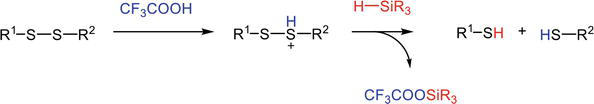Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
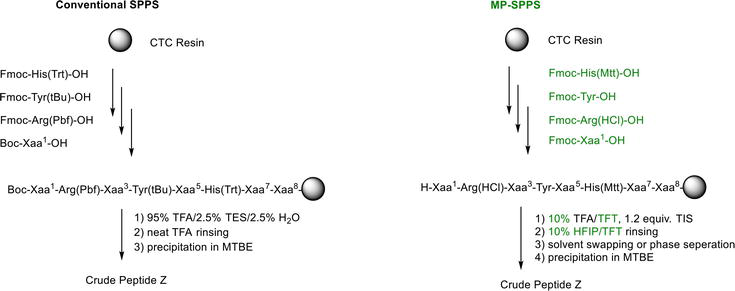
The conventional SPPS (solid-phase peptide synthesis) requires protecting the nucleophilic side chains of amino acids to prevent undesired modifications. A large volume of TFA (trifluoroacetic acid) is generally needed to remove these protecting groups post the peptide assembly. Such a process significantly lowers the productivity of the subject peptide manufacturing and is deemed contradictory to green chemistry concepts. Enabling the side-chain unprotected amino acid couplings should break through such a bottleneck in peptide production and drastically increase productivity. This aspiration creates the concept of MP-SPPS (Minimal-Protection Solid-Phase Peptide Synthesis), accomplished in peptide API (Peptide Z) manufacturing. Side-chain unprotected arginine and tyrosine have been successfully incorporated into the target peptide chain on solid supports. The target peptide Z could be readily obtained by treating the peptidyl resin with a diluted acid solution, that is, 10% TFA/TFT (trifluorotoluene), and precipitating the solid product in a radically reduced volume of anti-solvent. The MP-SPPS process achieves a 5.3-fold increase in peptide Z manufacturing and complies with the philosophy of green chemistry.
Chemical Development, Global Pharmaceutical R&D, Ferring Pharmaceuticals A/S, Denmark
*Address all correspondence to: yi.yang@ferring.com
1. Introduction
To date, green chemistry has focused on improving the safety and environmental sustainability of the manufacturing process, including the organic solvents commonly used in peptide manufacturing. Due to the inherent advantages of their delicate balance to solubilize various compounds, including amino acid building blocks, coupling reagents, reactive intermediates, and by-products, and to properly swell peptidyl resins, DMF (dimethylformamide) and NMP (N-methylpyrrolidone) are routinely utilized as default organic solvents for SPPS (Solid-Phase Peptide Synthesis). Moreover, DMF and NMP lower the occurrence of prevalent side reactions such as amino acid racemization, aspartimide formation, DKP formation, N-terminus capping, and premature peptide cleavage [1]. Despite these merits, DMF and NMP are not deemed green solvents, and alternative organic solvents for peptide synthesis have been widely pursued [2]. No green solvents to date can ensure high purity and excellent yield for any peptide synthesis, which represents one main impediment to their regular utilization for peptide manufacturing [3]. For instance, the target peptide sequence and the inherent side reactions induced during peptide synthesis dictate the choice of the optimal solvent. Consequently, lowering the amount of solvent used in SPPS and developing green solvent applications in peptide synthesis deserves in-depth investigation and synergic contributions from academia and industrial domains. Many insightful papers have been published describing the green solvent performances in peptide chemistry [4, 5, 6, 7]. The utilization of authentic green solvents for peptide manufacturing has yet to become routine in the industry. Given this reality, chemists continuously develop and seek incremental green chemistry strategies.
One major domain to exploit green chemistry concepts in peptide chemistry is to enhance peptide manufacturing productivity. The conventional SPPS strategy adopts stepwise assembly of the side-chain protected amino acid building blocks to solid supports, detaches the assembled peptide off the resin, and removes the side-chain protecting groups with a highly concentrated acid solution. The treated resin is removed, and the crude product is obtained by precipitating the peptide in a large volume of anti-solvent. Such a classic process is frequently restricted by small batch size and low productivity and is deemed contradictory to green chemistry concepts. An innovative MP-SPPS (Minimal-Protection Solid-Phase Peptide Synthesis) process is developed to address the root causes of the low productivity inherent to conventional SPPS. Radical productivity increase is accomplished on a peptide API manufacturing through the MP-SPPS process.
One of the most evident peptide manufacturing issues entangled in adverse ecology effects is its relatively low productivity and high PMI (Product Mass Intensity). A large volume of organic solvents is generally utilized in SPPS, including those applied in the amino acid coupling, resin rinsing, peptide cleavage/global deprotection, and workup.
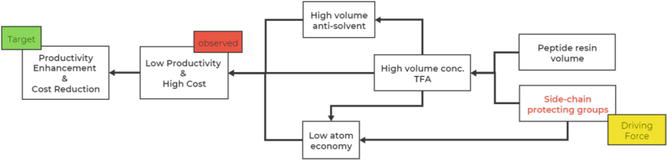
For example, small batch sizes of SPPS leading to poor productivity speaks for a common restriction in peptide industrial manufacturing, which frequently gives rise to the high costs of peptide API manufacturing. A QbD (Quality by Design) tool of the root-cause tree reveals that one of the prime reasons for the low productivity/high cost of peptide manufacturing originates from the amino acid side-chain protection (Figure 1).
Figure 1.
Root-cause tree of the low productivity/high-cost analysis from the conventional SPPS.
Highly concentrated TFA solution is generally applied in the Fmoc-SPPS to cleave the peptides from the solid supports through acidolysis and remove the side-chain protecting groups simultaneously. The physical volume of peptidyl resin and the presence of relatively stable protecting groups, for example, Pbf (2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl) [8], often require 200 equivalent of TFA (sometimes much more) relative to the equivalent of peptide molecule. Industrial setting involving peptide manufacturing precludes the standard evaporation technologies, such as rotary and thin-film evaporation, of highly concentrated TFA mixture, due to the operability, safety, and stability restrictions. The large volume of the derived peptide TFA solution at the step of peptide cleavage/global deprotection thus poses a considerable challenge to the subsequent workup processes. It requests an even greater amount of anti-solvent, such as MTBE (methyl tert-butyl ether), to sufficiently precipitate the crude peptide product from its TFA solution. Generally, the efficient precipitation of the peptide product requisites 3–7 volumes of anti-solvent cf. peptide TFA solution. This setup poses an evident bottleneck to the whole process due to handling the extraordinarily large volume of solutions, and it pronouncedly reduces the productivity of the affected manufacturing, as revealed by Figure 1. Such demands pose a serious burden to the industries. Moreover, the TFA-labile side-chain protecting group’s introduction-removal strategy adversely affects the atom economy of peptide synthesis.
Skipping the side-chain global deprotection step represents one of the potentially viable strategies to minimize the TFA quantity and the volume of the peptide solution prior to the product precipitation. It is not uncommon that the peptide cleavage/global deprotection step poses a bottleneck to the whole process, considerably lowering the batch size and limiting productivity. This artifact predominantly relates to the large volume of TFA applied at the step of peptide cleavage/global deprotection.
The acid-labile side-chain protecting groups requires a sufficiently high concentration of TFA in order to remove them from the masked functional groups quantitatively, and the physical volume of the peptidyl resin brings about the consumption of huge excess of TFA input (generally more than 200 equiv.) at the cleavage/global deprotection step. Incorporating side-chain unprotected residues in SPPS could significantly lower the TFA concentration required to detach the peptide molecules from the super acid-labile resins such as CTC, Sieber, and Ramage resins. Supplementing auxiliary low-boiling point solvents to the cleavage solution could decrease the TFA concentration and ease the following workup by stripping off the solvent under reduced pressure prior to the product precipitation. The resulting concentrated peptide mixture reduces the volume needed for the peptide precipitation step. Such a method improves peptide synthesis productivity and allows much greater batch sizes, which overall lowers manufacturing costs and aligns with green chemistry principles.
Peptide synthesis generally requires the protection of the nucleophilic Nα and the side chain of the amino acid building block. The Nα-protecting groups, generally Fmoc in Fmoc/tBu SPPS strategy, are temporarily used and removed post the amino acid coupling. The side-chain protecting groups, on the contrary, remain anchored on the protected functional groups during the peptide solid-phase synthesis and are simultaneously removed during or post peptide cleavage [9].
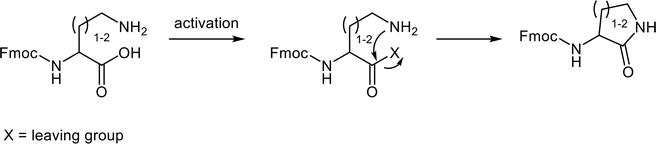
Protecting groups shield the nucleophilic reactivity from the side chains’ functional group on the peptide residues or the incoming reactive amino acid intermediate. Such nucleophilic groups could react with the electrophilic coupling agent or the incoming reactive amino acid intermediate present during the peptide bond formation. Moreover, an intramolecular reaction could occur if the side-chain nucleophilic group remains unmasked. This undesired conversion readily affects the Cγ- and Cδ-substituted amino acid derivatives such as Dab (2,4-Diaminobutyric acid), Orn, and Arg (Figure 2).
Figure 2.
Intramolecular cyclization of side-chain unprotected Cγ- and Cδ-substituted amino acid derivatives after carboxylate-activation.
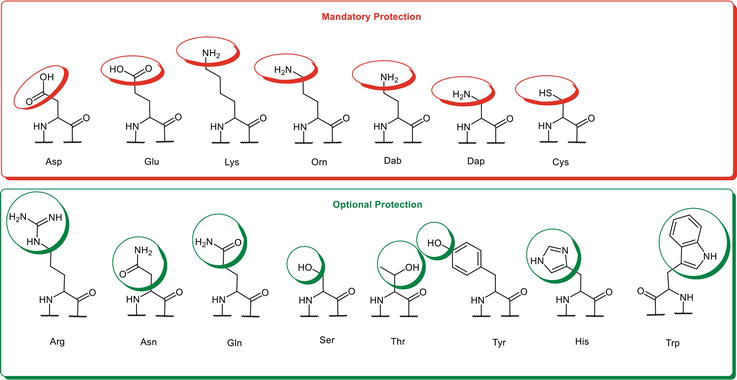
Consequently, mandatory protection applies to Nα, Lys-Nε, Orn-Nδ, Dab-Nγ, Dap (2,3-Diaminopropionic acid)-Nβ, COOHβ-Asp, COOHγ-Glu, Sβ-Cys, and COOHC−terminal (in case of amino acid side-chain immobilization) to avoid undesired side reactions that predominantly occur during the peptide coupling step (Figure 3). The subject carboxylate, amino, and thiol groups from these amino acids constitute the reactive functional group in question. If not properly masked, the Fmoc-SPPS chemistry will challenge their integrities, yielding a complex mixture of by-products.
Figure 3.
Differentiation of the side-chain protection strategy in SPPS.
On the contrary, other relevant nucleophilic functional groups, even though traditionally protected in peptide synthesis, could survive the coupling reactions without protection under proper conditions. These tolerable functional groups encompass Nδ,Nω,Nω′-Arg, the carboxamide group from Asn/Gln, Oβ-Ser/Thr, the phenolic group from Tyr, Nim-His, and Nindole-Trp (Figure 3).
By taking advantage of such tolerable functional groups, the minimal-protection SPPS (MP-SPPS) strategy aims to utilize amino acid building blocks without side-chain protecting groups. In other words, MP-SPPS make use of Fmoc-Asn-OH, Fmoc-Gln-OH, Fmoc-Ser-OH, Fmoc-Thr, Fmoc-His-OH, Fmoc-Arg-OH, and Fmoc-Trp-OH without side-chain protection unless the occurrence of unforgiving and invincible side reactions.
Besides the aforementioned benefits related to the productivity boost, MP-SPPS enables the following improvements:
Enabling considerably milder peptide cleavage conditions. Harsh conditions of peptide resin acid treatment, like an increased amount of TFA, the addition of strong acids such as trifluoromethanesulfonic acid, elevated temperature, and prolonged cleavage time might be wielded in case relatively stabler protecting groups could not be effectively removed under standard acidolysis conditions. In addition to representing a safer alternative, the implementation of MP-SPPS could preclude the harsh deprotection condition and lower the risk of acid-catalyzed side reactions such as Asn/Gln deamidation, aspartimide formation, and peptide backbone hydrolysis [10].
Outrooting a plethora of underlying side reactions inherent to the protecting groups, such as incomplete protecting group removal, Pbf, in particular, tert-butylation, sulfonation (formation of [M+80] impurities induced by Pbf degradation), trifluoroacetylation, and so on [11].
Eliminating the scavengers, like silane, that entraps the released protecting group cations. The electrophilic scavengers could complicate the deprotection reaction and induce undesired side reactions such as disulfide breaking. Exclusion of the scavengers could therefore cement the stability of certain labile moieties, like disulfide bonds (Figure 4) [12].
Improving the COGS (Cost of Goods Sold) by reducing TFA, anti-solvent, and scavenger, improving the atom economies and PMI.
Lowering the negative ecological impact (E-factor enhancement).
Strengthening manufacturing safety.
Figure 4.
TFA/Silane-induced disulfide reduction.
The concept of minimal-protection SPPS has been introduced previously. Strategies for assembling the target peptide products, including side-chain unprotected amino acids, have been reported hitherto, including ultrasound-facilitated peptide synthesis [13], amino acid couplings by restrainedly activated carboxylate species like p-nitrophenyl ester [14], or special building modalities such as iso-acyl dipeptide building block Boc-Ser/Thr(Fmoc-Xaa)-OH [15]. Side-chain unprotected amino acids could be utilized as the starting materials in these strategies for assembling the target peptide products. Nevertheless, the strategies apply mostly for small-scale syntheses or peptide derivatives with very short sequences, such as di- or tri-peptide. The development of MP-SPPS strategies fully compatible with large-scale industrial peptide production, including its applicability scopes, restrictions, and corresponding solutions, has yet to be fully elaborated. Overall, MP-SPPS subscribes to the growing interest in protecting group-free organic synthetic methodology [16].
In addition, functional groups, including Nα, Lys-Nε, Orn-Nδ, Dab-Nγ, Dap-Nβ, COOHβ-Asp, COOHγ-Glu, and Sβ-Cys, still require proper protection under MP-SPPS.
3.2 Side-chain unprotected arginine
Arg stands out as the most influential residue for the MP-SPPS among the non-mandatory side-chain protection amino acid species. Arg’s default side-chain protecting group Pbf is notoriously known for its acidolytic stability. A peptide of interest rich in Arg residues exacerbates the problem. Such situations call for the use of a high concentration of TFA. Furthermore, auxiliary stronger acid such as TFMSA (trifluoromethanesulfonic acid) is occasionally needed to boost the Pbf cleavage. Given the challenges inherent to Pbf acidolysis, MP-SPPS should target incorporating side-chain unprotected Fmoc-Arg-OH as a prime task.
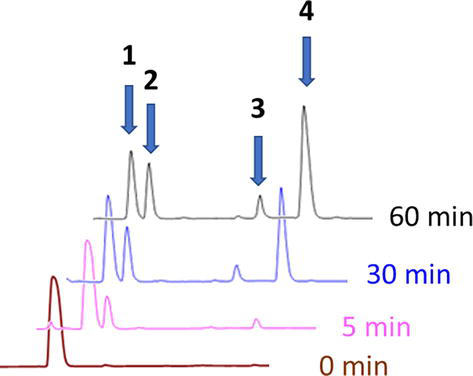
The extremely low conversion rate of Fmoc-Arg-OH coupling impedes its utilization as a side-chain unprotected building block when assembled to a peptidyl resin Xaa3-Tyr-Xaa5-His-Xaa7-Xaa8-2-chlorotrityl resin (Xaa3,5,7,8 are unreactive aliphatic or aromatic amino acids without nucleophilic side chains). In this case, the use of 1.5 equiv. Fmoc-Arg-OH with 1.5 equiv. DIC (N,N′-diisopropylcarbodiimide)/HOSu (N-hydroxysuccinimide) in DMF does not lead to the desired product formation even after 3 h of reaction. Alternative couplings additives Oxyma (Ethyl cyano(hydroxyimino)acetate) or HOBt (N-hydroxybenzotriazole) in DMF or NMP do not facilitate the subject coupling. Furthermore, various combinations of coupling additives and organic solvents could not form the desired peptide product. RP-HPLC analysis of the Fmoc-Arg-OH/DIC/Oxyma coupling solution reveals a rather complex mixture (Figure 5).
Figure 5.
RP-HPLC traces of the solutions of Fmoc-Arg-OH activation by DIC/Oxyma in DMF [1. Fmoc-Arg-OH; 2. Fmoc-Arg-lactam; 3. Fmoc-Arg(Fmoc-Arg)-OH; 4. Fmoc-Arg(Fmoc-Arg)-lactam].
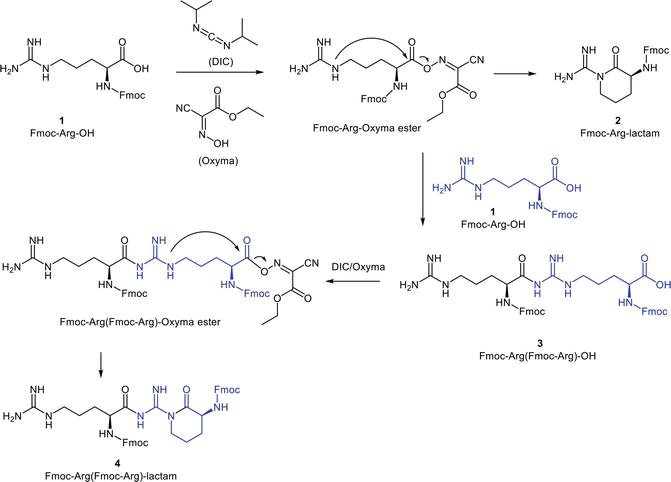
LC/MS reveals the cyclization and dimerization of the Fmoc-Arg-OH upon the carboxyl activation. Concretely speaking, Fmoc-Arg-OH (1) transforms to Fmoc-Arg-lactam (2), Fmoc-Arg(Fmoc-Arg)-OH (3), and the dimeric lactam (4) (Figures 5 and 6).
Figure 6.
Fmoc-Arg-OH transformation by DIC/Oxyma treatment.
Apparently, the undesired side reactions addressing Fmoc-Arg-OH prior to its condensation with the peptide Nα-group explain the low coupling yield. The guanidino functional groups are stereotypically deemed inert under common reaction conditions. However, the guanidino group of arginine, equating a Cδ-derivatized ornithine, enables the intramolecular cyclization depicted in Figures 2 and 6. The side-chain unprotected Fmoc-Arg-OH undergoes a kinetically favored six-membered ring cyclization with its activated carboxylate, yielding an unreactive lactam. The arginine guanidino group is not as inert as generally believed. Actually, freebase guanidine compounds constitute strong nucleophilic species and are capable of nucleophilic additions [17]. Arginine is generally tagged as unreactive simply because its guanidino side chains are mostly protonated under acidic to mild basic conditions thanks to its high pKa value (>12). Nonetheless, when the guanidino group is partially deprotonated, it can initiate a nucleophilic attack on the proper electrophilic substrates, as indicated in Figure 6. Therefore, Fmoc-Arg(Pbf)-OH is commonly used in Fmoc-SPPS. The bulky protecting group Pbf minimizes the risk of the aforementioned side reaction.
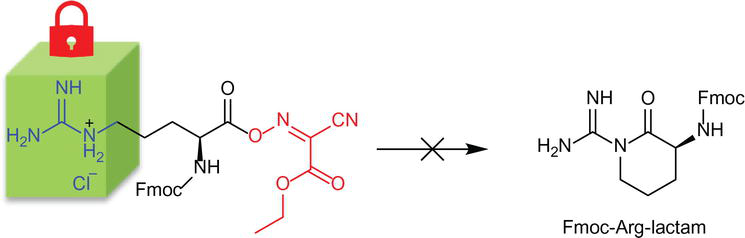
Nevertheless, the inherent issues centered on Fmoc-Arg-OH could be circumvented by using Fmoc-Arg(HCl)-OH, which undermines the Fmoc-Arg-OH unprotected guanidino side’s reactivity. The salt formation of Fmoc-Arg-OH simultaneously protonates the nucleophilic guanidino group and introduces steric interference by the chloride counterion. Furthermore, using a carbodiimide-based coupling agent does not require the utilization of an auxiliary base catalyst. With the undesired intramolecular cyclization minimized, the target amino acid coupling is cemented (Figure 7). Indeed, quantitative coupling of 1.5 equiv. Fmoc-Arg(HCl)-OH is accomplished by DIC/Oxyma in DMF, validating the utility of Fmoc-Arg(HCl)-OH as arginine synthon for the MP-SPPS strategy.
Figure 7.
Strategy to suppress the undesired lactam formation during Fmoc-Arg-OH carboxylate activation.
3.3 Side-chain unprotected histidine
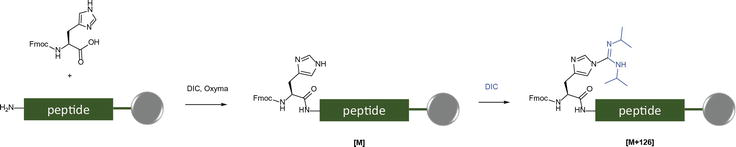
The side-chain unprotected histidine Fmoc-His-OH undergoes considerable imidazolyl modification by DIC during MP-SPPS, thanks to the pronounce nucleophilicity of imidazolyl group toward carbodiimide, resulting in an [M + 126] impurity (Figure 8).
Figure 8.
Modification of Nim-His by DIC.
Unlike the arginine cyclization side reaction delineated in Figure 6, the DIC-induced imidazolyl modification is progressive. It intensifies through the MP-SPPS process once the subject histidine residue is incorporated into the immobilized peptide chain. This artifact precludes utilizing Fmoc-His-OH as the building block when carbodiimide-based coupling reagents orchestrate the MP-SPPS processes.
Phosphonium salt coupling reagents such as PyBOP do not modify the histidine imidazolyl side chain under work conditions during the peptide assembly. This merit cements the intrinsic advantages of phosphonium coupling reagents which are inert to the Nα functional group during peptide synthesis.
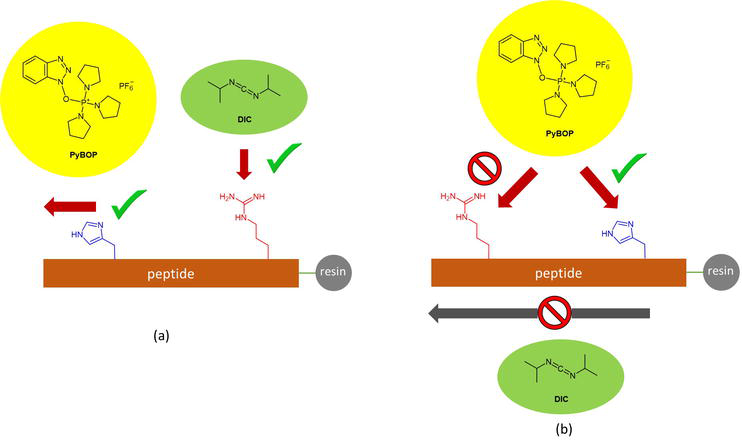
Incorporating side-chain unprotected Fmoc-Arg(HCl)-OH and Fmoc-His-OH could proceed well when the histidine is on the N-flank of the arginine. The assembly of Fmoc-Arg(HCl)-OH and the subsequent residues could be mediated by DIC/Oxyma strategy prior to the Fmoc-His-OH incorporation (Figure 9a). Then, the coupling reagents mixture uses PyBOP/base combination for all the following amino acid couplings. Otherwise, DIC will modify the imidazolyl side chain of the histidine residue already incorporated into the peptide chain attached to the solid support. On the contrary, an arginine N-terminal to the histidine, not necessarily in direct proximity, creates a dilemma for the MP-SPPS due to the reactivity of the histidine imidazolyl group toward carbodiimide coupling reagents and the requirement of the utilization of phosphonium salt to mediate all the amino acid couplings N-flanking to the subject histidine (Figure 9b). In this scenario, PyBOP will constitute the coupling reagent of choice to mediate all the amino acid couplings from the Fmoc-His-OH onwards, including the Fmoc-Arg(HCl)-OH. However, incomplete Fmoc-Arg(HCl)-OH incorporation might arise as a consequence. PyBOP is incapable of driving the Fmoc-Arg(HCl)-OH coupling into completion in the subject peptide assembly.
Figure 9.
Coupling reagent suitability for various Arg/His topologies: (a) the unprotected His is N-flank to the unprotected Arg; (b) the unprotected His is C-flank to the unprotected Arg.
The incorporation of a side-chain unprotected Fmoc-Arg(HCl)-OH in a -Arg-(Xaa)n-His- peptide topology will necessitate the usage of an alternative histidine synthon to satisfy the MS-SPPS demand. Because the major positive impact of MP-SPPS depends predominantly on reducing the amount and concentration of TFA during side-chain deprotection, the utilization of side-chain unprotected Arg should be prioritized. In other words, the reconciliation is made in favor of Fmoc-Arg(HCl)-OH relative to Fmoc-His-OH. In this scenario, one could resort to more acid-sensitive protecting groups for histidine, such as Mtt (methyltrityl) or Mmt (monomethoxytrityl). The acid-lability is ranked in the following incremental order of Trt < Mtt < Mmt. Moreover, a case study reveals that Fmoc-His(Mtt)-OH could be readily incorporated into the immobilized peptide chain by DIC/Oxyma chemistry with reduced racemization relative to that of Fmoc-His(Trt)-OH during peptide coupling [18]. More importantly, 10% TFA/DCM with 1.2 equiv. TIS (triisopropylsilane), as the Mtt+ scavenger, removes the Mtt-protecting group effectively and sufficiently.
3.4 Side-chain unprotected serine/threonine
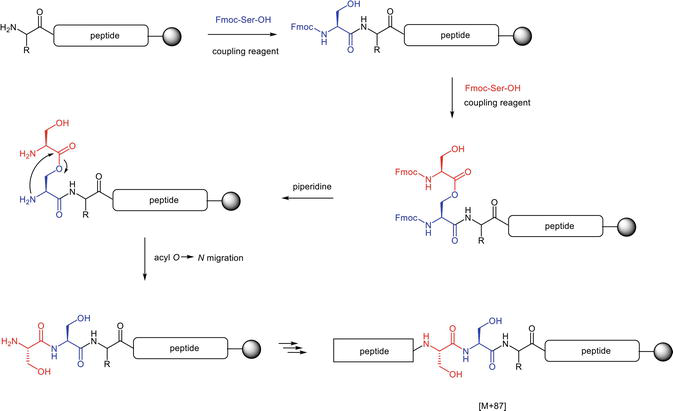
The nucleophilicity of the hydroxyl group-bearing amino acids couplings, like Fmoc-Ser-OH, Fmoc-Thr-OH, and Fmoc-Tyr-OH, represents an inherent challenge for MP-SPPS. Fmoc-Ser-OH, as a Cβ-hydroxy substituted amino acid, could mediate a double insert of serine residue and the resultant formation of endo-Ser impurity. The process is depicted in Figure 10.
Figure 10.
Formation of endo-Ser impurity from Fmoc-Ser-OH coupling.
A second Fmoc-Ser-OH could add to the unprotected hydroxyl group of another serine, either post or prior to the immobilization on the solid phase but during the coupling reaction. After removing the Fmoc protecting group from the Fmoc-Ser(Fmoc-Ser)-OH residue of the peptide chain attached to the solid support, the side-chain serinyl moiety could migrate to the peptide backbone through a favored five-membered ring intermediate initiated by the Nα-group (Figure 10). This acyl O→N shift is particularly facilitated in the basic milieu and results in the endo-Ser impurity formation with a molecular weight increase of 87 Da. Without the acyl O→N shift, the residual serinyl ester bond will be hydrolyzed at the subsequent piperidine treatment. The liberated hydroxyl group might further mediate ester bond formation at the subsequent amino acid coupling step. However, the formed ester impurity will unlikely lead to an acyl O→N shift due to the steric disadvantages of the greater ring size, which precludes the insertion of an extra residue in the peptide sequence. Fmoc-Thr-OH could, in principle, also accommodate this process, but the extent is much restrained due to the steric hindrance of the Cβ-methyl group.
In order to address the undesired serine side-chain esterification, process parameters such as amino acid pre-activation temperature, pre-activation time, coupling temperature, pH, solvent, coupling reagent, and coupling additive could be screened and tuned to minimize the endo-Ser impurity formation.
3.5 Side-chain unprotected tyrosine
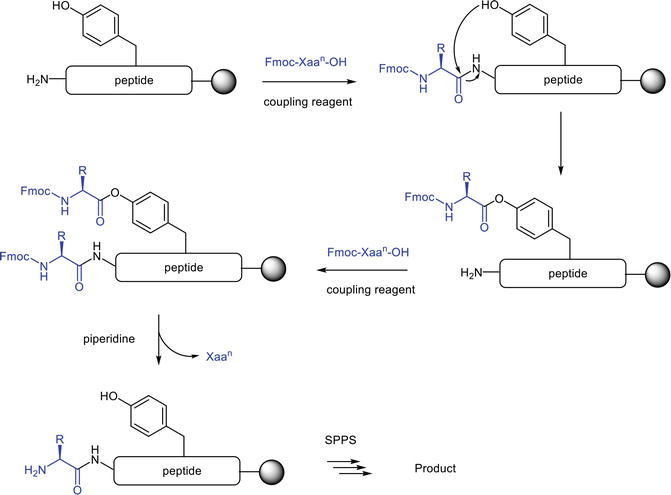
The phenolic group from the tyrosine side chain could also be subjected to phenyl ester formation in the MP-SPPS process but to a much-reduced extent due to the weaker nucleophilicity of phenolic hydroxyl group relative to the aliphatic hydroxyl group on serine (Figure 11). The undesired function between tyrosine’s phenolic group with the coupling reagent has also been reported [19].
Figure 11.
Putative mechanism of tyrosine esterification in MP-SPPS.
Notably, the susceptibility of tyrosine to esterification might be sequence dependent. In this study, the Tyr(Fmoc-Xaan) impurity formation occurs exclusively at a specific Fmoc-Xaan-OH coupling step without being detected at other coupling steps [17]. Spatial alignment might come into play to facilitate the tyrosine-induced esterification through acyl N→Ophenolic shift. Meanwhile, with this undesirable transformation taking place, the excess Fmoc-Xaan-OH in the reaction solution can acylate the liberated Nα-group to yield a derivative of [Tyr(Fmoc-Xaan)] peptide intermediate (Figure 11). The piperidine treatment removes the Nα-Fmoc protecting group and the Xaan ester from the tyrosine side chain, regenerating the targeted peptide sequence. Side-chain phenyl ester is facilely hydrolyzed in this process, and the target intermediate is subject to the following SPPS steps to yield the target peptide product. This process is schematized in Figure 11.
3.6 Side-chain unprotected asparagine/glutamine
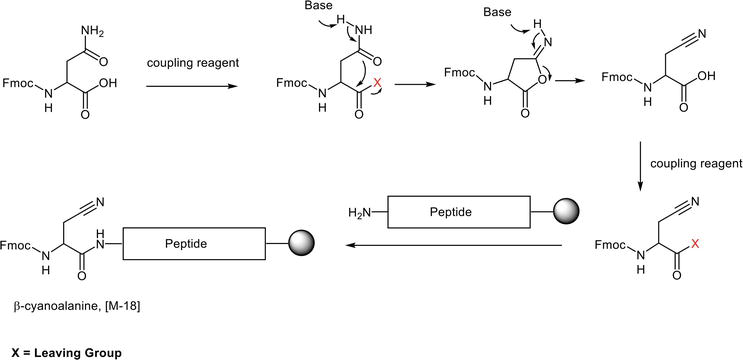
Side-chain unprotected Fmoc-Asn/Gln-OH also sustains the MP-SPPS. The most prevalent side reaction arising during the Asn activation and assembly is the dehydration and the resulting formation of a β-cyano side chain. The process involves the unprotected amide side chain attacking the activated carboxylate in the presence of a base, resulting in a five-membered ring intermediate iminohydrofuranone. The latter undergoes ring opening in basic conditions and gives rise to the formation of Fmoc-Ala(CN)-OH (Figure 12) [20]. The coupling reagent present in the reactive mixture can activate the resulting Fmoc-Ala(CN)-OH by-product, which incorporates into the peptide chain instead of the desired Fmoc-Asn-OH and yield an [M-18] impurity.
Figure 12.
Formation of β-cyanoalanine impurity through side-chain unprotected Asn activation and dehydration.
This undesired conversion occurs only at the Fmoc-Asn-OH activation step when incorporated into the growing peptide chain. No further dehydration will process once the side-chain unprotected Asn is part of the peptide chain. The activation reaction and the existence period of the activated Fmoc-Asn-OH species dictate the extent of the Fmoc-Asn-OH dehydration. Process parameters, including coupling reagent, additive, solvent, amino acid activation pattern, pre-activation temperature, pre-activation time, and coupling temperature, might impact the propensity of the Fmoc-Asn-OH dehydration.
The incorporation of side-chain unprotected Arg, Tyr, and Fmoc-His(Mtt)-OH during the SPPS on CTC resin of an octa-peptide of angiotensin II analog called peptide Z has been reported [17]. Peptide Z bears a sequence of H-Xaa1-Arg-Xaa3-Tyr-Xaa5-His-Xaa7-Xaa8-OH (Xaa1,3,5,7,8 are unreactive aliphatic or aromatic amino acids without nucleophilic side chains). The couplings of 1.5 equiv. Fmoc-Arg(HCl)-OH, Fmoc-His(Mtt)-OH, and Fmoc-Tyr-OH by DIC/Oxyma in DMF go quickly and quantitatively without forming any detectable related impurities. Interestingly, completing Fmoc-Arg(HCl)-OH coupling takes 2.5 h, as attested by a negative ninhydrin test, whereas all the other couplings are entirely completed within 1.5 h. The MP-SPPS strategy achieves quantitative couplings without the occurrence of major side reactions/incomplete coupling.
The peptide cleavage is pivotal to the MP-SPPS strategy as the following criteria should be gratified:
The acid cleavage solution should quantitatively release the immobilized peptide molecules from the solid supports.
The acid treatment should quantitatively deprotect the acid-labile side-chain protecting groups on the peptide.
The cleavage and deprotection solution should require the lowest possible acid concentration to accomplish the quantitative reactions.
The method should easily concentrate the peptide solution post the cleavage and deprotection without compromising the product integrity.
The peptide solution should be miscible with the anti-solvent at the product workup step; otherwise, solvent swapping should be performed before the product precipitation.
The solvent for the acid cleavage solution should comply with the industrial request. The low OEL (Occupational Exposure Limit) and high toxicity (confirmed animal carcinogen and probable carcinogen to humans) of DCM disqualify its utilization [21].
To address the MP-SPPS demands during peptide release from the solid support and deprotection, 10% TFA/DCM with 1.2 equiv. TIS (as the scavenger for the Mtt cation) could accomplish the above criteria except for the use of DCM. As a result, peptide Z is fully cleaved from the 2-chlorotrityl resin, and the Mtt protecting group on the His side chain is quantitatively removed simultaneously after 1 h at the ambient temperature.
To comply with all the MP-SPPS criteria, a solvent screen has been conducted to find the replacement for DCM. Among ACN, 2-MeTHF, IPA, EtOH, EtOAc, and TFT (trifluorotoluene), only TFT enables the successful replacement of DCM as the bulky solvent in conjunction with TFA for the one-pot peptide deprotection and release from the solid support.
Various reactions such as acylation, tosylation, silylation, Swern oxidation, and Dess-Martin oxidation have been accomplished by replacing DCM with TFT as the organic solvent [22]. TFT has a much higher boiling point (102°C) than DCM (40°C), granting improved safety profiles relative to DCM in the industries. Consequently, 10% TFA/TFT/1.2 equivalents of TIS (17 equivalents of TFA cf. peptide) at room temperature for 2 h gives a quantitative peptide Z cleavage and Mtt removal. Notably, the hydrophilic peptide Z could not fully solubilize in the relatively unpolar 10%TFA/TFT/TIS cleaving/deprotecting solution, even though the solution enables the quantitative cleavage of the peptide Z from the resin. Nonetheless, rinsing the cleaved resin three times with 10% HFIP (hexafluoroisopropanol)/TFT drastically enhanced the product recovery by effectively solubilizing the peptide Z in HFIP.
Interestingly, the peptide Z TFA/HFIP/TFT solution undergoes phase separation prior to the concentration step. The TFT phase contains nearly no product but only the by-product methyl triphenylmethane derived from Mtt cation quenched by TIS. The salting-out effect might explain the phase separation of the inherently miscible TFT and HFIP [23], which has been exploited by SALLE (salting-out assisted liquid-liquid extraction) for sample analysis [24]. The serendipitous phase separation affords a viable methodology to isolate peptide Z through tandem procedures of phase separation, facile condensation of the peptide HFIP solution (optional), and product precipitation. Despite such potential imparted by the phase separation, the combined peptide cleavage /rinsing solution in this study is subjected to solvent swapping from TFT to HFIP. No product degradation is detected in this process. The peptide Z precipitates with cold MTBE once added to the concentrated peptide/HFIP mixture to obtain a quantitative uncorrected total yield of SPPS/cleavage/isolation.
The LC/MS analysis reveals a crude product purity of 91.7%, comparable to those from various industrial batches (90–94%).
Figure 13 and Table 1 compare the conventional SPPS and MP-SPPS in terms of process and performance index, respectively.
Figure 13.
Comparison of peptide Z synthetic scheme from conventional SPPS and MP-SPPS.
Index
SPPS
MP-SPPS
Atom economy (amino acid)
22.3%
23.9%
Overall TFA (cleavage + rinsing)
25.6 L/mol
1.3 L/mol
TFT
0
23.9 L/mol
HFIP
0
7.8 L/mol
Concentration prior precipitation
0.038 M
0.2 M
MTBE as anti-solvent
78.9 L/mol (3 vol.)
15 L/mol (3 vol.)
Total volume (sum of MTBE and Peptide solution) of precipitation
105 L/mol (3 vol. MTBE)
20 L/mol (3 vol. MTBE)
Crude purity
90–94%
92%
Uncorrected yield (crude product)
quantitative
quantitative
Table 1.
Comparison of the major indices between conventional SPPS and MP-SPPS for peptide Z synthesis.
MP-SPPS could have a 5.3-fold of the conventional SPPS productivity.
The most striking attributes inherent to MP-SPPS are the radically reduced TFA consumption (by 95%) and the increased peptide concentration prior to the precipitation (by 4.3 folds). These two key indices enable appreciable increases in peptide manufacturing productivity through MP-SPPS. The overall precipitation volume (combination of peptide solution and the anti-solvent) enhances productivity 5.3-fold when taken as a baseline since it poses the bottleneck to the whole manufacturing process (Table 1). Overall, the quality and yield of the crude peptide Z produced from MP-SPPS are comparable to conventional SPPS.
The MP-SPPS strategy reduces the hazardous elements of SPPS. It represents an environmentally friendlier method and enhances manufacturing productivity when using the side-chain unprotected amino acids such as Arg, His, Ser, Thr, Tyr, Asn, and Gln. Because the targeted peptide sequence may affect the Fmoc-Arg(HCl)-OH coupling efficacity, thoughtful planning should include a pilot study. Nevertheless, excluding the Pbf protecting group alleviates the global deprotection processes appreciably, which generally request highly concentrated TFA, occasional supplementation of stronger acids, and prolonged reaction time. Furthermore, lowering the TFA concentration allows for recovering a peptide solution with a higher concentration, which in turn eases the peptide isolation by precipitation.
Consequently, this procedure enhances the productivity of peptide manufacturing and fully outroots the frequent Pbf-related sulfonation impurity [M+80]. In addition, MP-SPPS lowers the occurrence of highly concentrated TFA-induced impurities such as trifluoroacetylation [M+96], backbone hydrolytic cleavage, acid-induced aspartimide formation [M-17/18], acid-catalyzed disulfide bond reduction by silane [M+2] [9], and deamidation of carboxamide moieties [M + 1]. Deamidation impurity represents a serious general concern as chromatographic purification techniques frequently fail to remove them from the desired peptide products.
Notably, the efficacy of Fmoc-Arg(HCl)-OH coupling might be sequence-dependent. The MP-SPPS strategy does not provide a solution to the use of indispensable side-chain protection (such as Lys, Asp, and Glu) if the targeted peptide sequence includes such residues. However, adopting alternative orthogonal protecting groups could diminish the highly concentrated TFA mixture application. For instance, Pd0-catalyzed allyl ester cleavage removes the allyl ester protecting group from a carboxylate side chain of Asp and Glu.
In summary, the MP-SPPS process addresses the high demand for developing greener chemical processes and lowering peptide manufacturing costs. MP-SPPS aligns with important Green Chemistry Principles [25], including atom economy (improved PMI), less hazardous chemical syntheses (TFA), safer solvents/auxiliaries (TFT vs. DCM), design for energy efficiency (skipping peptide solution concentration through phase separation), reduce derivatives (elimination of the unnecessary protecting group introduction-elimination), and inherently safer chemistry for accident prevention (no TFA evaporation, no handling of bulky highly concentrated TFA solution). Attainable superiorities in productivity, cost-effectiveness, and E-factor (environmental factor) constitute the three pivotal pillars for applying the MP-SPPS in industries.
The author would like to thank Antoine Blanc, Ph.D., from The State University of New York College at Oneonta, for reviewing the manuscript.
References
1.Jadhav S, Martin V, Egelund PHG, Castro HJ, Krüger T, Richner F, et al. Replacing DMF in solid-phase peptide synthesis: Varying the composition of green binary solvent mixtures as a tool to mitigate common side-reactions. Green Chemistry. 2021;23:3312-3321
2.Ferrazzano L, Catani M, Cavazzini A, Martelli G, Corbisiero D, Cantelmi P, et al. Sustainability in peptide chemistry: Current synthesis and purification technologies and future challenges. Green Chemistry. 2022;24:975-1020
3.Wegner K, Barnes D, Manzor K, Jardine A, Moran D. Evaluation of greener solvents for solid-phase peptide synthesis. Green Chemistry Letters and Reviews. 2020;14:153-164
4.Ferrazzano L, Corbisiero D, Martelli G, Tolomelli A, Viola A, Ricci A, et al. Green solvent mixtures for solid-phase peptide synthesis: A dimethylformamide-free highly efficient synthesis of pharmaceutical-grade peptides. ACS Sustainable Chemistry & Engineering. 2019;7:12867-12877
5.Martin V, Egelund PHG, Johansson H, Le Quement ST, Wojcik F, Pedersen DS. Greening the synthesis of peptide therapeutics: an industrial perspective. RSC Advances. 2020;10:42457-42492
6.Jaradat DMM, Al Musaimi O, Albericio F. Advances in solid-phase peptide synthesis in aqueous media (ASPPS). Green Chemistry. 2022;24:6360-6372
7.Al Musaimi O, de la Torre BG, Albericio F. Greening Fmoc/tBu solid-phase peptide synthesis. Green Chemistry. 2020;22:996-1018
8.Carpino LA, Shroff H, Triolo SA, Mansour E-SME, Wenschuh H, Albericio F. The 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl group (Pbf) as arginine side chain protectant. Tetrahedron Letters. 1993;34:7829-7832
9.Isidro-Llobet A, Álvarez M, Albericio F. Amino acid-protecting groups. Chemical Reviews. 2009;109:2455-2504
10.Yang Y. Peptide fragmentation/deletion side reactions, intramolecular cyclization side reactions. In: Side Reactions in Peptide Synthesis. Academic Press; 2015. pp. 1-28, 119-156
11.Yang Y. Peptide global deprotection/scavenger-induced side reactions. In: Side Reactions in Peptide Synthesis. Academic Press; 2015. pp. 63-67
12.Yang Y, Hansen L, Badalassi F. Investigation of on-resin disulfide formation for large-scale manufacturing of cyclic peptides: A case study. Organic Process Research and Development. 2020;24:1281-1293
13.Anuradha MV, Ravindranath B. Acylation of unprotected amino acids using ultrasound. Tetrahedron. 1997;53:1123-1130
14.Gagnon P, Huang X, Therrien E, Keillor JW. Peptide coupling of unprotected amino acids through in situ p-nitrophenyl ester formation. Tetrahedron Letters. 2002;43:7717-7719
15.Arenzo HB, Bingenheimer W, Blanchette R, Morgans JRDJ, Robinson J III. Temporary serine protection in solid phase synthesis of LH-RH analogs. International Journal of Peptide Protein Research. 1993;41:342-346
17.Taylor JE, Bulla SD, Williams JMJ. Amidines, isothioureas, and guanidines as nucleophilic catalysts. Chemical Society Reviews. 2012;41:2109-2121
18.Yang Y, Hansen L, Ryberg P. Side-chain unprotected Fmoc-Arg/His/Tyr-OH couplings and their application in solid-phase peptide synthesis through a minimal-protection/green chemistry strategy. Organic Process Research and Development. 2022;26:1520-1530
19.Vrettos EI, Sayyad N, Mavrogiannaki EM, Stylos E, Kostagianni AD, Papas S, et al. Unveiling and tackling guanidinium peptide coupling reagent side reactions toward the development of peptide-drug conjugates. RSC Advances. 2017;7:50519-50526
20.Stroup AN, Cole LB, Dhingra MM, Gierasch LM. Synthesis and crystal structures of Boc-L-Asn-L-Pro-OBzl·CH3OH and dehydration side product, Boc-β-cyano-L-alanine-L-Pro-OBzl. International Journal of Peptide and Protein Research. 1990;36:531-537
21.Agency for Toxic Substances and Disease Registry Division of Toxicology and Environmental Medicine. Addendum to the Toxicological Profile for Methylene Chloride. Atlanta; 2010
22.Ogawa A, Curran DP. Benzotrifluoride: A useful alternative solvent for organic reactions currently conducted in dichloromethane and related solvents. The Journal of Organic Chemistry. 1997;62:450-451
23.Hyde AM, Zultanski SL, Waldman JH, Zhong Y-L, Shevlin M, Peng F. General principles and strategies for salting-out informed by the hofmeister series. Organic Process Research and Development. 2017;21:1355-1370
24.Tang YQ , Weng N. Salting-out assisted liquid–liquid extraction for bioanalysis. Bioanalysis. 2013;5:1583-1598
25.Anastas PT, Warner JC. Green Chemistry: Theory and Practice. New York: Oxford University Press; 1998. p. 30
Written By
Yi Yang
Submitted: 08 June 2023Reviewed: 15 June 2023Published: 11 August 2023
 Open access peer-reviewed chapter
Open access peer-reviewed chapter