
Abstract
Insulin resistance is the most important characteristic of both metabolic syndrome (MetS) and type 2 diabetes mellitus (T2D). It is estimated that MetS has a prevalence of up to 35% in the general population, rising up to 72% in individuals with T2D. Furthermore, insulin resistance promotes lipotoxicity through an increased free fatty acids flux, leading to both liver and heart disorders. Thus, recent studies have proven the association of metabolic dysfunction-associated fatty liver disease (MAFLD) with MetS and T2D. Interestingly, recent data incriminate the same mechanism for the development of metabolic cardiomyopathy, also known as cardiometabolic heart failure. The aim of this paper is to review the most important data regarding the association of T2D with the classic and the newer elements of the MetS, as well as to emphasize the molecular mechanisms that are accountable for this linkage and the possible therapeutic prospects that could influence these pathologies.
Keywords
- type 2 diabetes
- metabolic syndrome
- insulin resistance
- fatty liver disease
- metabolic cardiomyopathy
- atherogenic dyslipidemia
- cardiovascular risk
1. Introduction
Metabolic syndrome (MetS) is a condition that associates metabolically related traits, that increase the risk of type 2 diabetes (T2D) as well as atherosclerotic cardiovascular disease [1, 2]. The components of MetS are central obesity, high blood pressure, dyslipidemia (elevated triglycerides, decreased levels of high-density lipoprotein (HDL) cholesterol) and carbohydrate intolerance, the presence of at least 3 of these impairments defining MetS [3]. MetS is considered a major public health issue, both for its complications and high prevalence. Although worldwide there are several definitions still in use, regardless of the criteria used for the diagnosis, the global estimated prevalence of MetS in the adult population is 20–45%, with a predicted prevalence of 53% by 2035 [4, 5]. A more worrisome problem is the increase in obesity observed in children and adolescents, which was associated with a prevalence of MetS in the pediatric population of about 16% [6, 7].
The prevalence of overweight, obesity and T2D have also reached epidemic proportions. The World Health Organization (WHO) appreciated that the prevalence of obesity worldwide has nearly tripled since 1975, with an estimated prevalence of overweight of 39%, respectively 13% in the adult population in 2016 [8]. The International Diabetes Federation estimated that in 2021 537 million adults were living with diabetes, number that is projected to increase to 643 million by 2030, respectively to 783 million by 2045 [9].
Persons with MetS have a significantly higher risk of developing T2D, insulin resistance (IR) playing an important role in this association. Insulin is the main hormone involved in maintaining the glucose homeostasis. In addition to promoting glycogen synthesis, downregulating gluconeogenesis and glucose uptake through GLUT-4 receptor, insulin is an active participant in promoting nitric oxide and endothelin release in the endothelial cells, leading to vasodilation and/or vasoconstriction, in order to improve the blood glucose uptake of different organs [10]. In insulin resistant states, such as obesity, MetS and T2D, the insulin-dependent tissues undergo an inadequate response to insulin, with a decreased activation of endothelial nitric oxide synthase at the level of endothelial cells, with impaired insulin signaling pathways [10]. Due to the role played by IR in many noncommunicable disease (T2D, MetS, fatty liver, cardiovascular disease, Alzheimer’s disease, etc.), many fundamental and clinical studies have tried to assess the molecular mechanisms of this impairment [10].
The first author that has thoroughly studied IR was Reaven [11]. He suggested that IR, revealed by hyperinsulinemia, is the main factor responsible for both atherogenic dyslpidemia and dysfunctional glucose metabolism, as well as for an increase in arterial blood pressure [11, 12]. The clinical evaluation of IR and insulin sensitivity in vivo is a delicate issue, as the gold standard method is considered the euglycemic-hyperinsulinemic clamp, which in the clinical practice has limited use do to its sophisticated and technical nature [12, 13]. This is the reason why in the clinical practice other markers of insulin resistance are currently utilized. Among these, the homeostasis model assessment of insulin resistance (HOMA-IR) [14] is the main index used for estimating IR.
Another index that was proposed for assessing insulin sensibility/IR is the quantitative insulin sensitivity check index (QUICKI), which presents significantly lower values in persons that present insulin resistant states, such as MetS, prediabetes and T2D [15].
A biomarker which is easy to calculate, the triglyceride-glucose (TyG) index was also proposed for the assessment of IR [16]. A recent study, performed in over 15,000 subjects, concluded that although both HOMA-IR and TyG index were predictors of incident MetS, the TyG index had a higher predictive power [16]. Many other biomarkers are proposed as tools for assessing IR. However, there is still need for further studies in order to conclude the utility of these equations in the clinical practice, in order to improve the early diagnosis of metabolic complications such as fatty liver disease, obstructive sleep apnea, metabolic cardiomyopathy, neoplasms, etc.
Subjects with T2D that present MetS are at a higher risk of developing metabolic complications, as well as at higher risk for all-cause mortality [17]. The association of MetS and T2D is a frequent occurrence in the clinical practice, both disorders having as important risk factors the unhealthy lifestyle and obesity [18]. The aim of this paper is to gather information about the complications of MetS and T2D, in order to improve the early diagnosis and the therapeutic options for these persons.
2. From metabolic syndrome to type 2 diabetes: physiopathological mechanisms
MetS is one of the most common risk factors for developing T2D, these subjects having a 5 times higher risk than the general population [18]. One of the most important physiopathological mechanism that leads to the development of T2D in individuals with MetS is the pancreatic β-cell dysfunction [1]. The metabolic impairments (increased free fatty acids (FFA) and blood glucose) present in MetS, together with the proinflammatory status that characterizes this disorder, represent an adverse environment to β cells [1, 19].
Under these conditions, two possible scenarios might happen. In the first instance, the β cell must undergo compensatory functional changes, respectively insulin hypersecretion, to respond to the increased metabolic demands [1]. However, sometimes this hypersecretion of insulin may lead to IR, in the end resulting in impaired β cells (loss of β cell function and β cell mass) [1, 20]. On the other side, the metabolic stressor may first diminish insulin response in insulin – dependent tissues (liver), leading to IR which will promote insulin hypersecretion that may progress towards β cell dysfunction [1]. The exact mechanism involved in these two scenarios are not completely elucidated, however the ethnic factor seems to play an important role, the dysfunction of the β cells being more pronounced in individuals with Asian origins compared to Caucasians [1, 21]. In order to develop viable therapeutic intervention to prevent the progression from MetS to T2D, it is important to try to understand the intracellular mechanisms that are responsible for the shift in β cell adaptive response responsible for this progression, regardless if IR and/or nutrient response set off this complex process.
Subjects with MetS that in time develop T2D present only transient compensatory adaptation of the pancreatic β cells. The hypersecretion at cellular level leads to dysfunctions of the endoplasmic reticulum, translated into improper synthesis, folding, trafficking and, in the end, decreased insulin secretion, the β cells being unable to further sustain the enlarged workload necessary for maintaining normal blood glucose level. Animal and human studies [1, 22, 23, 24, 25, 26, 27, 28] likewise have described these same molecular endoplasmic reticulum and mitochondria defects, that over time result in β cells apoptosis.
However, it also must be noted that emerging studies have described two mechanisms through which the pancreatic β cells respond to environmental stress, respectively dedifferentiation and transdifferentiation, in order to avoid significant islet cell mass loss [1, 29]. In a 2012 study by Talchai et al. [30] aiming to assess the role of decreased β cell mass or impaired β cell function in animal models with diabetes, concluded that under stress conditions, not all dysfunctional cells were eliminated through apoptosis, some of the β cells suffering dedifferentiation process, reverting to a progenitor-like state. Interestingly, this study demonstrated that some of the differentiated cells are able to produce pancreatic hormones [30]. In a recent review, Bensellam et al. [31] proposed a novel hypothesis, concluding that dedifferentiation of β cells is a protective mechanism under stress conditions, such as MetS. This theory supported by the results obtained in many other animal and human models of prolonged insulin demand [32, 33, 34, 35, 36] is still incompletely understood, the characterization of both the trigger and the molecular pathways involved in β cells dedifferentiation requiring further studies [1, 31].
Recent studies [1, 37, 38, 39] have proposed transdifferentiation of β cells, defined as the loss of the β cell identity and converting into cell expressing different hormones, suggesting that these cells are prone to reprogramming. Furthermore, in the anatomopathological study by Sun et al. [34] on islet samples from both type 2 and type 1 diabetes, it was evidenced that β cells can express multiple hormones, which suggests their ability to lose their cell identity. Nonetheless, there is need for lineage analysis in order to demonstrate that these polymorphal cells emerged from monohormonal pancreatic β cells [1]. Figure 1 summarizes the pathological mechanisms leading to the development of T2D in persons with obesity and MetS.
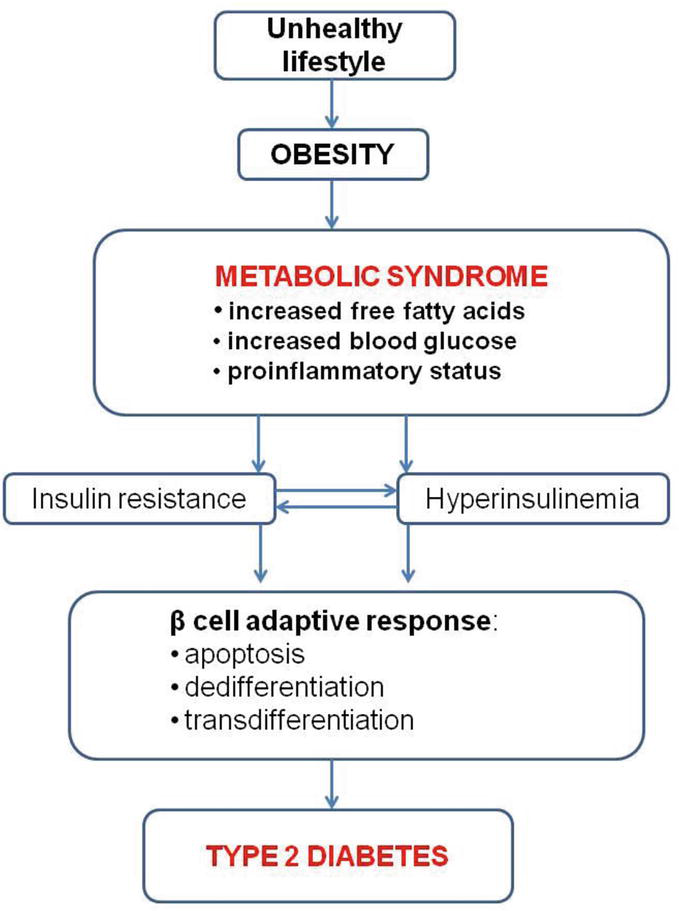
Figure 1.
Dysfunctions of pancreatic β cells that lead from MetS to T2D (adapted from [
IR is universally accepted as the central trait of MetS. However, animal and human studies have proposed hyperinsulinism as the initiating event leading to MetS [40, 41, 42]. In one of these studies [42] it was postulated that basal insulin secretion is increased while glucose stimulated insulin secretion is decreased as the subjects move from normal glucose tolerance (lean individuals) to obesity to impaired glucose tolerance, with glucose stimulated insulin secretion being less than half of total insulin secretion in persons with T2D [1].
All these data support the concept of early intervention in terms of lifestyle optimization in individuals with MetS and prediabetes. Indeed, two recent interventional studies [43, 44] have proved the role of diet and physical activity in regulating β cell function and preventing T2D in populations at risk.
3. Insulin resistance: the link between metabolic syndrome, type 2 diabetes and fatty liver disease
With a worldwide prevalence estimated at 25%, nonalcoholic fatty liver disease (NAFLD) is the most common liver disease, representing a major public health concern [45, 46]. From an anatomopathological point of view, the term NAFLD includes a diversity of hepatic lesions, such as steatosis, nonalcoholic steatohepatitis (NASH), fibrosis and cirrhosis, in subjects that have as a common trait the absence of alcohol consumption [47, 48, 49].
Studies [48, 50, 51] have revealed the association between NAFLD and metabolic disorders, a meta-analysis performed by Ballestri et al. demonstrating that persons with NAFLD have a 2-fold increased risk for the development of MetS and T2D over a median follow-up of 5 years [50]. Another meta-analysis form 2016 demonstrated that NAFLD was associated with MetS in 42.54% of the cases and with T2D in 22.51% of the cases, while obesity was the metabolic disease with the highest frequency in subjects with NAFLD (51.34%) [52]. In a study [53] that aimed to assess the frequency of steatosis and NASH in individuals with MetS or T2D that presented elevated liver enzymes, it was demonstrated that NAFLD had a prevalence of 94.82% in persons with MetS, while all the persons with T2D presented NAFLD. Furthermore, NASH was present in 58.52% in the case of people with MetS, while it had a prevalence of 96.82% in the individuals with T2D. This study concluded that NASH might be an early complication of T2D, IR being the link between the two pathologies [53].
Taking into consideration the high frequency of NAFLD in subjects with metabolic diseases, in 2020, an international panel of experts proposed the new terminology, metabolic dysfunction-associated fatty liver disease (MAFLD) [54]. This definition no longer restricts alcohol consumption or other secondary liver diseases, while introducing an important diagnosis criterion: the presence of a metabolic disorder [55, 56, 57]. This new term emphasizes the importance of metabolic factors in fatty liver pathophysiology, as well as facilitates patient – physician communication, making easier for the patient to understand the relationship between this syndrome and metabolic health [58].
IR is the main mechanism linking NAFLD/MAFLD and metabolic disorders. It is well documented the fact that systemic IR present in MetS and T2D is responsible for an increase in the flux of FFA from the visceral adipose cells towards the liver, followed by increased lipogenesis, that on one hand leads to fatty liver disease and on the other hand aggravates liver IR [59]. Moreover, continuous body of evidence supports the association of MAFLD with increased cardiovascular risk [59]. A possible explanation for this association comes from the fact that in MAFLD the hepatic tissue releases bioactive mediators that further increase liver and systemic IR, while also promoting atherogenic dyslipidemia and low grade inflammation [59, 60, 61]. It is important for the individuals with MetS and T2D to be screened for the presence of MAFLD, as these people have three major mortality causes: cardiovascular disease, extrahepatic malignancies and liver related complications (cirrhosis and hepatocellular carcinoma) [59, 60, 62, 63].
Among the etipathogenic factors involved in MAFLD, the most important remain lifestyle (unhealthy diet, increased alcohol consumption, etc.), microbiota dysregulations, ethnicity and genetic predisposition [64]. The role of unhealthy diet in the development of both MAFLD and metabolic disease is well recognized. High sugar and fructose intake as well as increased hepatic diacylglycerols levels lead to liver fat accumulation [59]. At the level of the hepatocyte, unhealthy dietary choices are translated into hepatic IR, oxidative stress and chronic inflammation, with subsequent development and progression of fatty liver [59].
Gut microbiota is another important factor for the maintenance of metabolic health. Intestinal dysbiosis was associated with both fatty liver and metabolic disorders development [65]. The analysis of gut microbiota in subjects with MAFLD showed a decrease in Ruminococcaceae family compared to healthy controls, while subjects with NASH presented an increase in
One of the mechanisms proposed to explain the role of impaired gut microbiota in fatty liver disease is the type and amount of short chain fatty acids (SCFAs), precursors of both gluconeogenisis and lipogenesis, which are produced by the fermentation of carbohydrates at the level of the gut, and therefore, are directly affected by gut dysbiosis [59]. SCFAs can regulate gluconeogenesis and lipogenesis, as they have a short bowel transit and increase the rate of nutrient absortion [59, 68].
Dysbiosis is also responsible for an increased intestinal permeability as well as for flaws in intestinal tight junctions, impairments described in individuals with MAFLD [69]. Other proposed mechanisms involve dysfunctions regarding choline, fasting-induced adypocite factor secretion (which induces fat accumulation in both adipocytes and hepatocytes), as well as increased lipopolysaccharide and bile acid production (which promote low grade inflammation) [59]. In summary, impaired gut microbiota is a promoter of MetS, T2D and MAFLD through all these pathways.
4. Cardiovascular risk in persons with insulin resistance, metabolic syndrome and type 2 diabetes
Cardiovascular disease (CVD) accounts for 46.2% of deaths due to non-communicable diseases, being the primary cause of disability and premature death [70]. CVD is the main cause of death in persons with MetS and T2D [71]. By definition, the individuals with MetS have an increased cardiovascular risk, as all components of this disorder are independent risk factors for CVDs; what is more, in MetS the combination of these risk factors further increases both the rates and the severity of CVDs [72]. So, it is not surprising that subjects with MetS and T2D are at risk for coronary atherosclerosis, myocardial infarction, microvascular dysfunctions, cardiac dysfunction and heart failure [72, 73].
Studies showed that persons with MetS often present CVDs. In a study from 2005 [74], it was observed that MetS was a frequent comorbidity in patients with acute myocardial infarction (46%). However, although the medical literature recognizes the high cardiovascular risk of individuals with MetS, there still exists controversy regarding the impact of this syndrome on subjects with CVDs [70]. In this regard, some studies concluded that persons with MetS remain at high cardiovascular risk independent of active management [75], other researches evidenced that the main prognosis factor for myocardial infarction in individuals with MetS is the presence of heart failure [76], while the study by van Kuijk et al. [77] concluded that MetS is not associated with an increase in mortality in individuals with CVDs. Starting from these premises and taking into account that the majority of the studies have assessed the impact of MetS on the prognosis and progression of CVDs, a meta-analysis from 2022 aimed to evaluate the associations between MetS, using different definitions, and the risk of cardiovascular events, cardiovascular death and all cause mortality [70]. The analysis of cardiovascular events in persons with MetS was analyzed based on the results reported by 23 studies totalizing a number of 77,125 subjects and proved that the risk of myocardial infarction is statistically significant higher in individuals with MetS [RR = 1.460, 95% CI (1.242, 1.716), P = 0.000] [70]. The risk of stroke, analyzed in 60,297 subjects, was also higher for the persons with MetS (RR = 1.435, 95% CI (1.131, 1.820), P = 0.000) [70]. Regarding all cause mortality, the results of this meta-analysis including over 145,000 subjects with MetS demonstrated an increase of all cause mortality in these individuals (RR = 1.220, 95% CI (1.103, 1.349), P = 0.000) [70].
Persons with T2D have a high risk of developing CVDs, that have higher prevalence rates in T2D compared with the general population and, what is more, there is an increase in the cardiovascular risk parallel to the fasting plasma glucose levels, starting from the stage of prediabetes/impaired fasting glucose [78, 79]. Fortunately, although the prevalence of T2D is increasing, in the last two decades there was observed a decrease in the incidence of CVDs and cardiovascular mortality in these individuals, compared to the general population [78]. A declining pattern was observed in many countries (Canada, United States, Sweden, United Kingdom, Korea) [80, 81, 82, 83, 84, 85]. However, even though the overall prevalence of CVDs in persons with T2D is decreasing, a new phenomenon was observed in these subjects, an increase in the prevalence of heart failure [78, 86]. A possible explanation might be the increase in life expectancy that came with the advances in diabetes therapeutic options [78, 86, 87].
The physiopathological mechanisms that link MetS, T2D and CVDs are complex and multifactorial, including IR, low grade inflammation and neurohormonal activation [88, 89]. The epicardial fat accumulation observed in persons with obesity is accountable for lipolysis, FFA release and reactive inflammation [88]. Additionally, obesity is also associated with increased oxidative stress, mitochondria being the main source of reactive oxygen species (ROS), mechanisms through which MetS leads to the development and progression of T2D, atherosclerosis and CVDs [90, 91]. MetS is characterized by a chronic increase in adiposity which can lead to important neuro-hormonal changes in the cardiovascular system, including activation of the renin-angiotensin-aldosterone system, increased proinflammatory cytokines, altered adipokines levels, as well as activation of the sympathetic nervous system [72]. The latter can lead to vasoconstriction, tachycardia, as well as metabolic abnormalities, such as excess lipolysis with an increase in FFA, peripheral and hepatic IR [72].
The relationship between IR and cardiovascular risk is well recognized by many studies, being considered as a major predictor of atherosclerotic disease [78, 92, 93, 94]. Recent studies [95] describe the role of IR at the myocyte level as a main contributor to the appearance of cardiomyopathy in persons with MetS and T2D, giving this way a possible explication to the increase in heart failure observed in persons with T2D.
Insulin signaling is mainly described in insulin dependent cells, as the activation of these pathways play important roles in cell metabolism, as well as cell growth and survival. Although the myocytes are not insulin dependent, insulin signaling is involved in maintaining normal heart functions, as studies have shown that these pathways are important under stress conditions, transcending the metabolic effects of insulin [95, 96]. Systemic IR, increased circulating FFA, both characteristic of MetS and T2D are described as major contributors to metabolic cardiomyopathy [97, 98, 99, 100]. Metabolic cardiomyopathy is characterized by hypertrophy and diastolic dysfunction that can progress to overt heart failure, the most frequent being heart failure with preserved ejection fraction (HFpEF) [97, 98, 99]. Although diabetic cardiomyopathy makes the object of many studies, the fact that at the moment there is no accurate definition for this disease, makes the interpretation of different studies more difficult; however, the prevalence of heart failure in persons with T2D was estimated at 10–30%, compared to 12% in the general population [101, 102]. Furthermore, in subjects with heart failure, the prevalence of diabetes was estimated at about 30% and it was observed that these people are generally younger and also the presence of T2D is associated with a worse prognosis in the case of heart failure with reduced ejection fraction (HFrEF), having a 75% higher mortality and increased risk of hospitalization [101, 102, 103, 104]. For the clinical practice it is very important to know that a good metabolic control can reduce the risk of developing heart failure, studies showing that a reduction of HbA1c of only 1% can lead to a 16% reduction of developing heart failure in persons with T2D [105].
5. Prevention strategies and therapeutic options
The main cause for the continuous increase in the prevalence of metabolic diseases (obesity, MetS, T2D) is the unhealthy lifestyle, consisting of high caloric intake and sedentarism. Therefore, the optimization of lifestyle can be an effective tool in preventing or delaying the onset of metabolic diseases. In individuals with T2D, weight loss of about 3–10% can bring benefits on the glycemic control [106]. Furthermore, weight loss can be associated with T2D remission, which makes dietary intervention very important for the correct managing of T2D [107]. Currently, many diets were proposed for weight control, amidst them Mediterranean diet, continuous energy-restricted diet (CERD), low carbohydrate diet, ketogenic diet, vegetarian diet, intermittent fasting (IF), etc. A type of diet still controversial in persons with T2D, despite studies showing improvements in metabolism, is CERD, as most guidelines recommend balanced diets with moderate calorie restriction [108, 109, 110].
One of the most discussed diets in chronic non-communicable disease is calorie restriction (CR), without malnutrition, this type of diet has increased life span and has impeded the development of age-related disorders in many animal models, including primates [111]. CR is defined by the reduction of caloric intake by 25–30%, maintaining all the essential nutrients [111]. In humans, moderate CR studies assessing longevity are still ongoing, while many other benefits are already recognized [112, 113]. Table 1 summarizes the benefic effects of CR in subjects with metabolic disorders.
| Parameter | Effects |
|---|---|
| Body mass |
|
| Body composition |
|
| Glycemic metabolism |
|
| Lipid metabolism |
|
| Inflammation |
|
| Other benefits |
|
Table 1.
In a recent meta-analysis that compared the safety of IF and CERD in subjects with MeTS and T2D, including four studies with a total of 355 individuals, it was demonstrated that the two diets had similar effects in term of glycemic control, while IF had a better effect on weight loss [107]. The authors concluded that IF represents a safe diet pattern for persons with MetS and T2D, which could be implemented as a part of these individuals’ management [107].
Lifestyle optimization must also address sedentarism. Observational and interventional studies alike have proved the importance of physical activity in mitigating MetS, as each of its components can be favorably influenced by physical exercise [114]. Regular physical activity can induce weight loss, together with a decrease in arterial blood pressure and improvements of lipid and glycemic profiles, through a reduction of IR [115, 116, 117, 118]. Due to all these benefits, persons with MetS and T2D should be encouraged to practice physical activity on a regular basis.
For subjects that require more than lifestyle interventions, there are two more therapeutic options that can help with weight loss and reduction in metabolic complication: bariatric surgery and pharmacological agents. Newer antidiabetic drugs, such as SGLT2 inhibitors and GLP-1 receptor agonists have demonstrated cardiovascular benefits beyond glycemic control in these persons [119, 120].
6. Conclusion
Metabolic diseases, particularly MetS and T2D have continuous increasing prevalence rates and have negative impact on the individual’s quality of life, bearing in the same time an important social and economic burden for the society. Therefore, it is imperious that prevention measures must be taken globally, as well as developing new strategies for the better control and delayed progression of these disorders, through lifestyle optimization and, when necessary, pharmacologic interventions.
References
- 1.
Hudish LI, Reusch JE, Sussel L. β cell dysfunction during progression of metabolic syndrome to type 2 diabetes. The Journal of Clinical Investigation. 2019; 129 :4001-4008. DOI: 10.1172/JCI129188 - 2.
Gemeda D, Abebe E, Duguma A. Metabolic syndrome and its associated factors among type 2 diabetic patients in Southwest Ethiopia, 2021/2022. Journal Diabetes Research. 2022; 2022 :8162342. DOI: 10.1155/2022/8162342 - 3.
Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: A joint interim statement of the international diabetes federation task force on epidemiology and prevention; National Heart, Lung, and Blood Institute; American Heart Association; world heart federation; international atherosclerosis society; and International Association for the Study of obesity. Circulation. 2009; 120 :1640-1645. DOI: 10.1161/CIRCULATIONAHA.109.192644 - 4.
de Siqueira Valadares LT, de Souza LSB, Salgado Júnior VA, de Freitas BL, de Macedo LR, Silva M. Prevalence of metabolic syndrome in Brazilian adults in the last 10 years: A systematic review and meta-analysis. BMC Public Health. 2022; 22 :327. DOI: 10.1186/s12889-022-12753-5 - 5.
Engin A. The definition and prevalence of obesity and metabolic syndrome. Advances in Experimental Medicine and Biology. 2017; 960 :1-17. DOI: 10.1007/978-3-319-48382-5_1 - 6.
Weihe P, Weihrauch-Blüher S. Metabolic syndrome in children and adolescents: Diagnostic criteria, therapeutic options and perspectives. Current Obesity Reports. 2019; 8 :472-479. DOI: 10.1007/s13679-019-00357-x - 7.
Weghuber D, Zelzer S, Stelzer I, Paulmichl K, Kammerhofer D, Schnedl W, et al. "metabolically healthy" phenotype in juvenile obesity - neck subcutaneous adipose tissue and serum uric acid are clinically relevant. Experimental and Clinical Endocrinology & Diabetes. 2013; 121 :384-390. DOI: 10.1055/s-0033-1341440 - 8.
World Health Organization. Obesity and Overweight [Internet]. 2021. Available from: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight [Accessed: June 6, 2023] - 9.
International Diabetes Federation. IDF Diabetes Atlas. 10th ed. Brussels, Belgium: International Diabetes Federation; 2021 - 10.
Onyango AN. Cellular stresses and stress responses in the pathogenesis of insulin resistance. Oxidative Medicine and Cellular Longevity. 2018; 2018 :4321714. DOI: 10.1155/2018/4321714 - 11.
Reaven GM. Pathophysiology of insulin resistance in human disease. Physiological Reviews. 1995; 75 :473-486. DOI: 10.1152/physrev.1995.75.3.473 - 12.
Ighbariya A, Weiss R. Insulin resistance, prediabetes, metabolic syndrome: What should every pediatrician know? Journal of Clinical Research in Pediatric Endocrinology. 2017; 9 :49-57. DOI: 10.4274/jcrpe.2017.S005 - 13.
Kim JK. Hyperinsulinemic-euglycemic clamp to assess insulin sensitivity in vivo. Methods in Molecular Biology. 2009; 560 :221-238. DOI: 10.1007/978-1-59745-448-3_15 - 14.
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985; 28 :412-419. DOI: 10.1007/BF00280883 - 15.
Hrebícek J, Janout V, Malincíková J, Horáková D, Cízek L. Detection of insulin resistance by simple quantitative insulin sensitivity check index QUICKI for epidemiological assessment and prevention. The Journal of Clinical Endocrinology and Metabolism. 2002; 87 :144-147. DOI: 10.1210/jcem.87.1.8292 - 16.
Son DH, Lee HS, Lee YJ, Lee JH, Han JH. Comparison of triglyceride-glucose index and HOMA-IR for predicting prevalence and incidence of metabolic syndrome. Nutrition, Metabolism, and Cardiovascular Diseases. 2022; 32 :596-604. DOI: 10.1016/j.numecd.2021.11.017 - 17.
Earnest CP, Johannsen NM, Swift DL, Gillison FB, Mikus CR, Lucia A, et al. Aerobic and strength training in concomitant metabolic syndrome and type 2 diabetes. Medicine and Science in Sports and Exercise. 2014; 46 :1293-1301. DOI: 10.1249/MSS.0000000000000242 - 18.
Regufe VMG, Pinto CMCB, Perez PMVHC. Metabolic syndrome in type 2 diabetic patients: A review of current evidence. Porto Biomedical Journal. 2020; 5 :e101. DOI: 10.1097/j.pbj.0000000000000101 - 19.
Gupta D et al. Temporal characterization of β cell-adaptive and -maladaptive mechanisms during chronic high-fat feeding in C57BL/6NTac mice. The Journal of Biological Chemistry. 2017; 292 :12449-12459. DOI: 10.1074/jbc.M117.781047 - 20.
Halban PA et al. β-Cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care. 2014; 37 :1751-1758. DOI: 10.2337/dc14-0396 - 21.
Yabe D, Seino Y, Fukushima M, Seino S. β cell dysfunction versus insulin resistance in the pathogenesis of type 2 diabetes in east Asians. Current Diabetes Reports. 2015; 15 :602. DOI: 10.1007/s11892-015-0602-9 - 22.
Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. The Journal of Clinical Investigation. 2006; 116 :1802-1812. DOI: 10.1172/JCI29103 - 23.
Chen C, Cohrs CM, Stertmann J, Bozsak R, Speier S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Molecular Metabolism. 2017; 6 :943-957. DOI: 10.1016/j.molmet.2017.06.019 - 24.
Rabhi N, Salas E, Froguel P, Annicotte JS. Role of the unfolded protein response in beta cell compensation and failure during diabetes. Journal Diabetes Research. 2014; 2014 :795171. DOI: 10.1155/2014/795171 - 25.
Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocrine Reviews. 2008; 29 :42-61. DOI: 10.1210/er.2007-0015 - 26.
Marchetti P, Bugliani M, De Tata V, Suleiman M, Marselli L. Pancreatic beta cell identity in humans and the role of type 2 diabetes. Frontiers in Cell and Development Biology. 2017; 5 :55. DOI: 10.3389/fcell.2017.00055 - 27.
Supale S, Li N, Brun T, Maechler P. Mitochondrial dysfunction in pancreatic β cells. Trends in Endocrinology and Metabolism. 2012; 23 :477-487. DOI: 10.1016/j.tem.2012.06.002 - 28.
Gerber PA, Rutter GA. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxidants & Redox Signaling. 2017; 26 :501-518. DOI: 10.1089/ars.2016.6755 - 29.
Remedi MS, Emfinger C. Pancreatic β-cell identity in diabetes. Diabetes, Obesity & Metabolism. 2016; 18 :110-116. DOI: 10.1111/dom.12727 - 30.
Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012; 150 :1223-1234. DOI: 10.1016/j.cell.2012.07.029 - 31.
Bensellam M, Jonas JC, Laybutt DR. Mechanisms of β-cell dedifferentiation in diabetes: Recent findings and future research directions. The Journal of Endocrinology. 2018; 236 :R109-R143. DOI: 10.1530/JOE-17-0516 - 32.
Ediger BN et al. LIM domain-binding 1 maintains the terminally differentiated state of pancreatic β cells. The Journal of Clinical Investigation. 2017; 127 :215-229. DOI: 10.1172/JCI88016 - 33.
Neelankal John A, Ram R, Jiang FX. RNA-Seq analysis of islets to characterise the dedifferentiation in type 2 diabetes model mice db/db. Endocrine Pathology. 2018; 29 :207-221. DOI: 10.1007/s12022-018-9523-x - 34.
Sun J et al. β-Cell dedifferentiation in patients with T2D with adequate glucose control and nondiabetic chronic pancreatitis. The Journal of Clinical Endocrinology and Metabolism. 2019; 104 :83-94 - 35.
Diedisheim M et al. Modeling human pancreatic beta cell dedifferentiation. Molecular Metabolism. 2018; 10 :74-86. DOI: 10.1016/j.molmet.2018.02.002 - 36.
Cinti F et al. Evidence of β-cell dedifferentiation in human type 2 diabetes. The Journal of Clinical Endocrinology and Metabolism. 2016; 101 :1044-1054. DOI: 10.1210/jc.2015-2860 - 37.
Gutierrez GD, Gromada J, Sussel L. Heterogeneity of the pancreatic beta cell. Frontiers in Genetics. 2017; 8 :22. DOI: 10.3389/fgene.2017.00022 - 38.
Swisa A et al. PAX6 maintains β cell identity by repressing genes of alternative islet cell types. The Journal of Clinical Investigation. 2017; 127 :230-243. DOI: 10.1172/JCI88015 - 39.
Gao T et al. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metabolism. 2014; 19 :259-271. DOI: 10.1016/j.cmet.2013.12.002 - 40.
Nolan CJ, Prentki M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: Time for a conceptual framework shift. Diabetes & Vascular Disease Research. 2019; 16 :118-127. DOI: 10.1177/1479164119827611 - 41.
Erion K, Corkey BE. β-Cell failure or β-cell abuse? Frontiers in Endocrinology (Lausanne). 2018; 9 :532. DOI: 10.3389/fendo.2018.00532 - 42.
Ferrannini E, Gastaldelli A, Miyazaki Y, Matsuda M, Mari A, DeFronzo RA. β-Cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: A new analysis. The Journal of Clinical Endocrinology and Metabolism. 2005; 90 (1):493-500. DOI: 10.1210/jc.2004-1133 - 43.
Malin SK, Kullman EL, Scelsi AR, Godin JP, Ross AB, Kirwan JP. A whole-grain diet increases glucose-stimulated insulin secretion independent of gut hormones in adults at risk for type 2 diabetes. Molecular Nutrition & Food Research. 2019; 63 :e1800967. DOI: 10.1002/mnfr.201800967 - 44.
Malin SK et al. Impact of short-term exercise training intensity on β-cell function in older obese adults with prediabetes. Journal of Applied Physiology. 2018; 125 :1979-1986. DOI: 10.1152/japplphysiol.00680.2018 - 45.
Kaya E, Yilmaz Y. Metabolic-associated fatty liver disease (MAFLD): A multi-systemic disease beyond the liver. Journal of Clinical and Translational Hepatology. 2022; 10 :329-338. DOI: 10.14218/JCTH.2021.00178 - 46.
Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic Steatohepatitis. Hepatology. 2019; 69 :2672-2682. DOI: 10.1002/hep.30251 - 47.
Amzolini AM, Forţofoiu MC, Barău Abu-Alhija A, Vladu IM, Clenciu D, Mitrea A, et al. Triglyceride and glucose index: A useful tool for non-alcoholic liver disease assessed by liver biopsy in patients with metabolic syndrome? Romanian Journal of Morphology and Embryology. 2021; 62 :475-480. DOI: 10.47162/RJME.62.2.13 - 48.
Davis TME. Diabetes and metabolic dysfunction-associated fatty liver disease. Metabolism. 2021; 123 :154868. DOI: 10.1016/j.metabol.2021.154868 - 49.
Micu ES, Amzolini AM, Barău Abu-Alhija A, Forţofoiu MC, Vladu IM, Clenciu D, et al. Systemic and adipose tissue inflammation in NASH: Correlations with histopathological aspects. Romanian Journal of Morphology and Embryology. 2021; 62 :509-515. DOI: 10.47162/RJME.62.2.17 - 50.
Ballestri S, Zona S, Targher G, Romagnoli D, Baldelli E, Nascimbeni F, et al. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. Journal of Gastroenterology and Hepatology. 2016; 31 :936-944. DOI: 10.1111/jgh.13264 - 51.
Muzurović E, Mikhailidis DP, Mantzoros C. Non-alcoholic fatty liver disease, insulin resistance, metabolic syndrome and their association with vascular risk. Metabolism. 2021; 119 :154770. DOI: 10.1016/j.metabol.2021.154770 - 52.
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016; 64 :73-84. DOI: 10.1002/hep.28431 - 53.
Masarone M, Rosato V, Aglitti A, Bucci T, Caruso R, Salvatore T, et al. Liver biopsy in type 2 diabetes mellitus: Steatohepatitis represents the sole feature of liver damage. PLoS One. 2017; 12 :e0178473. DOI: 10.1371/journal.pone.0178473 - 54.
Eslam M, Sanyal AJ, George J. International consensus panel. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 2020; 158 :1999-2014.e1 - 55.
Efrem IC, Moța M, Vladu IM, Mitrea A, Clenciu D, Timofticiuc DCP, et al. A study of biomarkers associated with metabolic dysfunction-associated fatty liver disease in patients with type 2 diabetes. Diagnostics (Basel). 2022; 12 :2426. DOI: 10.3390/diagnostics12102426 - 56.
Sakurai Y, Kubota N, Yamauchi T, Kadowaki T. Role of insulin resistance in MAFLD. International Journal of Molecular Sciences. 2021; 22 :4156. DOI: 10.3390/ijms22084156 - 57.
Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. Journal of Hepatology. 2020; 73 :202-209 - 58.
Yilmaz Y, Byrne CD, Musso G. A single-letter change in an acronym: Signals, reasons, promises, challenges, and steps ahead for moving from NAFLD to MAFLD. Expert Review of Gastroenterology & Hepatology. 2021; 15 :345-352. DOI: 10.1080/17474124.2021 - 59.
Lim S, Kim JW, Targher G. Links between metabolic syndrome and metabolic dysfunction-associated fatty liver disease. Trends in Endocrinology and Metabolism. 2021; 32 :500-514. DOI: 10.1016/j.tem.2021.04.008 - 60.
Lim S, Taskinen MR, Borén J. Crosstalk between nonalcoholic fatty liver disease and cardiometabolic syndrome. Obesity Reviews. 2019; 20 :599-611. DOI: 10.1111/obr.12820 - 61.
Lim S, Meigs JB. Links between ectopic fat and vascular disease in humans. Arteriosclerosis, Thrombosis, and Vascular Biology. 2014; 34 :1820-1826. DOI: 10.1161/ATVBAHA.114.303035 - 62.
Vilar-Gomez E, Calzadilla-Bertot L, Wai-Sun Wong V, Castellanos M, Aller-de la Fuente R, Metwally M, et al. Fibrosis severity as a determinant of cause-specific mortality in patients with advanced nonalcoholic fatty liver disease: A multi-National Cohort Study. Gastroenterology. 2018; 155 :443-457.e17. DOI: 10.1053/j.gastro.2018.04.034 - 63.
Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: Current concepts and future challenges. Nature Reviews. Gastroenterology & Hepatology. 2019; 16 :411-428. DOI: 10.1038/s41575-019-0145-7 - 64.
Cotter TG, Rinella M. Nonalcoholic fatty liver disease 2020: The state of the disease. Gastroenterology. 2020; 158 :1851-1864. DOI: 10.1053/j.gastro.2020.01.052 - 65.
Sharpton SR, Schnabl B, Knight R, Loomba R. Current concepts, opportunities, and challenges of gut microbiome-based personalized medicine in nonalcoholic fatty liver disease. Cell Metabolism. 2021; 33 :21-32. DOI: 10.1016/j.cmet.2020.11.010 - 66.
Raman M, Ahmed I, Gillevet PM, Probert CS, Ratcliffe NM, Smith S, et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clinical Gastroenterology and Hepatology. 2013; 11 :868-75.e1-3. DOI: 10.1016/j.cgh.2013.02.015 - 67.
Mouzaki M, Comelli EM, Arendt BM, Bonengel J, Fung SK, Fischer SE, et al. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology. 2013; 58 :120-127. DOI: 10.1002/hep.26319 - 68.
Aguirre M, Bussolo de Souza C, Venema K. The gut microbiota from lean and obese subjects contribute differently to the fermentation of arabinogalactan and inulin. PLoS One. 2016; 11 :e0159236. DOI: 10.1371/journal.pone.0159236 - 69.
Mao JW, Tang HY, Zhao T, Tan XY, Bi J, Wang BY, et al. Intestinal mucosal barrier dysfunction participates in the progress of nonalcoholic fatty liver disease. International Journal of Clinical and Experimental Pathology. 2015; 8 (4):3648-3658 - 70.
Li X, Zhai Y, Zhao J, He H, Li Y, Liu Y, et al. Impact of metabolic syndrome and It's components on prognosis in patients with cardiovascular diseases: A meta-analysis. Frontiers in Cardiovascular Medicine. 2021; 8 :704145. DOI: 10.3389/fcvm.2021.704145 - 71.
Bornachea O, Vea A, Llorente-Cortes V. Interplay between epicardial adipose tissue, metabolic and cardiovascular diseases. Clinic and Research in Arteriosclerosis. 2018; 30 :230-239. DOI: 10.1016/j.arteri.2018.03.003 - 72.
Tune JD, Goodwill AG, Sassoon DJ, Mather KJ. Cardiovascular consequences of metabolic syndrome. Translational Research. 2017; 183 :57-70. DOI: 10.1016/j.trsl.2017.01.001 - 73.
Bajaj NS, Osborne MT, Gupta A, Tavakkoli A, Bravo PE, Vita T, et al. Coronary microvascular dysfunction and cardiovascular risk in obese patients. Journal of the American College of Cardiology. 2018; 72 :707-717. DOI: 10.1016/j.jacc.2018.05.049 - 74.
Zeller M, Steg PG, Ravisy J, Laurent Y, Janin-Manificat L, L'Huillier I, et al. Prevalence and impact of metabolic syndrome on hospital outcomes in acute myocardial infarction. Archives of Internal Medicine. 2005; 165 :1192-1198. DOI: 10.1001/archinte.165.10.1192 - 75.
Boulon C, Lafitte M, Richeboeuf V, Paviot B, Pradeau V, Coste P, et al. Prevalence of metabolic syndrome after acute coronary syndrome and its prognostic significance. The American Journal of Cardiology. 2006; 98 :1429-1434. DOI: 10.1016/j.amjcard.2006.07.025 - 76.
Selcuk H, Temizhan A, Selcuk MT, Sen T, Maden O, Tekeli S, et al. Impact of metabolic syndrome on future cardiovascular events in patients with first acute myocardial infarction. Coronary Artery Disease. 2009; 20 :370-375. DOI: 10.1097/MCA.0b013e32832ed31e - 77.
van Kuijk JP, Flu WJ, Chonchol M, Bax JJ, Verhagen JM, Poldermans HD. Metabolic syndrome is an independent predictor of cardiovascular events in high-risk patients with occlusive and aneurysmatic peripheral arterial disease. Atherosclerosis. 2010; 210 :596-601. DOI: 10.1016/j.atherosclerosis.2009.12.018 - 78.
Yun JS, Ko SH. Current trends in epidemiology of cardiovascular disease and cardiovascular risk management in type 2 diabetes. Metabolism. 2021; 123 :154838. DOI: 10.1016/j.metabol.2021.154838 - 79.
Danaei G, Lawes CM, Vander Hoorn S, Murray CJ, Ezzati M. Global and regional mortality from ischaemic heart disease and stroke attributable to higher-than-optimum blood glucose concentration: Comparative risk assessment. Lancet. 2006; 368 :1651-1659. DOI: 10.1016/S0140-6736(06)69700-6 - 80.
Rawshani A, Rawshani A, Franzén S, Eliasson B, Svensson AM, Miftaraj M, et al. Mortality and cardiovascular disease in type 1 and type 2 diabetes. The New England Journal of Medicine. 2017; 376 :1407-1418. DOI: 10.1056/NEJMoa1608664 - 81.
Booth GL, Kapral MK, Fung K, Tu JV. Recent trends in cardiovascular complications among men and women with and without diabetes. Diabetes Care. 2006; 29 :32-37. DOI: 10.2337/diacare.29.01.06.dc05-0776 - 82.
Gregg EW, Cheng YJ, Srinivasan M, Lin J, Geiss LS, Albright AL, et al. Trends in cause-specific mortality among adults with and without diagnosed diabetes in the USA: An epidemiological analysis of linked national survey and vital statistics data. Lancet. 2018; 391 :2430-2440. DOI: 10.1016/S0140-6736(18)30314-3 - 83.
Lind M, Garcia-Rodriguez LA, Booth GL, Cea-Soriano L, Shah BR, Ekeroth G, et al. Mortality trends in patients with and without diabetes in Ontario, Canada and the UK from 1996 to 2009: A population-based study. Diabetologia. 2013; 56 :2601-2608. DOI: 10.1007/s00125-013-3063-1 - 84.
Jung CH, Chung JO, Han K, Ko SH, Ko KS, Park JY, et al. Improved trends in cardiovascular complications among subjects with type 2 diabetes in Korea: A nationwide study (2006-2013). Cardiovascular Diabetology. 2017; 16 :1. DOI: 10.1186/s12933-016-0482-6 - 85.
Park JH, Ha KH, Kim BY, Lee JH, Kim DJ. Trends in cardiovascular complications and mortality among patients with diabetes in South Korea. Diabetes and Metabolism Journal. 2021; 45 :120-124. DOI: 10.4093/dmj.2020.0175 - 86.
Cheng YJ, Imperatore G, Geiss LS, Saydah SH, Albright AL, Ali MK, et al. Trends and disparities in cardiovascular mortality among U.S. adults with and without self-reported diabetes, 1988-2015. Diabetes Care. 2018; 41 :2306-2315. DOI: 10.2337/dc18-0831 - 87.
Wright AK, Suarez-Ortegon MF, Read SH, Kontopantelis E, Buchan I, Emsley R, et al. Risk factor control and cardiovascular event risk in people with type 2 diabetes in primary and secondary prevention settings. Circulation. 2020; 142 :1925-1936. DOI: 10.1161/CIRCULATIONAHA.120.046783 - 88.
Martinelli I, Tomassoni D, Moruzzi M, Roy P, Cifani C, Amenta F, et al. Cardiovascular changes related to metabolic syndrome: Evidence in obese Zucker rats. International Journal of Molecular Sciences. 2020; 21 :2035. DOI: 10.3390/ijms21062035 - 89.
Rochlani Y, Pothineni NV, Kovelamudi S, Mehta JL. Metabolic syndrome: Pathophysiology, management, and modulation by natural compounds. Therapeutic Advances in Cardiovascular Disease. 2017; 11 :215-225. DOI: 10.1177/1753944717711379 - 90.
Marseglia L, Manti S, D’Angelo G, Nicotera A, Parisi E, Di Rosa G, et al. Oxidative stress in obesity: A critical component in human diseases. International Journal of Molecular Sciences. 2014; 16 :378-400. DOI: 10.3390/ijms16010378 - 91.
Steven S, Frenis K, Oelze M, Kalinovic S, Kuntic M, Bayo Jimenez MT, et al. Vascular inflammation and oxidative stress: Major triggers for cardiovascular disease. Oxidative Medicine and Cellular Longevity. 2019; 2019 :7092151. DOI: 10.1155/2019/7092151 - 92.
Koenen M, Hill MA, Cohen P, Sowers JR. Obesity, adipose tissue and vascular dysfunction. Circulation Research. 2021; 128 :951-968. DOI: 10.1161/CIRCRESAHA.121.318093 - 93.
Franks PW, Hanson RL, Knowler WC, Sievers ML, Bennett PH, Looker HC. Childhood obesity, other cardiovascular risk factors, and premature death. The New England Journal of Medicine. 2010; 362 :485-493. DOI: 10.1056/NEJMoa0904130 - 94.
Lee MJ, Wu Y, Fried SK. Adipose tissue heterogeneity: Implication of depot differences in adipose tissue for obesity complications. Molecular Aspects of Medicine. 2013; 34 :1-11. DOI: 10.1016/j.mam.2012.10.001 - 95.
Abel ED. Insulin signaling in the heart. American Journal of Physiology. Endocrinology and Metabolism. 2021; 321 :E130-E145. DOI: 10.1152/ajpendo.00158.2021 - 96.
Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. The Journal of Clinical Investigation. 2002; 109 :629-639. DOI: 10.1172/JCI13946 - 97.
Capone F, Vettor R, Schiattarella GG. Cardiometabolic HFpEF: NASH of the heart. Circulation. 2023; 147 :451-453. DOI: 10.1161/CIRCULATIONAHA.122.062874 - 98.
Nakamura M, Sadoshima J. Cardiomyopathy in obesity, insulin resistance and diabetes. The Journal of Physiology. 2020; 598 :2977-2993. DOI: 10.1113/JP276747 - 99.
Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: A hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia. 2018; 61 :21-28. DOI: 10.1007/s00125-017-4390-4 - 100.
Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nature Reviews. Endocrinology. 2016; 12 :144-153. DOI: 10.1038/nrendo.2015.216 - 101.
Seferović PM, Petrie MC, Filippatos GS, Anker SD, Rosano G, Bauersachs J, et al. Type 2 diabetes mellitus and heart failure: A position statement from the heart failure Association of the European Society of cardiology. European Journal of Heart Failure. 2018; 20 :853-872. DOI: 10.1002/ejhf.1170 - 102.
Lorenzo-Almorós A, Cepeda-Rodrigo JM, Lorenzo Ó. Diabetic cardiomyopathy. Revista Clínica Española (Barc). 2022; 222 :100-111. DOI: 10.1016/j.rceng.2019.10.012 - 103.
Nichols GA, Hillier TA, Erbey JR, Brown JB. Congestive heart failure in type 2 diabetes: Prevalence, incidence, and risk factors. Diabetes Care. 2001; 24 :1614-1619. DOI: 10.2337/diacare.24.9.1614 - 104.
Sarma S, Mentz RJ, Kwasny MJ, Fought AJ, Huffman M, Subacius H, et al. Association between diabetes mellitus and post-discharge outcomes in patients hospitalized with heart failure: Findings from the EVEREST trial. European Journal of Heart Failure. 2013; 15 :194-202. DOI: 10.1093/eurjhf/hfs153 - 105.
Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): Prospective observational study. BMJ. 2000; 321 :405-412. DOI: 10.1136/bmj.321.7258.405 - 106.
Reber E, Norman K, Endrich O, Schuetz P, Frei A, Stanga Z. Economic challenges in nutritional management. Journal of Clinical Medicine. 2019; 8 :1005. DOI: 10.3390/jcm8071005 - 107.
Wang X, Li Q , Liu Y, Jiang H, Chen W. Intermittent fasting versus continuous energy-restricted diet for patients with type 2 diabetes mellitus and metabolic syndrome for glycemic control: A systematic review and meta-analysis of randomized controlled trials. Diabetes Research and Clinical Practice. 2021; 179 :109003. DOI: 10.1016/j.diabres.2021.109003 - 108.
Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009; 325 :201-204. DOI: 10.1126/science.1173635 - 109.
Das SK, Roberts SB, Bhapkar MV, Villareal DT, Fontana L, Martin CK, et al. Body-composition changes in the comprehensive assessment of long-term effects of reducing intake of energy (CALERIE)-2 study: A 2-y randomized controlled trial of calorie restriction in nonobese humans. The American Journal of Clinical Nutrition. 2017; 105 :913-927. DOI: 10.3945/ajcn.116.137232 - 110.
Johnson ML, Distelmaier K, Lanza IR, Irving BA, Robinson MM, Konopka AR, et al. Mechanism by which caloric restriction improves insulin sensitivity in sedentary obese adults. Diabetes. 2016; 65 :74-84. DOI: 10.2337/db15-0675 - 111.
Pignatti C, D’Adamo S, Stefanelli C, Flamigni F, Cetrullo S. Nutrients and pathways that regulate health span and life span. Geriatrics. 2020; 5 :95. DOI: 10.3390/geriatrics5040095 - 112.
Fontana L. The scientific basis of caloric restriction leading to longer life. Current Opinion in Gastroenterology. 2009; 25 :144-150. DOI: 10.1097/MOG.0b013e32831ef1ba - 113.
Redman LM, Fontana L. Calorie restriction in humans: An update. Ageing Research Reviews. 2017; 39 :36-45. DOI: 10.1016/j.arr.2016.08.005 - 114.
Myers J, Kokkinos P, Nyelin E. Physical activity, cardiorespiratory fitness, and the metabolic syndrome. Nutrients. 2019; 11 :1652. DOI: 10.3390/nu11071652 - 115.
Pucci G. Sex- and gender-related prevalence, cardiovascular risk and therapeutic approach in metabolic syndrome: A review of the literature. Pharmacological Research. 2017; 120 :34-42. DOI: 10.1016/j.phrs.2017.03.008 - 116.
Sallis RE, Matuszak JM, Baggish AL, Franklin BA, Chodzko-Zajko W, Fletcher BJ, et al. Call to action on making physical activity assessment and prescription a medical standard of care. Current Sports Medicine Reports. 2016; 15 :207-214. DOI: 10.1249/JSR.0000000000000249 - 117.
Berra K, Rippe J, Manson JE. Making physical activity counseling a priority in clinical practice: The time for action is now. Journal of the American Medical Association. 2015; 314 :2617-2618. DOI: 10.1001/jama.2015.16244 - 118.
Omura JD, Bellissimo MP, Watson KB, Loustalot F, Fulton JE, Carlson SE. Primary care providers’ physical activity counseling and referral practices and barriers for cardiovascular disease prevention. Preventive Medicine. 2018; 108 :115-122. DOI: 10.1016/j.ypmed.2017.12.030 - 119.
Brown E, Heerspink HJL, Cuthbertson DJ, Wilding JPH. SGLT2 inhibitors and GLP-1 receptor agonists: Established and emerging indications. Lancet. 2021; 398 :262-276. DOI: 10.1016/S0140-6736(21)00536-5 - 120.
Cowie MR, Fisher M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nature Reviews. Cardiology. 2020; 17 :761-772. DOI: 10.1038/s41569-020-0406-8