Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
We performed density functional theory (DFT) calculations to analyze the geometrical and electronic properties of eight differently substituted tetrathiafulvalene (TTF) units (some with two Se heteroatoms) with 0, +1 and +2 charges. We demonstrate that, by minor deformations from planarity in the uncharged units disappear with electron removal. The release of the electrons as well as the stacking between units may be related to the delocalized π character of the HOMO, which is prevailing antibonding between C and S (Se) atoms. Energy considerations suggest the TTF precursor is less susceptible to oxidation compared to its different derivatives. Additionally, a noteworthy correlation is observed between the organic molecules under study and established square planar bis-dithiolene d8 metal complexes, which also exhibit a tendency towards redox isomerism.
Laboratory of Materials Chemistry and the Living: Activity and Reactivity (LCMVAR), Chemistry Department, Faculty of Matter Science, University of Batna 1, Batna, Algeria
Abdelatif Messaoudi*
Laboratory of Materials Chemistry and the Living: Activity and Reactivity (LCMVAR), Chemistry Department, Faculty of Matter Science, University of Batna 1, Batna, Algeria
*Address all correspondence to: amessaoudi@univ-batna.dz
1. Introduction
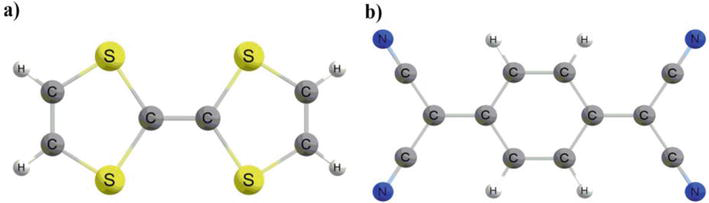
Both experimental [1] and theoretical data [2, 3] have established that tetrathiafulvalene (TTF in Figure 1a) and its derivatives possess exceptional donor properties. This assertion is further supported by the existence of stable mono-(TTF+•) and di-cationic (TTF2+) oxidized derivatives, which can be obtained through two sequential and reversible oxidation processes. While the first synthesis of a TTF derivative dates back to 1926, [4] genuine interest in TTF did not truly emerge until around 1972, following the synthesis of the chloride salt (TTF+, Cl−), [5] in 1973, Ferraris et al. made a groundbreaking discovery by demonstrating the formation of a highly conductive complex involving the electron-donating TTF and the electron-accepting tetracyano-p-quinodimethane (TCNQ) (Figure 1b) [6]. This novel compound exhibited not only metallic behavior across a broad temperature spectrum but also boasted the highest recorded electrical conductivity among all known organic compound σmax = 1.47 × 10−4 Ω−1 cm−1 at 66 K). Superconductivity was reported in (BEDT-TTF)4(ReO4)2 (BEDT-TTF = bis(ethylenedithiolo)tetrathiafulvalene) at 2 K in 1983 [7]. Since that time, there has been significant research conducted on TTF derivatives as potential components in charge transfer salts, with the aim of utilizing them as organic conductors [8] and superconductors [9], adducts with C60 [10, 11], conductive polymers [12], materials for non-linear optics (NLO) [13, 14], cationic sponges [15], ferromagnetic organic magnets [16], liquid crystals [17], dendrimers [18], molecular rotaxanes and catenanes [19].
Figure 1.
The neutral-state molecular structures of (a) TTF and (b) TCNQ.
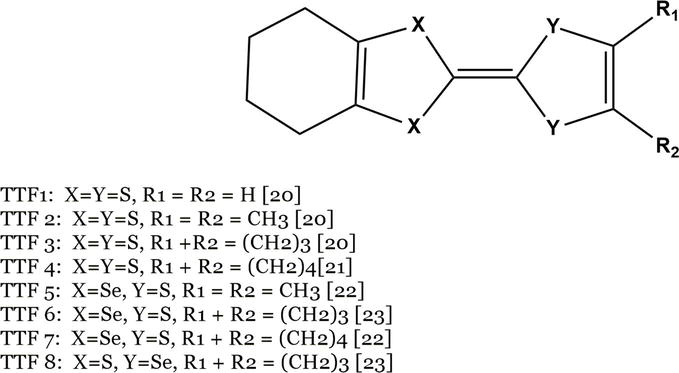
Because of our keen interest in organic materials, this paper presents a comprehensive computational investigation focusing on various structural components derived from the TTF framework, featuring specific chemical modifications. These modifications have the potential to induce electronic perturbations, resulting in varying tendencies for electron loss and potential implications for the π stacking mode. We have examined up to eight distinct units resembling TTF, utilizing either pure S or a combination of S/Se chalcogen atoms. Additionally, we have investigated alkyl substituents of varying chain lengths to explore potential molecular perturbations. All optimized species, labeled TTF1-TTF8 are depicted in Figure 2. Specifically, the combination of stacked TTF species with TCNQ in the crystal is anticipated to have diverse effects on the formation of TTF-TCNQ charge transfer compounds.
Figure 2.
Molecular structures of donor-substituted TTF molecules TTF1-TTF8.
Table 1 displays the chosen experimental characteristics of the mentioned group of compounds, as documented in the literature [20, 21, 22, 23, 24]. The compounds TTF1, TTF2, and TTF6 demonstrate conductive behavior, whereas TTF3, TTF5, TTF7, and TTF8 act as semiconductors, despite having different stacking modes with separated DDD-AAA successions. On the other hand, TTF4 appears to be an insulator, characterized by mixed stack structures and no charge transfer. It is worth noting that among the eight derivatives, only TTF6-TCNQ, which is di-seleniated, exhibits a higher degree of charge transfer compared to TTF-TCNQ. However, all eight derivatives are relatively poor conductors. Nevertheless, our analysis aimed to elucidate the effects of chemical variations rather than identifying the most efficient system.
We performed all computations using the Density Functional Theory (DFT) method with the B3LYP functional, which combines Becke’s three-parameter nonlocal exchange functional with the Lee-Yang-Parr correlation functions [25, 26]. The 6-311 + G(d,p) basis set provided by the Gaussian 16 package [27] was utilized. Our initial models were based on X-ray structures [9, 28, 29, 30, 31, 32] sourced from the Cambridge Database or electron diffraction studies [20]. All optimizations were conducted in the gas phase. The resulting minima were verified through frequency calculations and showed consistent agreement with established structural data, supporting our comparisons of energy and other chemical-physical properties [33].
Stable charge-transfer complexes (CTC) are generated when an electron π-donor like TTF is combined with an electron acceptor such as TCNQ. This interaction facilitates electron transfer between the stacked π units, leading to the development of cationic and anionic traits in these units, respectively. Specifically, our research has focused on demonstrating how different TTF-type molecules are inclined to engage in this described process.
3.1 Neutral-state molecular geometries TTF and its derivatives
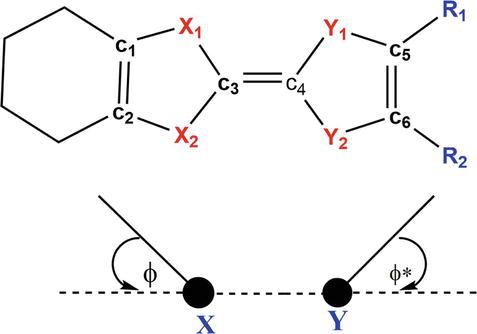
At the outset, we performed optimizations for all the neutral models presented in Figure 2. Our objective was to discern consistent patterns in the molecular geometry and stereochemistry influenced by both the chalcogen atom characteristics and the varying alkyl substituents with different chain lengths. Table 2 provides a comparison between the optimized bond lengths and angles, as well as the corresponding experimental values, for all the TTF-based derivatives, encompassing the original TTF precursor. And their labeling schemes are shown in Figure 3.
Calculated and experimental observed geometric properties of TTF, and its derivatives.
Figure 3.
Atomic labeling scheme for TTF and its derivatives.
Specifically, the designated dihedral angles ϕ and ϕ*, as defined in the Figure 3, offer insights into the level of planarity exhibited by the molecule. Interestingly, these angles, which typically range from 10 to 15°, indicate that no molecule can achieve perfect planarity. This is clearly evident from the images in Figure 4. Notably, all these molecules are expected to exhibit a planar geometry due to their formal count of 14 π electrons, indicative of aromaticity. However, in reality, they adopt a “boat”-like conformation, even the highly symmetric TTF precursor, which attains C2v symmetry. This particular symmetry is unattainable for the other derivatives, primarily due to the different chemical nature of their lateral groups. These groups typically introduce bending on one side of the central X2C〓CY2 skeleton, which inherently remains planar. Interestingly, the degree of deformation appears to be more pronounced when sulfur atoms are substituted with selenium atoms. This suggests that the pπ lone pairs of the chalcogen atoms might exhibit greater localization, potentially leading to repulsive interactions with the lateral groups and resulting in minor geometric adjustments.
Figure 4.
Side views of TTF and its derivatives.
Usually, the angles formed by the bonds around chalcogen atoms are approximately 90° (H▬S▬H angle in H2S is 92.1 and H▬Se▬H 91° in H2Se). However, the bond angles involving carbon and other atoms (denoted as C▬X▬C) tend to be larger, ranging from 92° to 96° as shown in Table 2. This is due to the presence of a ring structure. Additionally, the molecular constraint can cause some deviations from perfect planarity. Regardless, the noticeable geometric deviations are expected to incur a relatively modest energy penalty, potentially compensated for by the stabilizing effect of stacking the planar units within a crystal lattice. Recent examples have demonstrated instances where stacking directly impacts the internal geometry of a building block [35, 36, 37]. Table 2 displays variations among the three optimized C〓C double bonds within the TTF framework (C1〓C2, C3〓C4 and C5〓C6). These bond distances remain relatively similar across sulfur-containing species, including the original TTF and derivatives TTF1-TTF4. In this context, the central C3〓C4 bond is only around 0.01 Å longer compared to the adjacent bonds. Notably, in available crystal structures, this difference can be up to three times larger. Interestingly, in compounds TTF5-TTF8, where two selenium (Se) atoms replace sulfur (S) atoms, the disparities in C▬C distances become nearly negligible. This suggests the potential influence of altered π delocalization due to the substitution of S atoms with Se atoms.
3.2 Electron-related aspects
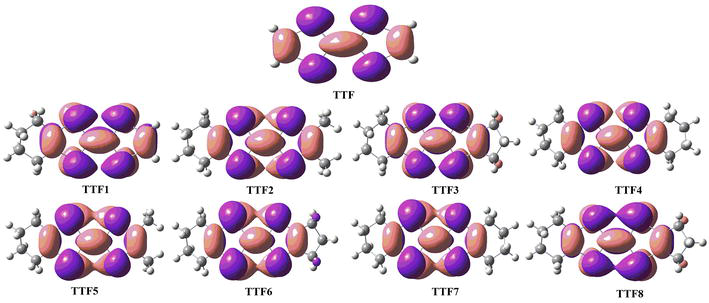
The preceding examination of the compounds reveals that, despite slight deviations from the flatness of the two C〓C bonded five-membered rings; these systems retain their fundamentally aromatic nature by being exclusively characterized by their 14 π-electron count. This count includes four electron pairs from the chalcogen atoms, contributing to the extension of π conjugation. Even the substitution of Se for S atoms (compounds TTF5-TTF8) does not introduce any significant alteration to the underlying electronic structure. Only a distinct pattern is discernible from the Highest Occupied Molecular Orbitals (HOMOs) depicted in Figure 5, which represent the highest pair of π electrons within the aromatic system.
Figure 5.
The HOMO level of the TTF and its derivatives.
In more detailed analysis, the Highest Occupied Molecular Orbital (HOMO) exhibits localized π-bonding characteristics at the three C〓C double bonds, but it predominantly assumes an antibonding nature due to interaction with the pπ lone pairs of the four chalcogen atoms. This likely accounts for its higher energy and the tendency towards depopulation. Additionally, Table 2 illustrates that the two lateral C▬S distances are marginally longer than the corresponding central ones, with differences of less than 0.02 Å, possibly indicating varying degrees of antibonding character in the HOMO. The sulfur-containing species (TTF1-TTF4) appear to possess an almost symmetric HOMO, akin to the TTF precursor, while the presence of two selenium atoms (models TTF5-TTF8) introduces some asymmetry. The C▬Se distances in Table 2 indeed measure approximately 0.13–0.16 Å larger than the C▬S distances, primarily due to different covalent radii. However, it remains plausible that this effect arises from an enhanced C▬Se π* character in the HOMO.
Additionally, the HOMO’s extended π character, along with its substantial π* interactions, foreshadows the potential loss of one or two electrons, leading to the formation of either a radical cation (TTF+) or a dication (TTF2+). The ensuing ramifications of this oxidation will be discussed in more detail.
3.3 Atomic Polarizability Tensor charges
The Atomic Polarizability Tensor (APT) is comprehended as the combination of the charge tensor and the charge flux tensor, forming a charge-flux model [38]. The associated atomic charges for both TTF and its derivatives are presented in Table 3.
TTF
TTF1
TTF2
TTF3
TTF4
TTF5
TTF6
TTF7
TTF8
C1
0.01
0.07
0.07
0.08
0.08
0.04
0.04
0.04
0.08
C2
0.01
0.07
0.07
0.08
0.08
0.04
0.04
0.04
0.08
C3
0.11
0.11
0.12
0.11
0.11
0.01
−0.01
−0.00
0.14
C4
0.11
0.12
0.11
0.11
0.11
0.15
0.16
0.16
0.01
C5
0.01
0.00
0.10
0.06
0.08
0.10
0.06
0.07
0.02
C6
0.01
0.00
0.10
0.06
0.08
0.10
0.06
0.07
0.02
X1
−0.14
−0.20
−0.21
−0.21
−0.21
−0.10
−0.10
−0.10
−0.22
X2
−0.14
−0.20
−0.21
−0.21
−0.21
−0.10
−0.10
−0.10
−0.22
Y1
−0.14
−0.15
−0.21
−0.19
−0.21
−0.22
−0.21
−0.22
−0.08
Y2
−0.14
−0.15
−0.21
−0.19
−0.21
−0.22
−0.21
−0.22
−0.08
Table 3.
Charges of the optimized TTF molecules and its derivatives TTF1-TTF8.
The charges of the carbon atoms involved in the π bonds of TTF and its derivatives typically hover around zero or exhibit a slight positive charge. Notably, there are exceptions; for instance, atom C3 displays a slightly negative charge in the TTF6 and TTF7 derivatives. In broad terms, the carbon atoms within the lateral C〓C bonds tend to be slightly more positively charged compared to their counterparts in the central region, implying that the chalcogen atoms exert a greater electron-withdrawing effect on the external C〓C bonds.
On the contrary, the chalcogen atoms consistently exhibit negative charges, a consequence of their higher electronegativity. Notably, the residual electron density of the selenium (Se) atoms appears to be nearly double that of the sulfur (S) atoms, implying smaller contributions from the Se pπ electron density. This observation aligns with the HOMO depictions, which also indicate a similar trend.
3.4 Oxidized species
TTF and its derivatives possess the ability to form stacked solid-state structures, wherein a distinct positive charge aligns with the corresponding negative charge of the stacked TCNQ counter ion. Unfortunately, we were unable to utilize the specific computational methods outlined in a referenced review article [39] for the species depicted in Figure 2. Despite this limitation, we found it valuable to examine the redox behavior of TTF-like units when one or two electrons are sequentially removed. This approach prevents a direct comparison with the stacked TTF system, where the electron density removed per unit is merely a fraction of what we considered. Nevertheless, the data in Table 4 facilitates the assessment of electron removal effects through a comparison of geometries with those of the neutral precursors listed in Table 3.
Calculated and experimental observed geometric properties of the mono- and bis-oxidized TTF, and its derivatives.
As mentioned, the delocalized HOMOs of TTF and its derivatives shown in Figure 4 appear to allow for potential electron vacancies due to the predominant antibonding nature of the level. As a result, any C▬X bond (where X represents a chalcogen atom) becomes stronger upon electron removal, while the strength of C〓C bonds decreases. This observation finds further support in the structural alterations observed in TTF-type units. For instance, the central C3〓C4 bond length increases by 0.05 Å and 0.05 Å in the monocation and dication, respectively. A similar trend can be observed in the lateral C〓C bonds, albeit with an inverted effect progression, ranging from 0.01 Å to 0.03 Å. In contrast, all C▬S bonds undergo significant shortening, with the most pronounced reduction occurring in the C▬S bond linked to the central C〓C linkage. In this instance, the average C▬S distance decreases by 0.03 Å in the monocation and 0.06 Å in the dication. The oxidation process also exerts a notable influence on the conformation of TTF and its derivatives. Upon the first oxidation, the radical cations adopt an almost planar conformation (ϕ = ϕ* ≈ 0). This change is likely driven by reduced electronic repulsion between the entire lateral groups and the less electron-rich chalcogen atoms. Table 5 presents the calculated Atomic Polarizability Tensor (APT) charge distribution for the TTF models in both their radical cation and dication states. Notably, whereas the chalcogen atoms were somewhat negatively charged in the neutral species (as shown in Table 3), they exhibit a gradual shift towards more positive charges in the singly and doubly oxidized species. This observation confirms a significant impact on the atoms under consideration.
TTF+• (TTF2+)
TTF1+• (TTF12+)
TTF2+• (TTF22+)
TTF3+• (TTF32+)
TTF4+• (TTF42+)
TTF5+• (TTF52+)
TTF6+• (TTF62+)
TTF7+• (TTF72+)
TTF8+• (TTF82+)
C1
−0.11 (−0.15)
−0.03 (0.03)
−0.04 (−0.01)
−0.05 (−0.02)
−0.05 (−0.02)
−0.05 (−0.02)
−0.06 (−0.03)
−0.06 (−0.03)
−0.06 (−0.03)
C2
−0.11 (−0.15)
−0.03 (0.03)
−0.04 (−0.01)
−0.05 (−0.02)
−0.05 (−0.02)
−0.05 (−0.02)
−0.06 (−0.03)
−0.06 (−0.03)
−0.06 (−0.03)
C3
0.20 (0.15)
0.06 (−0.17)
0.13 (0.06)
0.21 (0.08)
0.18 (0.08)
0.05 (−0.02)
0.13 (0.04)
0.1 (0.04)
0.26 (0.15)
C4
0.20 (0.15)
0.32 (0.40)
0.23 (0.11)
0.13 (0.08)
0.18 (0.08)
0.28 (0.20)
0.17 (0.11)
0.22 (0.11)
0.04 (0.01)
C5
−0.11 (−0.15)
−0.13 (−0.17)
−0.01 (0.02)
−0.06 (−0.02)
−0.05 (−0.02)
−0.02 (−0.01)
−0.07 (−0.03)
−0.06 (−0.03)
−0.07 (−0.03)
C6
−0.11 (−0.15)
−0.13 (−0.17)
−0.01 (0.02)
−0.06 (−0.02)
−0.05 (−0.02)
−0.02 (−0.01)
−0.07 (−0.03)
−0.06 (−0.03)
−0.07 (−0.03)
X1
0.12 (0.36)
0.09 (0.32)
0.07 (0.30)
0.07 (0.30)
0.06 (0.30)
0.13 (0.35)
0.12 (0.34)
0.12 (0.34)
0.06 (0.24)
X2
0.12 (0.36)
0.09 (0.32)
0.07 (0.30)
0.07 (0.30)
0.06 (0.30)
0.13 (0.35)
0.12 (0.34)
0.12 (0.34)
0.06 (0.24)
Y1
0.12 (0.36)
0.09 (0.31)
0.04 (0.31)
0.11 (0.33)
0.06 (0.30)
0.03 (0.31)
0.11 (0.32)
0.06 (0.28)
0.17 (0.39)
Y2
0.12 (0.36)
0.09 (0.31)
0.04 (0.31)
0.11 (0.33)
0.06 (0.30)
0.03 (0.31)
0.11 (0.32)
0.06 (0.28)
0.17 (0.39)
Table 5.
The charges of TTF and its derivatives TTF1-TTF8 computed for the radical cation and the dication, respectively.
In TTF+• and TTF2+, the majority of the positive charge is concentrated at the sulfur (S) atom sites, similar to the other models.
3.5 Quantitative energy values for the frontier orbital levels in the different redox isomers
Table 6 present the electronic energies associated with the HOMO, the single occupied molecular orbital (SOMO) and the lowest unoccupied molecular orbital (LUMO) of both TTF and its derivatives shown in Figure 2 (TTF1-TTF8). These data provide insights into the comparative susceptibility to oxidation across the series of compounds.
Neutral
Cation
Dication
HOMO
LUMO
SOMO
LUMO
HOMO
LUMO
TTF
−4.77
−1.17
−9.42
−5.53
−15.87
−12.65
TTF1
−4.68
−1.06
−9.00
−5.20
−14.80
−11.83
TTF2
−4.57
−1.01
−8.71
−5.00
−14.18
−11.35
TTF3
−4.55
−0.99
−8.68
−4.92
−14.05
−11.35
TTF4
−4.53
−0.99
−8.63
−4.91
−14.04
−11.27
TTF5
−4.66
−1.36
−8.70
−5.28
−13.92
−11.44
TTF6
−4.65
−1.35
−8.67
−5.21
−13.77
−11.41
TTF7
−4.62
−1.34
−8.62
−5.20
−13.76
−11.33
TTF8
−4.63
−1.33
−8.67
−5.19
−13.76
−11.39
Table 6.
Comparative electronic energy values (eV) of the HOMOs of the uncharged species and the SOMO and LUMO of the oxidized derivatives.
Among the uncharged species similar to TTF, the uncharged precursor appears to pose the greatest challenge for oxidation due to its lowest energy HOMO level (−4.77 eV). Introduction of alkyl substituents with larger ethylenic chains (TTF1-TTF4) results in an elevation of the HOMO energy to approximately −4.53 eV, thereby facilitating electron removal. Similarly, substituting Se for S atoms (TTF5-TTF8) exhibits a comparable yet slightly flatter trend following alkyl substitution (ranging from −4.66 to −4.63 eV). Furthermore, the data suggests that, in general, it is easier to remove the first electron than the second, as indicated by the correspondingly smaller ΔE values for HOMO-SOMO and SOMO-LUMO, respectively. For instance, in the case of the TTF precursor, these differences are −4.65 and −4.09 eV, signifying that the formation of the monocation is more favorable than the dication. The lower absolute energies of the HOMO, SOMO, and LUMO, in ascending order, can be attributed to an increasing positive charge, which progressively hinders electron removal. It’s worth noting that an accurate determination of the endothermic or exothermic nature of oxidation necessitates modeling the entire reaction, encompassing the oxidation reactant. In this discussion, we have primarily highlighted select aspects related to potential electron loss from isolated TTF-like units. In actual TTF-TCNQ charge transfer compounds, it is plausible that the π stacking energy of both the TTF and TCNQ components could sufficiently favor single electron oxidation.
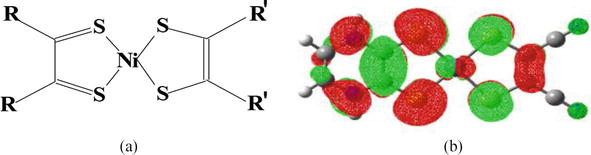
Lastly, we highlight a potential correlation between the current organic and planar components and square planar d8-transition metal complexes like bis-dithiolene-M (M (M = Ni(II), Pd(II), Pd(II)) [39]. In this context, the metal ion substitutes the central C〓C bond (see Figure 6). Essentially, the π* level of the latter exhibits a similar structure to a dπ metal orbital, enabling the creation of HOMOs with analogous topology. These HOMOs, in turn, facilitate diverse redox characteristics and also introduce additional non-linear optical (NLO) properties. This correlation suggests a significant connection between seemingly unrelated organic and inorganic compounds.
Figure 6.
(a) A Ni(II) bis-dithiolene complex with relevance to the current TTF derivatives; (b) the π-delocalized frontier molecular orbital (MO) of a metal species that, akin to the TTF1-TTF4 derivatives discussed here, supports a set of redox isomers (refer to [42] for further details).
We conducted an analysis using DFT/B3LYP/6-311 + G(d,p) calculations to examine the geometrical and electronic characteristics of a series of TTF-like species. This series includes the precursor and several of its derivatives with alkyl substitutions, as well as analogs containing two selenium (Se) atoms. These models provide valuable insights into both the structural and electronic characteristics of TTF. These attributes hold significant importance in the context of TTF/TCNQ solid-state compounds, where stacking interactions drive one-electron transfer processes. In each instance, the optimized series of redox isomers, encompassing the uncharged, monocationic, and dicationic derivatives, closely align with the available experimental geometries. The generally consistent nature of the Highest Occupied Molecular Orbitals (HOMOs), characterized by evident carbon/chalcogen π antibonding character, appears to facilitate depopulation (either partial or complete) and subsequently influences the redox chemistry. As a result, the oxidized molecules exhibit increasingly planar configurations. Comparative energy assessments indicate that, in all cases, the first oxidation step is more energetically favorable than the second. Furthermore, these organic species appear to share similarities with their inorganic counterparts, such as certain Ni(II) square planar complexes featuring two dithiene chelates, which exhibit comparable redox behaviors [42]. Additionally, the electron transfer properties foreshadow important connections between organic systems, particularly Organic Light-Emitting Devices (OLEDs), and metal complexes [43].
This research received funding from the Directorate General of Scientific Research and Technological Development (DGRSDT) and the Ministry of Higher Education and Scientific Research (MESRS) in Algeria. We are grateful to Dr. Rafik Nezzar Manager of High Performance Computing (HPC), University of Batna 2 for his invaluable technical support.
1.Wudl F, Smith GM, Hufnagel EJ. Bis- 1,3-dithiolium chloride: An unusually stable organic radical cation. Journal of the Chemical Society D: Chemical Communications. 1970;21:1453-1454. DOI: 10.1039/c29700001453
2.Midoune A, Messaoudi A. DFT/TDDFT computational study of the tetrathiafulvalene-1,3-benzothiazole molecule to highlight its structural, electronic, vibrational and non-linear optical properties. Comptes Rendus Chimie. 2020;23:143-158. DOI: 10.5802/crchim.12
3.Midoune A, Messaoudi A. DFT/TDDFT theoretical study of the structural, electronic and nbo analysis of cu(II) and cu(I) complexes that contain the tetrathiafulvalene-1,3-benzothiazole ligand. Inorganica Chimica Acta. 2021;516:120151. DOI: 10.1016/j.ica.2020.120151
4.Lebed AG. The Physics of Organic Superconductors and Conductors. 1st ed. Berlin: Springer-Verlag, Berlin; 2008. DOI: 10.1007/978-3-540-76672-8
5.Wudl F, Wobschall D, Hufnagel EJ. Electrical conductivity by the bis(1,3-dithiole)-bis(1,3-dithiolium) system. Journal of the American Chemical Society. 1972;94(2):670-672. DOI: 10.1021/ja00757a079
6.Bechgaard K. The Chemistry of TTF-TCNQ, Molecular Metals. Boston, MA: Springer; 1979. 1-6 p. DOI: 10.1007/978-1-4684-3480-4_1
7.Parkin SSP, Engler EM, Schumaker RR, Lagier R, Lee VY, Scott JC, et al. Superconductivity in a new family of organic conductors. Physical Review Letters. 1983;50(4):270-273. DOI: 10.1103/PhysRevLett.50.270
8.Laukhina E, Vidal-Gancedo J, Laukhin V, Veciana J, Chuev I, Tkacheva V, et al. Multistability in a BEDT-TTF based molecular conductor. Journal of the American Chemical Society. 2003;125(13):3948-3953. DOI: 10.1021/ja0280123
9.Martin L. Molecular conductors of BEDT-TTF with tris (oxalato) metallate anions. Coordination Chemistry Reviews. 2018;376:277-291. DOI: 10.1016/j.ccr.2018.08.013
10.Martín N, Sánchez L, Guldi DM. Stabilisation of charge-separated states via gain of aromaticity and planarity of the donor moiety in C60-based dyads. Chemical Communications. 2000;2:113-114. DOI: 10.1039/A908770B
11.Smeh A, Manef AR. DFT study of the competition between cycloaddition reactions type [2 +2] and [4 +2] applied to the fullerene molecule. Journal of Materials and Environmental Science. 2014;5:1683-1690. Available from: http://www.jmaterenvironsci.com
12.Roncali J. Linearly extended π-donors: When tetrathiafulvalene meets conjugated oligomers and polymers. Journal of Materials Chemistry. 1997;7:2307-2321. DOI: 10.1039 /A703956E
13.Karakas A, Karakaya M, Ceylan Y, El Kouari Y, Taboukhat S, Boughaleb Y, et al. Ab-initio and DFT methodologies for computing hyperpolarizabilities and susceptibilities of highly conjugated organic compounds for nonlinear optical applications. Optical Materials. 2016;56:8.17 DOI : 10.1016/j.optmat.2016.01.036
14.Ayadi A, Szukalski A, El-Ghayoury A, Haupa K, Zouari N, Myśliwiec J, et al. TTF based donor-pi-acceptor dyads synthesized for NLO applications. Dyes and Pigments. 2017;138:255-266. DOI: 10.1016/j.dyepig.2016.11.030
15.Hansen TK, Joergensen T, Stein PC, Becher J. Crown ether derivatives of tetrathiafulvalene. 1. The Journal of Organic Chemistry. 1992;57:6403-6409. DOI: 10.1021/jo00050a010
16.Coronado E, Galán-Mascarós JR, Giménez-Saiz C, Gómez-García CJ, Ruiz-Pérez C. Hybrid organic/inorganic molecular materials formed by tetrathiafulvalene radicals and magnetic trimeric clusters of dimetallic oxalate-bridged complexes: The series (TTF)4{MII(H2O)2[MIII(ox)3]2}· nH2O(MII= Mn, Fe, Co, Ni, Cu and Zn; MIII= Cr and Fe; ox= C2O42−). European Journal of Inorganic Chemistry. 2003;12:2290-2298. DOI: 10.1002/ejic.200200589
17.Ringsdorf H, Bengs H, Karthaus O, Wüstefeld R, Ebert M, Wendorff JH, et al. Induction and variation of discotic columnar phases through doping with electron acceptors. Advanced Materials. 1990;2:141-144. DOI: 10.1002/adma.19900020306
18.Bryce MR, Devonport W, Goldenberg LM, Wang C. Macromolecular tetrathiafulvalene chemistry. Chemical Communications. 1998;9:945-952. DOI: 10.1039/A800536B
19.Asakawa M, Ashton PR, Balzani V, Credi A, Hamers C, Mattersteig G, et al. A chemically and electrochemically switchable [2] catenane incorporating a tetrathiafulvalene unit. Angewandte Chemie International Edition. 1998;37:333-337. DOI: 10.1002/(SICI)1521-3773(19980216)37:3<333::AID-ANIE333>3.0.CO;2-P
20.Hargittai I, Brunvoll J, Kolonits M, Khodorkovsky V. Tetrathiafulvalene: Gas-phase molecular structure from electron diffraction. Journal of Molecular Structure. 1994;317:273-277. DOI: 10.1016/0022-2860(93)07873-U
21.Abbaz T, Bendjeddou A, Gouasmia A, Regainia Z, Villemin D. Synthesis and electrochemical proprieties of novel unsymmetrical bis-tetrathiafulvalenes and electrical conductivity of their charge transfer complexes with tetracyanoquinodimethane (TCNQ). International Journal of Molecular Sciences. 2012;13(7):7872-7885. DOI: 10.3390/ijms13077872
22.Sánchez-Vergara ME, Leyva-Esqueda M, Alvárez-Bada JR, García-Montalvo V, Rojas-Montoya ID, Jiménez-Sandoval O. Optical and electrical properties of TTF-MPcs (M= Cu, Zn) interfaces for optoelectronic applications. Molecules. 2015;20:21037-21049. DOI: 10.3390/molecules 201219742
23.Fabre JM, Amouroux J, Giral L, Chaseau D. Synthesis and studies of new materials based on dithiadiselenafulvalenes. Synthetic Metals. 1991;42:2049-2052. DOI: 10.1016/0379-6779(91)92012-7
24.El Kacemi K, Lamache M. Electrochimie du tetrathiofulvalene-tetracyanoquinodimethane ed d'autres complexes a transfert de charge. Electrochimica Acta. 1986;31:1197-1204. DOI: 10.1016/0013-4686(86)80134-7
25.Becke AD. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. The Journal of Chemical Physics. 1993;98:5648-5652. DOI: 10.1063/1.464913
26.Lee C, Yang W, Parr RG. Accurate and simple analytic representation of the electron-gas correlation energy. Physical Review B. 1988;37:785-789. DOI: 10.1103/PhysRevB.37.785
28.Cambridge Structural Database System, Version 1.16. Cambridge, CB2 1EZ, UK: Cambridge Crystallographic Data Centre; 2013
29.Cooper WF, Kenny NC, Edmonds JW, Nagel A, Wudl F, Coppens P. Crystal and molecular structure of the aromatic sulfur compound, bis-1, 3-dithiole: Evidence for d-orbital participation in bonding. Chemical Communication. 1971;1:889-890. DOI: 10.1039/c29710000889
30.Rosokha SV, Lu J, Han B, Kochi JK. Unusual structural effects of intermolecular π-bonding in the tetracyanopyrazine (ion-radical) dimer. New Journal of Chemistry. 2009;33:545-553. DOI: 10.1039/B812829D
31.Tashiro S, Umeki T, Kubota R, Shionoya M. Simultaneous arrangement of up to three different molecules on the pore surface of a metal–macrocycle framework: Cooperation and competition. Angewandte Chemie International Edition. 2014;53:8310-8315. DOI: 10.1002/anie.201404179
32.Zeller M, Azov VA. 2-(1,3-Dithiol-2-ylidene)-1,3-dithiole-4-carbaldehyde. Acta Crystallographica Section E Structure Reports Online. 2013;69:o1157-o1157. DOI: 10.1107/s160053681301711x
34.Midoune A, Messaoudi A, Boumedjane Y. DFT study of a series of tetrathiafulvalene species and their redox isomer. Inorganic Chemistry Communications. 2019;100:118-124. DOI: 10.1016/j.inoche.2018.12.026
35.Canossa S, Bacchi A, Graiff C, Pelagatti P, Predieri G, Ienco A, et al. Hierarchy of supramolecular arrangements and building blocks: Inverted paradigm of crystal engineering in the unprecedented metal coordination of methylene blue. Inorganic Chemistry. 2017;56:3512-3516. DOI: 10.1021/acs.inorgchem.6b02980
36.Grirrane A, Pastor A, Galindo A, Ienco A, Mealli C, Rosa P. First example of a tetra-carboxylate bridged dimanganese species. Chemical Communications. 2003;4:512-513. DOI: 10.1039/b211886f
37.Grirrane A, Pastor A, Galindo A, Álvarez E, Mealli C, Ienco A, et al. Thiodiacetate–manganese chemistry with N ligands: Unique control of the supramolecular arrangement over the metal coordination mode. Chemistry–A European Journal. 2011;17:10600-10617. DOI: 10.1002/chem.201100988
38.Ferreira MM, Suto E. Atomic polar tensor transferability and atomic charges in the fluoromethane series CHxF4-x. The Journal of Physical Chemistry. 1992;96:8844-8849. DOI: 10.1021/j100201a030
39.Ishibashi S. First-principles electronic-band calculations on organic conductors. Science and Technology of Advanced Materials. 2009;10:024311. DOI: 10.1088/1468-6996/10/2/024311
40.Katayama C, Honda M, Kumagai H, Tanaka J, Saito G, Inokuchi H. Crystal structures of complexes between hexacyanobutadiene and tetramethyltetrathiafulvalene and tetramethylthiotetrathiafulvalene. Bulletin of the Chemical Society of Japan. 1985;58:2272-2278. DOI: 10.1246/bcsj.58.2272
41.Ashton PR, Balzani V, Becher J, Credi A, Fyfe MC, Mattersteig G, et al. A three-pole supramolecular switch. Journal of the American Chemical Society. 1999;121:3951-3957. DOI: 10.1021/ja984341c
42.Curreli S, Deplano P, Faulmann C, Ienco A, Mealli C, Mercuri ML, et al. Electronic factors affecting second-order NLO properties: Case study of four different push-pull bis-dithiolene nickel complexes. Inorganic Chemistry. 2004;43:5069-5079. DOI: 10.1021/ic0496469
43.Qu W, Zhang F, Liu S, Wei D, Dong X, Yu B, et al. Efficient blue electroluminescence of iridium (III) complexes with oxadiazol-substituted amide ancillary ligands. Dyes and Pigments. 2017;145:116-125. DOI: 10.1016/j.dyepig.2017.05.042
Written By
Assia Midoune and Abdelatif Messaoudi
Submitted: 29 August 2023Reviewed: 19 September 2023Published: 23 April 2024