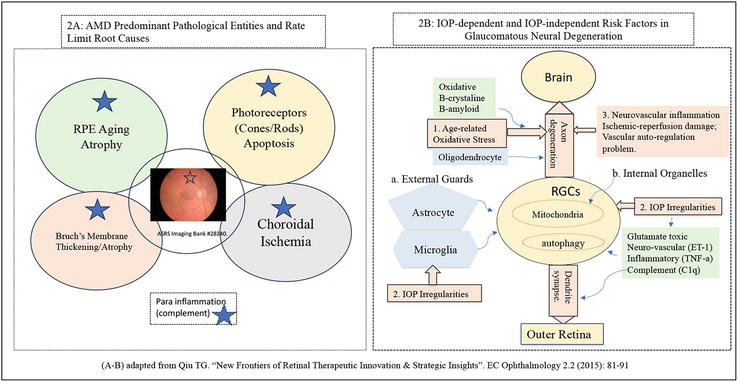
Figure 2.
Provides diagrams illustrating the rate limit disease root causes in AMD and glaucoma respectively. A) Illustrates the four key possible rate limit root causes associated with dry AMD: RPE aging oxidative damage, Bruch’s membrane thickening, or atrophic thinning, choroidal ischemia and cone photoreceptor driven photoreceptor cell loss (unknown reasons). Recent clinical trials of C3 inhibitor (pegcetacoplan) and C5 inhibitor (Avacincaptad pegol) showed that removing subretinal inflammation did not stop but only slowed down GA growth rate at about 20% at 24 months, suggesting that complement inflammation or Para inflammation is not the rate limit root cause of GA secondary to AMD. B) Summarizes the IOP dependent and IOP independent risk factors contributing to the retinal ganglion cell death (apoptosis) in glaucoma including hypertensive glaucoma and normal tensive glaucoma (subset). All rights reserved @ 2015, 2023 Dr. Guanting Tina Qiu [
2.2 Rate limit root causes in AMD
Complement therapy has taught us that removing subretinal para inflammation is not sufficient to stop the disease progression in AMD. We also have learned that from real world evidence of Rescula eye drops in treating small subset refractory glaucoma. Rescula is a B-K channel activator [10], and it acts by tightening the gate to block inflammatory molecules from entering into the anterior chamber, which alleviates active metabolic or para inflammation at the trabecular meshwork ™; however, it cannot treat or remove the existing pathological processes that cause uncontrolled intraocular pressure (IOP), because Rescula does not have the right mechanism of action (MOA) to repair the existing damage to the TM. Recent discoveries by Dr. GQ Qiu (PCT application No.: 18/356,874. 2023) has for the first time shed light on the pressure sensor (dysfunction or failure) as the rate limit root cause of IOP diurnal irregularities or failure in patients with glaucoma (Figure 2B) [7, 8]. She also found that matrix metalloproteinase (MMP) therapy can rejuvenate TM by removing ECM debris and improving the TM functionality, ultimately leading to patients being on an extended drug holiday, during which period patients preserve vision [8, 11, 12]. Pressure sensor is made up of two integral parts of Schlemn’s canal endothelium (sensing cells) and its underlying basement membrane (BM) in trabecular meshwork ™; their roles in TM outflow drainage share in common with RPE and Bruch’s membrane as a blood retina barrier and a water pump in the retina health. Micro pulse laser retinal rejuvenation (2RT) in drusen resolution in early-stage AMD is a clinical validation that Bruch’s membrane aging is a rate limit root cause [8]. Unlike the avascular TM outflow drainage system, retina has two vascular circulation systems and the complex neural circuitry; patients with GA or AMD undergo Muller glial driven subretinal remodeling processes, and the rate limit root causes of AMD could be much more complicated than Bruch’s membrane aging problem alone (thickening, rigidity, atrophy). For example, choroidal ischemia within the macular area has received increasing attention as a rate limit to hRPE graft survival and genetic transfer as bottleneck challenges. Any effective treatment has to be able to remove the rate limit root causes in order to truly prevent and stop the disease from deterioration toward the end stage with irreversible vision loss [13].
3. Stargardt’s macular degeneration (STGD)
3.1 Genetic epidemiology & clinical features
Stargardt’s macular degeneration was first recognized in 1909 by Stargardts, who described 7 patients with recessive inherited macular dystrophy; later Franceschetti coined the term as fundus flavimaculatus (FFM) [14]. It is a single ABCA4-gene-mutant autosomal recessive form of juvenile macular degeneration that occurred 1 in 10,000 population, the most prevalent inherited macular degeneration, which is diagnosed during the first 1–2 decades, or the early 50s in late onset disease. It is estimated of 65,000 patients in the US/EU, less prevalent than retinitis pigmentosa. Clinically, it features as progressive center macular visual loss, central scotoma, and delayed dark adaptation. ABCA4 gene mutation causes RPE and photoreceptor cell loss (apoptosis) as a result of toxic A2E accumulation primarily at the RPE level. In ophthalmic clinical exams, STGD patients have distinguished macular atrophy and yellow-white flecks at the posterior pole on color fundus photography (Figure 3) [14, 15]. Fluorescein angiography (FA) sometimes shows “dark” choroid in the macular area, possibly related to significant A2E deposits at the RPE level that absorbs the blue fluorescence light and its underlying choroidal capillary loss or ischemia (non or poor perfusion). SD-OCT shows progressive choroidal capillarity atrophy underneath the macular over time. By the age of 50 years old, a majority of patients become legally blind.
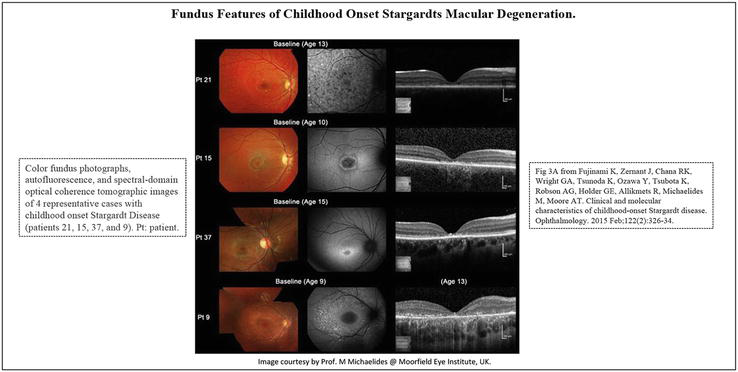
Figure 3.
Fundus features of childhood onset Stargardts macular degeneration in 4 representative cases (patients 21, 15, 37, and 9). Patient 21 has numerous flecks at the posterior pole without central atrophy (fundus grade: 2) and autofluorescence (AF) imaging demonstrates widespread multiple foci of high and low AF signal at the posterior pole with a heterogeneous background (AF type 2). SD-OCT identifies marked outer retinal loss at the central macula. Patient 15 has central atrophy without flecks (fundus grade: 3a) and AF imaging demonstrates a localized low AF signal at the fovea with a high signal edge surrounded by a homogeneous background (AF type: 1). SD-OCT detects marked outer retinal loss at the central macula. Patient 37 has central atrophy with macular flecks (fundus grade: 3b) and a localized low AF signal at the fovea surrounded by a homogeneous background with perifoveal foci of high signal (AF type: 1). SD-OCT shows outer retinal loss at the central macula. Patient 9 has central atrophy with peripheral flecks extending anterior to the vascular arcades (fundus grade: 3b) and a localized low AF signal at the macula surrounded by a heterogeneous background and widespread foci of high AF signal extending anterior to the vascular arcades (AF type: 2). SD-OCT reveals outer retinal disruption at the macula. Pt = patient. A) From Ref. [
3.2 Initial attempts & lessons learned
Gene replacement is a preferable choice for treating STGD, and early treatment warrants better vision preservation. However, ABCR4 gene is a large gene with 6.8kd, which exceeds the maximal of adeno-associated virus (AAV) gene payload (5.0 kb). Between 2010 and 2017, Sanofi Aventis/Oxford Biomedical conducted the first lentiviral-ABCA4 gene replacement (StarGen/SAR422459) clinical trials (Ph1/2a), which did not achieve measurable visual functional improvement across low-, mid-, and high-dose cohorts in a total of 26 patients; hence, the company did not proceed to Ph2b/3 clinical trials [16, 17, 18]. The first retinal gene therapy success (voretigene neparvovec or Luxturna) was not by a chance; AAV2-RPE65 gene transfection into the RPE monolayer in LCA2 is relatively easier in comparison, and clinical endpoints (mobility score and retinal sensitivity test) are feasible and practical at a relatively lower bar to the end-stage terminally ill-sighted patients. On the other hand, ABCA4 gene codes for the photoreceptor outer segment disc protein (rim protein) [15]. In a diseased setting, subretinal Muller glial cell remodeling entangles the remaining photoreceptor soma and inner segment [19]. No photoreceptor outer segment remains, which poses great challenges for genetic transfection toward a wrapped photoreceptor (hibernating cones) in the form of cell clusters or islands, never mentioned of reconstructing the outersegment discs where the rim proteins reside. Using the remaining RPE (ectopic gene therapy) to produce the rim protein may not be sufficient to rescue the photoreceptors with inherited genetic abnormalities. In order to succeed, surgical delivery parameter design and endpoint measures require in-depth surgical and medical expertise along with meticulous study planning.
3.3 Challenges for a large gene delivery
AAV-based mini-gene (Iveric Bio), dual AAV (Biogen), RNA, and nanoparticle-based genetic delivery (Saliogen) are actively being explored for treating STGD1; however, these technologies so far have not yielded encouraging results. The gene transfer efficiency in animal models is fairly low (less than 50%), and there seems still a long way to go. For other large gene-mutated IRD, such as Retinitis pigmentosa-1 (RP1, 7 kb), RP25 (2.0 Mb), and MYO7A/usher1B (7.0 kb), the field faces a similar challenge. Gene editing potentially could be an alternative; however, the low editing efficiency of the current CRISPR technology remains a bottleneck hurdle [20] . Pharmacotherapy by removing A2E is also being studied for STGD1 in phase 2/3 clinical trials with a promise (Alkeus Pharma, Belite Bio) [21]. Once a patient passes a therapeutic window where little photoreceptor and RPE remain at the central macular region, cell replacement and optogenetic gene delivery may offer better alternatives. In 2012–2017, Advanced Cell Technology (now Astellas Pharma) tested their first hESC-hRPE line (MA09-hRPE) in 12 patients with end-stage STGD in Ph1/2 clinical trials, which provided the first glimpse of hESC-derived RPE cell behaviors and its interplay with the host in patients with end-stage macular degeneration [4, 22].
3.4 Holistic approach to complex problems
From early-stage disease intervention to late-stage cell replacement, we need to take a holistic approach in order to develop better strategies to treat a terminally ill inherited retinal disease such as STGD and RP, namely through combinations of pharmacotherapy with gene replacement or photoreceptor/hRPE transplant to address the underlying disease pathologies such as choroidal ischemia. Figure 4 illustrates Cell & Gene Therapy Approach to Rare Genetic Disease in the eye care space. In principle, gene replacement needs to intervene early, whereas hRPE/photoreceptor transplant or Optogenetic treatment targets the advanced stage of AMD or STGD. Pharmacotherapy needs to gear toward disease staging and phenotypic pathological entities; for example, a STGD patient has extensive inflamed A2E depositions in the subretinal space, and a RP patient has extremely poor vascular circulation featuring ischemia inflammation (vascular sheath). A choroideremia patient suffers significant choroidal ischemia. These pathological entities pose significant inherited technical challenges for successful gene transfer and graft survival in transplant settings. Biogen failed CHM and XLRP gene therapy programs at pivotal phase 3 clinical trials in 2021, which could be attributed to such bottleneck hurdles, and clinical surgical delivery parameter design with improved tactics may or may not be able to circumvent.
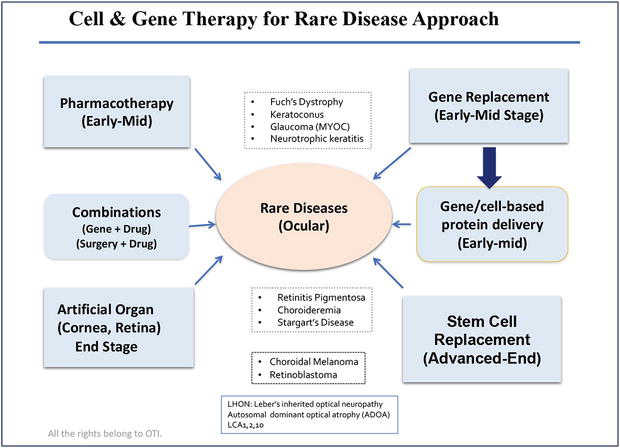
Figure 4.
Provides an overview of rare genetic disease approach, in principle, early intervention through gene replacement therapy in combination with pharmacological treatments are the key for preventing vision loss in inherited retinal disease or eye diseases. When disease advances to late stage or end stage where there are limited photoreceptor and RPE cells remained, artificial retina, cell transplant and optogenetic gene therapy offer alternative new hope. All rights reserved @ 2023 Dr. Guanting Tina Qiu.
4. Gene therapy for IRD and drug delivery bio factory
4.1 An evolving paradigm of retinal gene therapy
Since the first human genome project in 1990, Spark Therapeutics led by Prof. Jean Bennett and Dr. Katherine High has successfully developed the first retinal gene therapy, Luxturna (approved in late 2017), to treat LCA2 caused by RPE65 mutation, an early onset of RP with severe retinal photoreceptor and RPE degeneration. This success marks the beginning of gene therapy for retina and paves a clinical regulatory path forward; most importantly, it has validated AAV viral vector clinical safety and gene transfer efficiency in a diseased setting. Currently, there are hundreds of AAV-based gene therapies in clinical trials; dozens of AAV-based retinal gene therapies, including gene editing for IRD (CHM, LCA1, LCA10, STGD, achromatopsia, LCA5), are also in clinical trials, with XLRP being at the most advanced stage of phase 3 clinical development (Janssen Pharma/MeriaTGx, Syncona/AGTC). Using AAV-mediated protein delivery of therapeutic drugs, such as anti-VEGF, and complement factors are also being explored by Adverum, RegenxBio and Gyroscope/Novartis through subretinal, intravitreal, and suprachoroidal delivery routes in patients with wet AMD and GA, respectively. Optogenetic is another form of genetic medicine that uses genetic engineering to deliver photosensitive protein (e.g., channelrhodopsin) to the retinal ganglion cells or bipolar cells, for the purpose of replacing the photosensitive function of photoreceptors lost in the advanced disease of RP, AMD, or STGD [23]. GenSight Bio and Nanoscope have first reported encouraging results in early-stage clinical trials in patients with end-stage RP. Figure 5 highlights an evolving paradigm of genetic medicine.
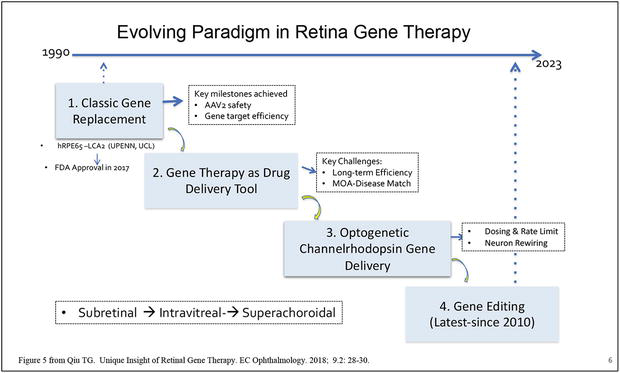
Figure 5.
Shows that retinal gene therapy has evolved from the classic gene replacement therapy (Luxtuna approved in 2017) to genetic based drug or protein delivery (bio factory) and optogenetic gene therapy to the latest gene editing technology. Gene delivery also has expanded from subretinal injection to office-based intravitreal injection and suprachoroidal gene delivery via microneedle (Clearside bio). So far, Luxtuna is the only FDA approved gene therapy product for retina. All rights reserved @ 2018 Dr. Guanting Tina Qiu,
4.2 Recent setbacks
In 2021, Biogen/Nightstar’s two leading gene therapy programs: cotoretigene toliparvovec (BIIB112) for XLRP @ Ph2/3 and timrepigene emparvovec (BIIB111/AAV2-REP1) for choroideremia (CHM) @ Phase 3, did not meet clinical endpoints [24, 25, 26], which was quite unexpected and became the first awakening call to the field. In 2022, ProQR’s Sepofarsen phase 3 RNA therapy for treating LCA10 with CEP290 p.Cys998X mutation also failed to meet the primary endpoint, despite that Ph2 clinical trials showed very promising results [27]. Editas Medicine recently also announced a pause of the world’s first gene editing program using an
4.3 Understanding the host environment
First, one should view gene therapy as a cell tissue transplant; genetic medicine has to work through host tissue cells, not a singular play like anti-VEGF drugs removing excessive proteins in the retina and vitreous cavity, which does not require host cells to do additional work. Gene transfer involves high energy consumption of living cells with metabolic competence. Secondly, genetic delivery differs from protein/drug delivery; it requires high concentration viral particles per targeted cell (1e6:1 ratio) at a given time (millisecond). Inner limiting membrane (ILM) and/or external limiting membrane (ELM) become a rate limit barrier for most AAV viral vector penetrating through the sensory retina (at speed and quantity) toward the RPE layer from the intravitreal delivery route. Subretinal gene delivery is limited to a small size of bleb area being rescued, thus affecting the long-term durability [28]. Thirdly, retinal remodeling in IRD involves significant Muller glial scar formation (details see next section). Fourthly, choroidal ischemia in IRD presents significant challenges for gene transfer, especially to the photoreceptor cells that completely depend upon its underlying choroidal capillary network to supply oxygen and nutrients. Functional compensation through adaption is limited in IRD patients.
4.4 Suprachoroidal gene delivery via microneedle
It represents a new direction for future gene delivery, which has only been validated as a bio factory drug delivery of anti-VEGF or complement factor. In order to produce sufficient proteins, targeted genes are often ubiquitous to multiple cell types (RPE and vascular endothelium or Muller glial). For specific gene transfer toward RPE or photoreceptor, superachoroidal delivery route has not been tested for its clinical efficiency in IRD patients, though preclinical evidence points toward extensive gene transfection to the photoreceptor cells and RPE layers in healthy pig models. In principle, gene transfer is easier to the monolayer RPE than multi-layer photoreceptors, which often form cluster islands in a diseased condition. In retinal degenerative condition, when photoreceptors (rods) die, it triggers Muller glial proliferation and migration, extending its processes toward defected RPE layer to “repair or amend” blood-retinal barrier. During the course, Muller glial entangles with the remaining cones and disrupts ELM integrity, where the cone and Muller cell are in a parallel and hand-in-hand relation (Figure 6) [29, 30, 31]. Additionally, bipolar and amacrine often migrate toward the outer layer of the retina by taking residence where the rods were lost. In response to the injuries, microglial and macrophages are mobilized to clean up the dying or dead cell debris. One could imagine that the subretinal microenvironment is very harsh in GA or STGD patients. When we design clinical trials for a given gene therapy, the doing and surgical delivery parameters are some of the key considerations, surgeons begin to learn to navigate around these disease pathological entities in each individual eye, in order to achieve desirable clinical outcome.
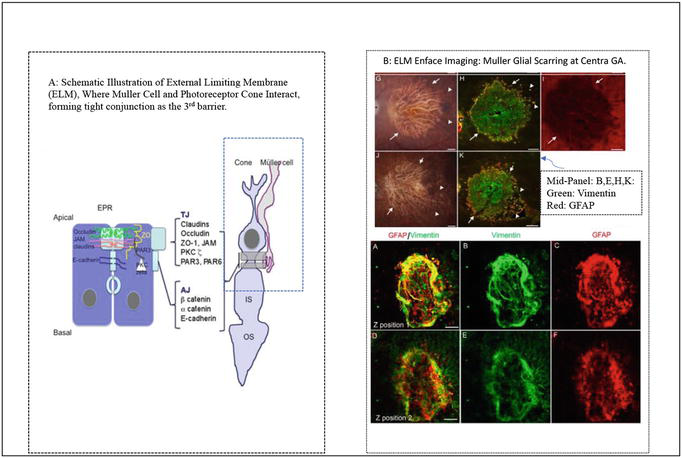
Figure 6.
(A-B): A) Shows a diagram of Muller glia and cone photoreceptor forming external limiting membrane (ELM) as the 3rd barrier of gene delivery toward the RPE and outer layer of the retina in human eye. B) Shows Muller glial scarring occupies and circles around the ELM platform, where remaining hibernating cone photoreceptor cells form islands or clusters, and being wrapped by the static Muller glial scars as the result of subretinal remodeling in patients suffering from aged related macular degeneration. A) From Ref. [
4.5 Technical challenges for future development
While the field holds exciting promises, we must realize that gene therapy is still at a very early stage, even for basic AAV-mediated gene replacement for treating diseases, such as XLRP, CHM, and LCA1. Current technology has not been able to help patients to regain vision in a way that they can live independently. The only exception perhaps is LUMEVEQ , an AAV-mediated gene therapy for treating Leber’s hereditary optic neuropathy (LHON ND4), a rare mitochondrial disease causing a sudden RGC cell loss in teens and young adults, and 98% of patients are bilateral with acute onset. The Ph2 and Ph3 clinical trials of LUMEVEQ have shown improvement in more than three lines, and patients have been able to back to school and engage in sports [32]. This case suggests that the host cell metabolic health determines the gene therapy outcome. Acute disease has better prognosis than chronic IRD. Genetic bio factory targeting AMD and DME will achieve better clinical outcomes than neural protection via gene transfer in chronic IRD with severe pathological conditions. In the future, we need more potent and safer viral vectors to enable office-based genetic medicine delivery, such as intravitreal or superachoroidal delivery via microneedles (Clearside Bio). Also, we need to develop novel genetic engineering technologies that will allow one genetic product to treat more than one gene mutations (gene agnostic approach).
5. Stem cell therapy for macular degeneration
5.1 Key problems
In transplant medicine, the quantity and quality of donor cells are of prerequisite condition. In retinal transplantation, the reconstruction of neural circuitry presents the most formidable challenge. Decades of scientific and clinical research efforts spending on fetal retinal sheet transplant have taught us many great lessons on this topic. For RPE suspension, selecting an appropriate cell seeding density (total dose needs to be based on the lesion size) is the key tactic to ensure a rapid graft survival without causing excessive metabolic burden to the remaining host RPE. Similar to cornea endothelium transplant (Aurion Bio) [33], hESC-RPE cell possesses its embryonic or intricate capacity to self-assembly, forming a monolayer RPE at the basement membrane following the subretinal transplantation in patients with GA; OpRegen transplant has demonstrated long-term survival (3–5 years) and visual functional improvement corresponding with the anatomic recovery, such as ELM reconstruction. The poor health of Bruch’s membrane (BM), choroidal ischemia within the macular area, subretinal Muller glial scar, as well as local immune-inflammation are the four key prognostic factors in the host retina, which affect graft survival rate [4]. In a preclinical rat model of retinal degeneration, E17-derived rat retinal progenitor cell suspension transplant also showed its intricate embryonic competence to self-reorganize and form multilayer laminal sheet without the need for extracellular matrix scaffolding [34].
5.2 hESC-derived hRPE transplant
hESC or induced pluripotent stem cell or iPSC-derived hRPE cell lines will become the mainstay hRPE donor cell for treating macular degeneration. One of the key criteria of an ideal donor hRPE cell line is to obtain proficient biological competence from a short-term
The clinical regulatory path for hESC- or iPSC-derived hRPE or human photoreceptor progenitor cell line validation differs from conventional drug development. The first hESC-derived RPE line, MA09-hRPE has been studied in 38 human subjects for up to 10 years now; there is no report of tumorigenesis. OpRegen has been studied in 25 patients for up to 6 years; there is no tumorigenesis reported. The ethical issue of using human ESC has a clearance. The key technical hurdle lies ahead of the cell biological competence, which could take many years to fine tune for its clinical proficiency. Figure 7 provides a brief summary of the hRPE cell line selection and validation process, between
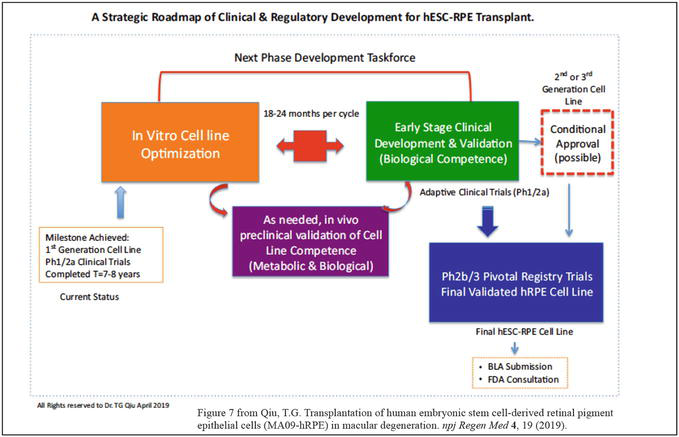
Figure 7.
Provides a strategic roadmap of clinical and regulatory development of hESC or iPSC derived RPE or photoreceptors as well as other lineage restricted cells for treating retinal degeneration or other human conditions, it may require back and forth between petri dish experimentation and early stage clinical studies in a small scale to validate the donor cell biological competences before moving toward pivotal clinical trials. All rights reserved @ 2019 author (Qiu GT).
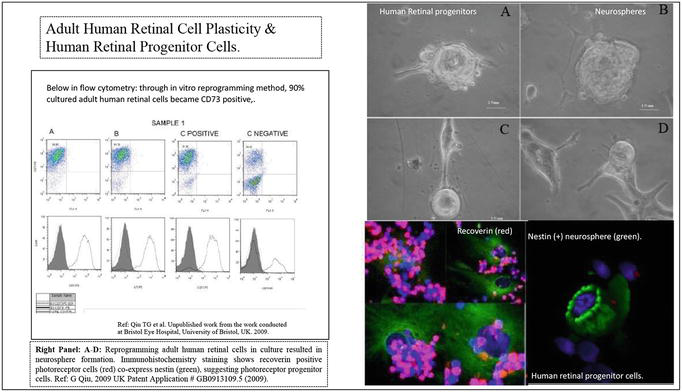
Figure 8.
(L-R): Left panel shows adult human retinal cells isolated from post-mortem human eyes are able to be reprogrammed in vitro cultural condition and regenerate clusters of neurospheres containing photoreceptor lineage specific progenitor cells, expressing nestin (green) and recoverin (red), showed in immunohistochemical assays. Right panel: Flow cytometry further confirmed that majorities of reprogrammed human retinal cells (90%) are possible photoreceptor lineage cells expressing CD73 biomarker, in correspondence with the results from immunohistochemical assays. This is the first time to challenge the century dogma of post mitotic human retinal cell plasticity. All rights @ 2009 author (Qiu GT).
5.3 Photoreceptor transplantation and cell reprogramming
hESC- or iPSC-derived photoreceptor progenitor transplant is largely at the preclinical stage. State-of-the-art research conducted by Prof. Robin Ali at Kings College London in collaboration with Prof. James Bainbridge at UCL in England has shed light on the possibility of generating cone photoreceptors from pluripotent ESC cell lines through 3D retinal organoids
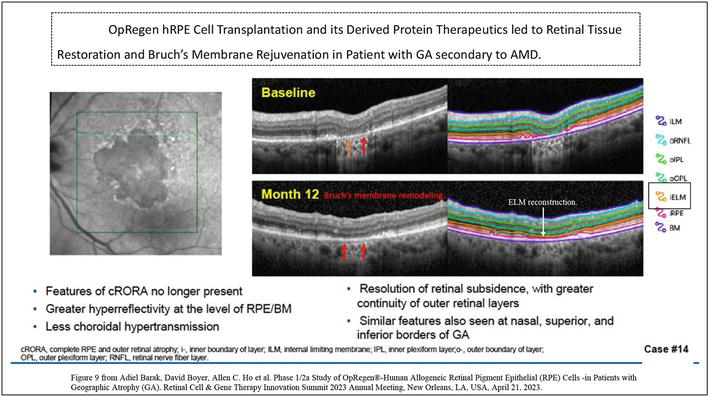
Figure 9.
(L-R). Shows an example of a female patient with GA/AMD receiving OpRegen hRPE transplant in her left eye, the grafted cells can self-assemble forming a monolayer RPE at GA lesion area and refurbish its underlying Bruch’s membrane with increased elasticity, her BCVA gained more than 2 lines at 12 months post-transplant. Left panel is AF imaging showing a large well demarcated GA lesion. Right panel is SD-OCT imaging of a section line at the GA border showing the structure improvement and tissue restoration at 12 months post-transplant compared to the baseline. Specifically, reconstructing the integrity and continuity of the ELM is seen at 12-month, labeled in orange line on SD-OCT. the ELM integrity is corresponding to the functional BCVA gain. Image courtesy by lineage cell therapeutics, USA.
6. Conclusion
Retinal photoreceptor cell loss (apoptosis) is the principle and common pathological pathway that leads to irreversible vision loss in almost all the retinal diseases such as AMD, STGD, diabetic retinopathy, and IRD. These devasting age-related or genetically caused retinal diseases are increasingly becoming significant socioeconomic burdens. The conventional concept that post-mitotic human photoreceptor cells lose the embryonic competence to regenerate during adulthood has been the central dogma of developmental biology for decades, yet we have just begun to understand what is possible through the lens of
Acknowledgments
Special thanks to Lineage Cell Therapeutic and CEO, Brian Culley, who gave me the opportunity to work on OpRegen hRPE transplant project in 2021 and have made important discoveries to the field of hESC derived RPE therapeutics. I am also grateful to Prof. Andrew Dick, MD at Bristol Eye Hospital, University of Bristol, UK for hosting me in 2009 as Research Fellow of National Eye Research Center, UK during my sabbatical to the UK, along with his team’s technical assistance on retinal stem cell research.
Conflict interest
None.
References
- 1.
Maguire AM, Russell S, Chung DC, et al. Durability of Voretigene Neparvovec for Biallelic RPE65-mediated inherited retinal disease: Phase 3 results at 3 and 4 years. Ophthalmology. 2021; 128 (10):1460-1468 - 2.
Hood L, Rowen L. The human genome project: Big science transforms biology and medicine. Genome Medicine. 2013; 5 :79 - 3.
Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998; 282 (5391):1145-1147 - 4.
Qiu TG. Transplantation of human embryonic stem cell-derived retinal pigment epithelial cells (MA09-hRPE) in macular degeneration. npj Regenerative Medicine. 2019; 4 :19 - 5.
Fleckenstein M, Mitchell P, Freund KB, et al. The progression of geographic atrophy secondary to age-related macular degeneration. Ophthalmology. 2018; 125 (3):369-390 - 6.
Wolf AT, Harris A, Oddone F, et al. Disease progression pathways of wet AMD: Opportunities for new target discovery. Expert Opinion on Therapeutic Targets. 2022; 26 (1):5-12 - 7.
Qiu TG. New Frontiers of Retinal Therapeutic Innovation & Strategic Insights. EC Ophthalmology. 2015; 2 (2):81-91 - 8.
Qiu TG. Methods and compositions for treatments of glaucoma and related conditions. PCT Application No.: 18/356,874. Application date: July 21, 2023. Designee: OphthalmicTherapeutic Innovation LLC; 2023 - 9.
Shughoury A, Sevgi DD, Ciulla TA. The complement system: A novel therapeutic target for age-related macular degeneration. Expert Opinion on Pharmacotherapy. Sep-Dec 2023; 24 (17):1887-1899 - 10.
Qiu TG. Revisit Rescula and cystoid macular Edema and refractory glaucoma. Journal of Clinical and Experimental Ophthalmology. 2015; 6 :473 - 11.
Qiu TG. Trabodenoson on trabecular meshwork rejuvenation: A comprehensive review of clinical data. Expert Opinion on Investigational Drugs. 2021; 30 (3):227-236 - 12.
Bacharach J, Tatham A, Ferguson G, et al. ARTEMIS 2 study group. Phase 3, randomized, 20-month study of the efficacy and safety of Bimatoprost implant in patients with open-angle glaucoma and ocular hypertension (ARTEMIS 2). Drugs. 2021; 81 (17):2017-2033 - 13.
Qiu TG. Defining the problem in ophthalmic drug development and clinical translation. EC Ophthalmology. 2016; 3 :314-319 - 14.
Fujinami K, Zernant J, Chana RK, et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology. 2015; 122 (2):326-334 - 15.
Tsybovsky Y, Molday RS, Palczewski K. The ATP-binding cassette transporter ABCA4: Structural and functional properties and role in retinal disease. Advances in Experimental Medicine and Biology. 2010; 703 :105-125 - 16.
Kong J, Kim SR, Binley K, et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Therapy. 2008; 15 (19):1311-1320 - 17.
Binley K, Widdowson P, Loader J, et al. Transduction of photoreceptors with equine infectious anemia virus lentiviral vectors: Safety and biodistribution of StarGen for Stargardt disease. Investigative Ophthalmology & Visual Science. 2013; 54 :4061-4071 - 18.
Parker MA, Erker LR, Audo I, et al. Three-year safety results of SAR422459 (EIAV-ABCA4) gene therapy in patients with ABCA4-associated Stargardt disease: An open-label dose-escalation phase I/IIa clinical trial, cohorts 1-5. American Journal of Ophthalmology. 2022; 240 :285-301 - 19.
Marc RE, Jones BW, Watt CB, Strettoi E. Neural remodeling in retinal degeneration. Progress in Retinal and Eye Research. 2003; 22 (5):607-655 - 20.
McCullough KT, Boye SL, Fajardo D, et al. Somatic gene editing of GUCY2D by AAV-CRISPR/Cas9 alters retinal structure and function in mouse and macaque. Human Gene Therapy. 2019;30 (5):571-589 - 21.
Charbel Issa P, Barnard AR, Herrmann P, et al. Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin a dimerization. Proceedings of the National Academy of Sciences of the United States of America. 2015; 112 (27):8415-8420 - 22.
Mehat MS, Sundaram V, Ripamonti C, et al. Transplantation of human embryonic stem cell-derived retinal pigment epithelial cells in macular degeneration. Ophthalmology. 2019; 125 (11):1765-1775 - 23.
Sahel JA, Boulanger-Scemama E, Pagot C, et al. Partial recovery of visual function in a blind patient after optogenetic therapy. Nature Medicine. 2021; 27 :1223-1229 - 24.
Abbouda A, Avogaro F, Moosajee M, et al. Update on gene therapy clinical trials for Choroideremia and potential experimental therapies. Medicina (Kaunas, Lithuania). 2021; 57 (1):64 - 25.
MacLaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: Initial findings from a phase 1/2 clinical trial. Lancet. 2014; 383 (9923):1129-1137 - 26.
MacLaren RE, Fischer MD, Gow JA, et al. Subretinal timrepigene emparvovec in adult men with choroideremia: A randomized phase 3 trial. Nature Medicine. 2023; 29 :2464-2472 - 27.
Russell SR, Drack AV, Cideciyan AV, et al. Intravitreal antisense oligonucleotide sepofarsen in Leber congenital amaurosis type 10: A phase 1b/2 trial. Nature Medicine. 2022; 28 (5):1014-1021 - 28.
Qiu TG. Unique insight of retina gene therapy. EC Ophthalmology. 2018; 9 (2):28-30 - 29.
Omri S, Omri B, Savoldelli M, Jonet L, Thillaye-Goldenberg B, Thuret G, et al. The outer limiting membrane (OLM) revisited: Clinical implications. Clinical Ophthalmology. 2010; 4 :183-195 - 30.
Edwards MM, McLeod DS, Bhutto IA, et al. Subretinal glial membranes in eyes with geographic atrophy. Investigative Ophthalmology & Visual Science. 2017; 58 (3):1352-1367 - 31.
Marshall J, Mellerio J. Disappearance of retino-epithelial scar tissue from ruby laser photocoagulations. Experimental Eye Research. 1971; 12 (2):173-174 - 32.
Newman NJ, Yu-Wai-Man P, Carelli V, et al. And LHON study group. Efficacy and safety of Intravitreal gene therapy for Leber hereditary optic neuropathy treated within 6 months of disease onset. Ophthalmology. 2021; 128 (5):649-660 - 33.
Numa K, Imai K, Ueno M, et al. Five-year follow-up of first 11 patients undergoing injection of cultured corneal endothelial cells for corneal endothelial failure. Ophthalmology. Apr 2021; 128 (4):504-514 - 34.
Qiu G, Seiler MJ, Mui C, et al. Photoreceptor differentiation and integration of retinal progenitor cells transplanted into transgenic rats. Experimental Eye Research. 2005; 80 (4):515-525 - 35.
Francois B, Hogge G, Rami S, Sha-Bat, A B, Jordi M, Eyal B, Qiu, TG. Methods and compositions for treating retinal diseases and conditions. PCT publication No.: 2021/242788A1. Designee: Lineage Cell Therapeutic, Inc. Patent publication date: 02 Dec 2021 - 36.
Ribeiro J, Procyk CA, West EL, et al. Restoration of visual function in advanced disease after transplantation of purified human pluripotent stem cell-derived cone photoreceptors. Cell Reports. 2021; 35 (3):109022 - 37.
Grisé KN, Coles BLK, Bautista NX, et al. Activation of adult mammalian retinal stem cells in vivo via antagonism of BMP and sFRP2. Stem Cell Research & Therapy. 2021; 12 :560 - 38.
Qiu TG. Retinal progenitor cells. UK Patent Application Number: GB0913109.5. 2009. Designee: University of Bristol, UK, patent application date: Aug 12 2009 - 39.
Koso H, Minami C, Tabata Y, et al. CD73, a novel cell surface antigen that characterizes retinal photoreceptor precursor cells. Investigative Ophthalmology & Visual Science. 2009; 50 (11):5411-5418 - 40.
Gagliardi G, Ben M'Barek K, Chaffiol A, et al. Characterization and transplantation of CD73-positive photoreceptors isolated from human iPSC-derived retinal organoids. Stem Cell Reports. 2018; 11 (3):665-680 - 41.
Barak A, Boyer D, Ho AC, et al. Phase 1/2a study of OpRegen®-human allogeneic retinal pigment epithelial (RPE). In: Cells -in Patients with Geographic Atrophy (GA). Retinal Cell & Gene Therapy Innovation Summit 2023 Annual Meeting. New Orleans, LA, USA: April 21, 2023