Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
Systemic sclerosis (SSc), also known as scleroderma, is a systemic autoimmune rheumatic disease characterized by dysregulation of the immune system, fibrosis of the skin and visceral organs, and vasculopathy. Inflammatory activation may be important in the initiation and progression of vasculopathy and fibrosis in response to homeostatic disturbance. Numerous factors trigger and enable sustained inflammation such as increased oxidative stress, involved in progressivity and disease severity. This chapter will focus on the role of inflammation and the involvement of multiple immune mediators contributing to autoimmune activity of SSc.
Division of Allergy and Clinical Immunology, Department of Internal Medicine, Andalas University School of Medicine/M. Djamil Hospital, Padang, West Sumatra, Indonesia
Raveinal Masri
Division of Allergy and Clinical Immunology, Department of Internal Medicine, Andalas University School of Medicine/M. Djamil Hospital, Padang, West Sumatra, Indonesia
*Address all correspondence to: dwityaelvira@med.unand.ac.id
1. Introduction
Systemic sclerosis or scleroderma (SSc) is a systemic autoimmune disease characterized by a pathogenic triad: microangiopathy, immune dysfunction and fibrosis of the skin and visceral organs, in which chronic inflammation plays a crucial role and leads to the severity of organic lesions [1, 2]. The degree of skin fibrosis, immunological profile, and microvascular dysfunction determine the clinical classification of the disease, consisting of limited cutaneous SSc (lSSc) and diffuse cutaneous SSc (dSSc) disease subtype. dSSc is a devastating and serious disease with significant morbidity and mortality [2]. Scleroderma is now conceived as a complex disease with multiple pathogenic pathways. In particular, the central role of immune system cells and inflammatory mediators, fibroblasts and other cells determining the regulation of the extracellular matrix (ECM) is now recognized [3, 4, 5].
Dysregulations of the immune system were one of the first mechanisms recognized to be responsible for the onset of SSc [3]. An inflammatory process characterized by immune cell infiltration may be crucial in the initiation and progression of vasculopathy and fibrosis in response to homeostatic disturbance. The oxidative stress also increases the susceptibility of the disease [4, 5, 6]. One quarter of patient present with inflammatory signature, characterized by increased C-reactive protein and ESR, especially in early inflammatory phase of the disease. These biomarkers correlated with disease activity, severity, poor pulmonary function, and shorter survival [7]. Despite the increased acute-phase reactants that does not alter in treated patients, the inflammatory reactions in SSc are not as “highly inflammatory” as other rheumatic autoimmune diseases such as rheumatoid arthritis (RA) or systemic lupus erythematosus (SLE), whereas SSc usually shows less classical inflammatory signs such as rubor, calor, or dolor. Mitev et al. studied differences between patients with inflammatory SSc and non-inflammatory SSc, characterized by continuously elevated inflammatory biomarkers (ESR and CRP). Higher risk for SSc with high inflammatory biomarkers is present in older males, higher modified Rodnan skin score (mRSS), lower lung function compared to non-inflammatory SSc. Based on their study, macrophages of SSc with higher inflammatory markers were shown less affected with immunosuppressant treatment, thus maintaining the inflammation activity in inflammatory SSc patients [7, 8].
A recent study demonstrated that Toll-like receptors (TLRs) are involved in SSc pathogenesis through two main mechanisms. The first mechanism is through increased of TLR expression due to persistent damage after viral infection with stimulation of interferon (IFN), transforming growth factors-β (TGF-β) and other mediators. The second mechanism is through the increased presence of damage-associated molecular patterns TLR agonists such as tenascin-C (TEN-C) and Fibronectin-EDA (FN-EDA) [9, 10, 11, 12, 13, 14].
Persistent damage after viral infection could increase chronic inflammation by activation of dendritic cells by TLRs which leads to the production of several pro-inflammatory cytokines, [2] particularly type I interferon (IFN) which have been shown to be overexpressed in peripheral blood in patients with systemic sclerosis. Viral infection such as Epstein Barr-viruses activate TLR activation as an antiviral response against γ-herpes virus, which induces type I IFN and cytokines response. Interferons are multifunctional cytokines responsible for inducing cellular resistance to viruses. There is evidence of a strong IFN signature in SSc where nearly 50% of patients with SSc show the so-called “interferon signature (IFN signature)” in peripheral blood and sera [6]. It appears that the IFN signature in SSc may observed in the early phase of the disease, even before the skin fibrosis is well established. In early SSc, type I IFNs were thought to be responsible for the inflammatory process and activated mediators of fibrosis such as B-cell activating factor (BAFF) expression and serum levels of propeptide N-terminal type III procollagen. Increased expression of this type I IFN affects the function of immune and endothelial cells such as monocytes, as well as increased differentiation, survival, proliferation, and activation of T, B, dendritic cells, and fibroblasts. Increased expression of MxA (marker of IFN type I activity) and interferon regulatory factors (IRFs) also occurs in endothelial cells of SSc patients, correlated with the presence of ischemic ulcers digital [2, 6, 9, 10, 11, 12].
The second mechanism on how TLR signaling may also be implicated in SSc pathogenesis is the presence of several agonists of TLR that are expressed in damaged cells of affected tissues and released into extracellular space. Tenascin-C (TEN-C) and Fibronectin-EDA (FN-EDA) were two of TLRs agonist identified as endogenous danger signal that drives TLRs signaling into upregulation of inflammation and fibrosis amplification in SSc. Tenascin-C is a multifactorial hexameric extracellular (ECM) glycoprotein consisting of 4 domains: a tenascin assembly (TA) domain, epidermal growth factor-like (EFGL) repeats, up to 17 FNIII-like repeats and a fibrinogen-like globe (FBG). Fibronectin-EDA (FN-EDA/fibronectin containing extra domain A) is cellular fibronectin expressed in endothelial cells that is associated with tissue remodeling, fibroblast differentiation, angiogenesis, and inflammation. Tenascin-C as a modulator of inflammatory response will stimulate the pro-inflammatory cytokine such as IL-6, IL-8, and TNF, meanwhile FN-EDA induced expression of multitude dermal fibroblast cytokines release from mast cells, such as CXCL-13, IL-8 and TNF-α. Tenascin-C and FN-EDA activate TLR4 activity in innate immune response, via TGF-B2 signaling lead to increase of fibrosis occurred in SSc [13, 14, 15, 16, 17, 18].
3. The transition from innate immunity to adaptive immunity
Innate lymphoid cells (ILCs) are recently discovered innate immune cells that serve as first-line defense resembling the Th1, Th2, and Th17 in adaptive immunity. There are three types of ILC that have been described so far: ILC1, ILC2 and ILC3. These ILCs express MHC class II and possess transcription factors and cytokine profiles reminiscent of Th cells; ILC1 produces IFNγ; ILC2 produces IL-5, IL-9 and IL-13; ILC3 produces IL-17A and IL-22 [5, 6]. These cells act as antigen presenting cells (APC) like dendritic cells [6]. The role of ILCs as transition factors between innate and adaptive immunity in the pathogenesis of SSc is still an interesting topic of research. Dendritic cells also play a key role in the transition from innate to adaptive immunity through their ability to identify antigens from pathogen- or damage-associated molecular models (PAMPS or DAMPS) using TLRs, NLRs, RIG-I-like receptors (RLRs), and receptors for advanced glycation end products (RAGEs), thus process this information to T cells via the MHC-II/binding receptor complex antigen [6, 19, 20].
In SSc patients, ILC2 interaction with TGF-β could trigger profibrotic mechanisms of cutaneous fibroblast through increased of TGF-β and downregulation of Killer Cell Lectin-Like Receptor G (KLRG1). Binding of KLRG1 and ILC2 in circulation could migrate to the skin and pass to pro-fibrotic form, KLRG1 - ILC2, causing imbalance of anti-fibrotic and pro-fibrotic mediators and activate fibrosis mechanisms in skin fibroblast. ILC-2 produces IL-9 that shown to be increased in SSc patients, along with the increased of Th-9 lymphocytes, through mast cell-Th9-ILC2s. Activation of ILC-2 in stimulating chronic inflammation involves the secretion of alarmins, such as IL-33 cytokines, which play a central role in the modulation of ILC-2 and fibrosis in response to the initial inflammation phase in SSc [6, 19, 20].
Several observations over several decades strongly imply a major role of the adaptive immune system in the pathogenesis of SSc. T cells are also present in the inflammatory tissue infiltrates of SSc patients which involve Th-1, Th-17 followed by a witch to Th-2 leading to irreversible fibrosis and ‘burn-out’ of disease. In early untreated dcSSc skin among CD4+ T cells, TH2 cells are detected along with TH17 cells, follicular helper T cells (Tfh), and regulatory T cells (Tregs). CD4 + CCR7-memory T cells produced IL-13, IL-4, and TNFα, particularly in dcSSc [14]. In addition, IL-13 overproducing CD8+ T cells and IL-4 overproducing CD4 + CD8+ double positive T cells were detected in SSc skin lesions. The role of T-regulatory cells in SSc still conflicting. Most studies have reported that there were decreased frequencies and impaired function of T-regs cells followed by an imbalance of Treg/Th17 with reduced levels of cytokines serum, TGF-β and IL-10. Tregs may act as an anti-inflammatory by releasing cytokines with dual function such as IL-10, thus provide a protective role against atypical immune activation. TGF-β as pro-inflammatory cytokine is involved in Tregs differentiation, but the stimulation is inhibited by Th-1 cytokine, IL-6. The balance between Th17 cells and Tregs is deregulated during SSc development [21, 22, 23].
Abnormal T cell activation is a crucial element in the initiation and progression of SSc. The subtle regulation between the interaction of Th1/Th2 cells has long been the focus of the step. Either by releasing soluble mediators like IFN-γ (Th1 cells), IL-4 and IL-13 (Th2 cells), or by directly contacting fibroblasts, Th1 cells are considered anti-fibrotic in inhibiting ECM deposition and promoting MMP secretion, while Th2 cells are the opposite [3, 5, 14]. Fibroblasts respond to fine control of Th1/Th2 cell stimulation, which is followed by secretion of mediator. Th1 cells secrete anti-angiogenic and anti-fibrotic mediators, while Th2 pro-fibrotic and pro-angiogenic. Fibroblasts act as “immune sentinels’ in a paracrine manner by releasing cytokines and having direct and indirect cell–cell interactions with immune cells. Continuous two-way communication between immune cells and fibroblasts was thought to be the primary driver of SSc. T cells contribute to endothelial dysfunction and cytokine-mediated macrophage and fibroblast/myofibroblast activation, while fibroblasts secrete ECM, collagens, glycosaminoglycans (GAGs), and fibronectin leading to the formation of fibrosis in SSc. Chizzolini et al. suggested that activated T cells are the dominant lymphocyte population in lesioned skin and in particular T cell infiltrates correlate with skin involvement suggesting an association between autoimmunity and fibrosis [3, 6, 21, 22, 23].
T-follicular helper cells were also studied for involvement in SSc pathomechanisms. Several studies have shown higher levels of follicular T cells (Tfh) in peripheral blood and skin, but the remains study has shown inconsistent results. Follicular T helper cells (Tfh) provide essential support for B cell differentiation and antibody production in the germinal centers of secondary lymphoid organs. They express inducible T cell co-stimulator (ICOS), programmed cell death protein 1 (PD) 1, transcription factor BCL6 and produce IL-21 [24]. SSc patients with elevated Tfh cells and plasmablasts more frequently progressed to a late nailfold capillaroscopy pattern that was correlated with internal organ involvement [25]. A recent study showed that early SSc correlated with increased of Tfh cells, particularly SSc with severe skin lesions. Morita et al. in his study reported that there was dysregulation on Tfh homeostasis, identified by increasing cTfh17 and cTh1 cells in SSc patients, associated with the increased plasma level of IL-17F [26]. cTfh17 stimulate pro-inflammatory and pro-fibrotic cytokines, and can induce B cell differentiation, resulting in an imbalance of B cells subsets. Naïve B cells and plasmablast were found increased, while memory B cells were reduced in SSc due to immunologic abnormalities in SSc [27].
Absolute count of B cells shown contradictory result in peripheral blood of SSc patients, as B cells may find decreased, increased, or similar with controls. The different results among these studies might be due to differences methods used, the threshold definition, and/or on treatment of glucocorticoids or immunosuppressants. B cells also contribute to SSc pathogenesis, as specific autoantibodies (ATA, ACA) are found in SSc and B cell secretome includes IL-6 and TGF-β. Increased levels of IL-6 were found in the early diffuse SSc (dSSc), that could stimulate fibroblast from affected skin areas. Therefore, the loop between fibroblast and B cells is partly dependent on IL-6. Other cytokines that were produced by circulating B cells were IL-8, IL-1β, BAFF and CXCL13 (pro-inflammatory cytokines and chemo-attractant). These cytokines polarized activity of CD4 T cells into Th2, Tfh and/or Th17. Increased of Th2 cytokines then enhanced autoantibody production from plasma cells and contribute to collagen production from fibroblast [28, 29, 30].
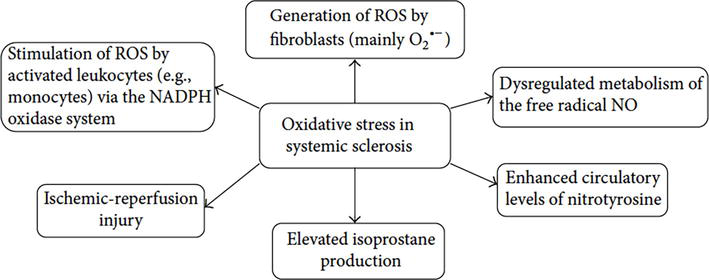
In SSc, oxidative stress, defined as an imbalance of oxidant and antioxidant states, is generated by fibroblasts, activated leukocytes, dysregulated metabolism of free radical nitric oxide and enhance of isoprostane and nitrotyrosine enzyme that induce oxidative stress, as seen in Figure 1 [4, 31, 32, 33].
Figure 1.
Patomechanisms of oxidative stress role in systemic sclerosis [4].
A recent meta-analysis on SSc patients described that several biomarkers of oxidative stress were found to be elevated in serum, such as malondialdehyde (MDA), nitric oxide and endogenous nitric oxide inhibitors compared to controls. Increased oxidative stress was also found in urine samples (8-Oxo-2′-deoxyguanosine (8-oxodG) and isoprostanes), lung and skin (increased H2O2 and nitric oxide). Fibroblasts extract from fibrotic skins of SSc patients shown higher ROS levels compared to control. These studies suggest that ROS play a role in SSc pathogenesis from earlier state of inflammation on SSc. In endothelial cells, higher levels of hydrogen peroxide were found especially in SSc patients with dcSSc compared to the lcSSc. A study in mice showed that increased levels of H2O2 in endothelial cells were able to create neoepitopes that may lead to the production of SSc autoantibodies, anti-DNA topoisomerase I Abs. Other autoantibodies such as anti-peroxiredoxin and anti-methionine sulfoxide reductase A (MRSA) were also detected at higher level in SSc serum. These autoantibodies induced 59% inhibition of peroxiredoxin I enzymatic activity and inactivation of oxidation of proteins by methionine reduction, which leads to chronically increased oxidative stress found in SSc patients. In animal and clinical studies, the local production of hypochlorous acid (HOCl) or hydroxyl radicals can stimulate the immune response and the production of anti-DNA topoisomerase-1 autoantibodies, while the generation of peroxynitrite triggers the production of anticentromere antibodies against centromere protein B. Another study on endothelial cells and fibroblast showed that there was no changes serum-induced ROS production or cell proliferation if autoantibodies were present. However, studies confirm that anti-PDGRF (anti-PDGFR Abs), can trigger the production of ROS and, thus, activate the development of SSc [4, 31, 32, 33].
Inflammasomes are large macromolecular cytoplasmic signaling complexes that control the proteolytic activation of two highly pro-inflammatory cytokines, interleukin-1 (IL-1), IL-1B and IL-18 under different signaling from danger to defend the host. Assembly and activation of inflammasomes are triggered by stimulation of the protein recognition receptor (PRR), which interacts with the adapter molecule and is bound to pro-caspase-1. With this caspase-1 is cleaved and activated so it can cleave pro-IL-1B and pro-IL-8, resulting in the active form of IL-1B and IL-8, respectively, which stimulate the inflammatory response. Activation of caspase-1 through the inflammasome pathway is also able to induce pyroptosis, a type of inflammatory cell death [34, 35, 36, 37].
Inflammasomes containing the NLR-3 family pyrin domain (NLRP3) are increasingly implicated in many inflammatory conditions. The involvement of NLRP3 on SSc was recognized only recently. The study of the BLM mouse model shows the importance of IL-1B and IL-18 in increasing the fibrosis score. The increase in these cytokines also induced in vivo activation of the NLRP3 inflammasome in a human study. Artlet et al. made a breakthrough by directly linking the upregulation of inflammasomes to the pathogenesis of SSc. In SSc dermal fibroblasts, Artlet et al. found a 5–7-fold increase in NLRP3 inflammasome protein expression, IL-1B, and IL-18, while treatment showed attenuation in levels of collagen and myofibroblast thickness with caspase-1 inhibitor. Upregulation of IL-1B/IL-18 signaling on SSc skin fibroblasts caused an increase in IL-6 and IL-1B 10 times higher after IL-1B treatment compared to fibroblast from healthy control. This result shows positive feedback mechanism and explains why modest initial stimulation might increase in chronic inflammation and subsequent ECM deposition [35, 36, 37].
Prolonged exposure of microvascular endothelial cells to IL-1B can induce permanent transformation into myofibroblasts. Chronic inflammation and inflammasomes lead to continuous cleavage of IL-1B in a positive feedback mechanism, which maintains a high level of active TGF-B1 protein, causing fibrosis. IL-18 is also capable of inducing fibroblast proliferation. The first report showing that inflammasomes are involved in SSc pathogenesis was obtained in animal model where IL-1B was shown to be crucial for establishment of bleomycin-induced pulmonary fibrosis. Inflammasomes NLRP3 and AIM2 were found to be increased in SSc lung and skin, while caspase-1 activity is also increased in SSc fibroblasts. SSc lesional fibroblasts secrete more IL-1B and IL-18 with increased clinical parameters of skin thickening suggests a potential role of these cytokines in SSc fibrotic complications.
7. Cytokines as fibrotic mediators in SSc pathogenesis
7.1 Interleukin-1 (IL-1) super family
The IL-1 family of cytokines consists of 11 cytokines, consisting of 7 ligands with agonist activity (IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36βand IL-36γ) as well as 4 ligands with antagonistic activity (IL-1Ra, IL-37 and IL-38). Interleukin-1 can also be classified into 3 subtypes, namely IL-1 type 1, type 2 and type 3. IL-1 cytokine members are involved in the innate response and adaptive immunity in SSc, as well as in the process of fibrosis. The IL-1 cytokines that are currently being discussed in connection with SSc are the IL-18 and IL-33. There are not many studies related to the role of IL-36, IL-37 and IL-38 in SSc so these three cytokines are still an interesting area for further research [38]. The potential role of the IL-1 superfamily can be seen in the following Table 1 [38].
Common name
Receptor
Potential roles in SSc or fibrosis
IL-1α
IL-1R1
Upregulated in lesional skin and serum. Induce production of IL-6 and PDGF. Promote viability of SSc fibroblasts [38].
IL-1β
IL-1R1 dan IL-1R2
Elevated in the serum, BAL and lesional skin. Induce IL-6 and TGF-B1 and promote Th17 cell differentiation [38].
IL-1Ra
IL-1R1
Upregulated in SSc-affected fibroblasts on skin and lung. Induce fibroblast differentiate into myofibroblast [38].
IL-18
IL-18Ra
Elevated in serum and BAL. Pro-fibrotic and anti-fibrotic effects were reported in fibrosis of skin and lung [38].
IL-33
ST2 (known as IL-1R4)
Downregulated in early SSc and upregulated in late SSc. Elevated in serum. Induce M2 macrophages and ILC2 to produce IL-13 and TGF-B [38].
Potential role of IL-1 super family cytokines in SSc and fibrosis [38].
Genetic studies of IL-1 in SSc also show that there is a relationship between IL-1 genetic variants and the risk of developing SSc, such as IL-1A, IL-18 and IL-33 variants. The IL-1β variant, SNP rs1143634, is thought to play a protective role in the occurrence of SSc, characterized by the low expression of this variant in SSc patients compared to healthy controls. IL-1 family allele variants and their relation to risk/protection against SSc can be seen in the following Table 2 [38].
IL-1 Family
SNP associated with SSc
Risk/Protection
IL-1A
rs1800587 rs17561
Risk
IL-1B
rs1143634 rs1143627 rss16944
Protection Risk Risk
IL-18
rs1946518 rs187238
IL-33
rs7044343 rs1157505 rs11792633 rs1929992
Risk
Table 2.
Risk or protection Allel variants of IL-1 super family cytokines [38].
Up to now, IL-1α was the most studied for the involvement in the pathogenesis of SSc from IL-1 cytokine family. Few studies reported that IL-1α is expressed in very high levels in the dermal fibroblasts of SSc patients as compared to normal. In addition, high levels of IL-1α are also associated with the incidence of digital ulcers, which shows a relationship with the incidence of obliterative vasculopathy. IL-1α also induces IL-6 which plays a role in the process of fibrosis through pro-fibrotic gene expression in vivo, which increases TGF-B1 production and regulates TGF-B receptors. TGF-B is a major regulatory cytokine of fibrosis through EMT stimulation, fibroblast proliferation, ECM product synthesis and MMP inhibition. IL-1 also induces PDGF, a chemotactic factor that induces the expression of N-cadherin and α-sma, both of which are markers for myofibroblast cells. This further strengthens the role of IL-1α as a cytokine that plays a role in fibroblast-myofibroblast differentiation, myofibroblast longevity which is the main key in the continuation of skin fibrosis in SSc [38, 39, 40].
7.2 Interleukin-18 (IL-18)
Interleukin-18 in SSc was found significantly increase compared to healthy control subjects. IL-18 was found to be increased both by serum and BAL examination in SSc with pulmonary disorders, indicating that IL-18 is involved in the fibrotic process that occurs in this disease. The study by Kitasato et al. showed that IL-18 mediates liver cell fibrosis by activating CD4 T cells, which can be suppressed by administration of anti-IL-18. In renal fibrosis, stimulation of the renal tubules with IL-18 can induce a-SMA, collagen-I and fibronectin which stimulate the process of fibrosis. However, another study by Nakatani-Okuda et al. showed opposite results, where IL-18 is also thought to have anti-fibrotic effects. This can be seen from a study they conducted on model mice with IL-18 deficiency, which experienced more severe fibrosis than wild-type mice. Another study by Kim et al. showed that IL-18 downregulation occurs in human dermal fibroblasts which also indicates an anti-fibrotic effect of IL-18 on SSc, so that the actual role of IL-18 as a pro-fibrotic or anti-fibrotic still requires further research [41, 42].
IL-18 has the capabilities of IL-1 cytokines in general, such as stimulating TGF-B1 expression, inducing type I and type III collagen and mediating collagen gel contraction. In myocardial tissue, administration of IL-18 as a recombinant therapy can lead to remodeling and interstitial myocardial fibrosis [42].
7.3 Interleukin-33 (IL-33)
Elevated serum IL-33 levels are found in SSc and are associated with the severity of skin fibrosis, reduced lung function, and peripheral vascular involvement, such as digital ulcers. An additional study also suggests that IL-33 may be a serum marker for vascular abnormalities in SSc. In the skin biopsies from early SSc patients, the expression of IL-33 protein was down regulated. by contrast, in patients with late stage SSc, IL-33 protein expression was constitutive found in most endothelial cells. Elevated serum IL-33 levels were also found in uterine fibrosis in women and were also found to be elevated by BAL (bronchoalveolar lavage) patients with IPF. IL-33 and its receptor ST2 are increased in fibrotic livers, and they were directly correlated to collagen expression [43, 44].
Studies on IL-33 also show that IL-33 has two types, namely full-length IL-33 and mature IL-33 which are functionally different. Patients with IPF showed a significantly higher increase in full-length IL-33 than mature IL-33. In brief, the critical role of IL-33 in SSc pathogenesis has been elucidating. However, more studies on the precise function of IL-33 in the process of immune dysfunction, vasculopathy, and fibrosis are required in SSc [45].
7.4 TNF superfamily
Although TGF-B is also a major factor in the pathogenesis of fibrosis, it is also known that the TNF superfamily (TNFSF) is involved as a mediator in the inflammatory process. TNFSF is a group of cytokines consisting of 19 ligands and 29 receptors. TNFSF plays a role not only in inflammation, but also in upregulation of the fibrotic process. Several TNFSF ligands are known to be associated with and involved in the process of pulmonary fibrosis, heart, skin, gastrointestinal tract, kidney, and liver [46].
In SSc it is known that the TNFSF involved in pathogenesis are LIGHT, BAFF, OX40L, CD70 and TRAIL which can directly stimulate direct fibrosis in stromal cells (epithelial, fibroblasts and smooth muscle cells). SSc patients are known to have an increase in soluble TNFR1 which is related to the degree of disease severity. Hugle et al. showed that there was upregulation of TNFR1 and TNFR2 in dermal T cells of patients with dcSSc, with TNFR2 being more related to skin thickening [46].
LIGHT is also associated with the risk of pulmonary fibrosis. LIGHT is also found increased? in skin biopsies of patients with SSc [46]. BAFF serum levels are also increased in SSc and can act directly on fibroblast and B cells. OX40L levels are usually associated with the severity or worsening of skin and lung fibrosis, so OX40L is thought to be a marker of fibrosis in the lung [46]. Recent studies have shown that OX40 L can affect the development and progression of fibrosis in various tissues and various degrees of fibrosis severity. CD70 is a ligand that is expressed on antigen-presenting cells (APC) and fibroblast cells. CD27:CD70 co-stimulation is a pathway that activates innate and adaptive immunity in the process of fibrosis because CD27 is expressed more in B cell and T cell subsets. In SSc, CD27 was correlated with the severity of fibrosis in skin biopsies showing that the CD27 pathway: CD70 is an important target for further research. Luo et al. demonstrated that CD70 expression in SSc is related to disease severity, however, further research is needed on the contribution of the CD27:CD70 pathway to the pathogenesis of SSc [46].
TRAIL, also known as TNFSF10, is also known to be associated with SSc, although studies have shown that TRAIL is more related to apoptosis of cancer cells and transformed cells. The results of a study using modified TRAIL as a therapy for skin fibrosis, showed that TRAIL can effectively destroy skin myofibroblasts and can restore the contours of the fibrous skin to close to normal skin in studies with SSc model mice. However, further research is needed to see which sensitive cell types are most associated with TRAIL sensitivity and possibly other pathways that contribute to TRAIL expression [46].
7.5 Interleukin-6 (IL-6) super family
IL-6 is a pleiotropic cytokine that plays a role in the inflammation that underlies SSc disease. IL-6 plays a role not only in the acute phase response, but also in chronic inflammation, autoimmunity, endothelial cell dysfunction and fibrogenesis. The IL-6 cytokine family is a group of cytokines consisting of IL-6, IL-11, ciliary neurotrophic factor (cTNF), leukemia inhibitory factor (LIF), oncostation M (OSM), cardiotrophin-1 (CT-1), cardiothropin- like cytidine (CLC) and IL-27. All of them belong to the same family because of the similarity of their receptor complexes containing the gp130 receptor subunit. IL-6 has a dominant pro-inflammatory activity, through IL-6 trans- signaling activity where IL6-binds to soluble IL-6R(sIL-6R) and then binds to gp130 expressing cell. Some of the effects of trans-signaling activity are the recruitment of MN cells, stimulation of endothelial cells, smooth muscle cells, inhibition of apoptosis by T cells and differentiation of T-reg cells. Meanwhile, classic IL-6 signaling on IL-6R expressing cells provides protective effects including IL-6 dependent regeneration, protection against bacterial infection. Cleaving of IL-6R to form soluble IL-6R is performed majority by ADAM17. In SSc, it is known that ADAM17 has increased activity which initiates the formation of sIL-6R, besides that in SSc there is also an increase in serum IL-8 which stimulates sIL-6R from neutrophils. As IL-6/sIL-6R can bind to the gp130 receptor which activates the STAT3 signal, which in turn increases the expression of the adhesion molecules ICAM-1, VCAM-1, IL-8, and MCP-1 release, accompanied by IL-6 [47, 48].
Interleukin-6 is associated with diffuse skin fibrotic involvement and early disease [49]. SSc patient fibroblasts were also found to express increased IL-6 compared to healthy controls, and IL-6R was also found in skin biopsies of limited cutaneous type SSc. Due to the biological nature of IL-6 trans-signaling, this cytokine can save T cells from cell apoptosis so that it will stimulate chronic inflammatory conditions through a positive autocrine feedback system. The trans-signaling signal is also able to increase autoantigen presentation by suppressing Na+2/K+ATPase by IL-6, it is also suspected that this IL-6/sIL-6R complexion can inapropriately change the signaling pathways of T-cell apoptosis thereby increasing cell persistence. T autoreactive [50].
7.6 Interleukin-17 (IL-17)
IL-17 family cytokines are cytokines expressed by Th-17 cells through the induction of the cytokine Il-23. In the early 2000s, genomic sequencing studies demonstrated that the IL-17 cytokine consists of 6 subtypes namely IL17A, IL17B, IL17C, IL17D, IL17E (also called IL-25), and IL17F. IL-17F is structurally similar to IL-17A (55%), and is more commonly expressed as IL-17A. IL-17B, IL-17C, and IL-17D have a structural similarity of 23–29% with IL-17A, while IL-17E, the most divergent of all IL-17 families, has a structural similarity of around 16%. In IL-17 family of cytokines, IL-17A plays the most important role as a pro-inflammatory cytokine. One of the effect is to maintains the stability of mRNA of TNF induced genes, so that it amplifies the effects of TNF [51, 52].
SSc, characterized by fibrosis of several organs, would be linked to a bias in favor of the activation of immune responses mediated by the Th17 pathway. Studies have shown that IL-17 is significantly higher in the serum of patients with SSc [51]. Increased IL-17A was also found in pulmonary fibrosis, kidney, heart, and skin of SSc patients, and IL-17A was also thought to be related to lung remodeling [51]. Animal studies have shown that IL17A plays a role in skin fibrosis through a TGF-B dependent pathway that induces collagen deposition in skin tissue [52].
Human studies show an increase in IL-17A+ expression in the dermis of the SSc patient skin and this is also followed by an increase in circulating Il-17A [51]. Expression of IL-17A is inversely proportional to the severity of SSc, IL-17A was also found to be associated with SSc fibroblast proliferation but its effects on collagen and ECM protein synthesis are still trivial. In lung tissue, the fibrotic effect of IL-17A has been proven through studies in experimental animals, but studies in humans have not been able to show a clear relationship between IL-17A and the progression of pulmonary fibrosis in SSc patients [51]. Xing et al’s study showed that IL-17A in SSc patients mediated endothelial cell inflammation via ERK1/2 phosphorylation. IL-17A can induce endothelial cells which in turn release cytokines and chemokines that activate neutrophil infiltration, also stimulate endothelial cell apoptosis which is thought to be related to endothelial cell dysfunction in abnormal inflammation [52].
SSc, or scleroderma, is a rare and complex autoimmune disease. Research advancement has improved knowledge on how important contribution of inflammation in disease pathogenesis. Oxidative stress leading to inflammatory cells infiltration, activation of ILC and dendritic cells, inflammasomes and Th cells subpopulations disbalance observed in blood and tissue of SSc are the proposed triggering events leading to early endothelial damage which leadsto vasculopathy and vicious circle of extracellular matrix deposition. Dysregulation of innate and adaptive immunity involve several pro-inflammatory cytokines and mediators that regulate fibroblast production of ECM. Several studies proposed the successful treatment of SSc by inhibiting cytokine mediators and inflammasomes, which led to new therapeutic concepts. Future research of these cytokines is warranted to strengthen their role as treatments and biomarkers of SSc in precision medicine.
References
1.Szabo I, Muntean L, Crisan T, Rednic V, Sirbe C, Rednic S. Novel concepts in systemic sclerosis pathogenesis: Role of miRNAs. Biomedicine. 2021;9(1471):1-18. DOI: 10.3390/biomedicines9101471
2.Benfaremo D, Svegliati S, Paolini C, Agarbati S, Moroncini G. Systemic sclerosis: From pathophysiology to novel therapeutic approaches. Biomedicine. 2022;10(163):1-21. DOI: 10.3390/biomedicines10010163
3.Sepulveda AS, Gonzalez AE, Suarez SA, Lima DE, Islas AE, Castaneda AR, et al. Systemic sclerosis pathogenesis and emerging therapies, beyond the fibroblast. BioMed Research International. 2019;2019:1-16. DOI: 10.1155/2019/4569826
4.Gorniak BG, Puszczewicz M. Oxidative damage and antioxidative therapy in systemic sclerosis. Mediators of Inflammation. 2014;2014:1-12. DOI: 10.1155/2014/389582
5.Suhee K, Park HJ, Lee SI. The microbiome in systemic sclerosis: Pathophysiology and therapeutic potential. International Journal of Molecular Sciences. 2022;23(16154):1-23. DOI: 10.3390/ijms232416154
6.Pattanaik D, Brown M, Postlethwaite BC, Postlethwaite AE. Pathogenesis of systemic sclerosis. Frontiers in Immunology. 2015;6(272):1-40. DOI: 10.3389/fimmu.2015.00272
7.Mitev A, Christ L, Feldman D, Binder M, Moller K, Kanne AM, et al. Inflammatory stays inflammatory: A subgroup of systemic sclerosis characterized by high morbidity and inflammatory resistance to cyclophosphamide. Arthritis Research & Therapy. 2019;21(262):1-10. DOI: 10.1186/s13075-019-2057-x
8.Muangchan C, Harding S, Khimdas S, Bonner A, Barron M, Pope J, et al. Association of C-reactive protein with high disease activity in systemic sclerosis: Results from the Canadian Scleroderma Research Group. Arthritis Care & Research. 2012;64(9):1405-1444. DOI: 10.1002/acr.21716
9.Liu J, Zhang H, Su Y, Zhang B. Application and prospect of targeting innate immune sensors in the treatment of autoimmune diseases. Cell & Bioscience. 2022;12(68):1-19. DOI: 10.1186/s13578-022-00810-w
10.Truchetet ME, Brembilla NC, Chizzolini C. Current concepts on the pathogenesis of systemic sclerosis. Clinical Reviews in Allergy and Immunology. 2021;2021:1-22. DOI: 10.1007/s12016-021-08889-8
11.Frasca L, Lande R. Toll-like receptors in mediating pathogenesis of systemic sclerosis. Clinical and Experimental Immunology. 2020;201(1):14-24. DOI: 10.1111/cei.13426
12.O’Reilly S. Toll-like receptors in systemic sclerosis: An emerging target. Immunology Letters. 2018;195:2-8. DOI: 10.1016/j.imlet.2017.09.001
13.Zhao K, Kong C, Shi N, Jiang J, Li P. Potential angiogenic, immunomodulatory, and anti-fibrotic effects of mesenchymal stem cell derived extracellular vesicles in systemic sclerosis. Frontiers in Immunology. 2023;14(112527):1-13. DOI: 10.3389/fimmu.2023.1125257
14.Mouawad JE, Feghali-Bostwick C. The molecular mechanisms of systemic sclerosis-associated lung fibrosis. International Journal of Molecular Sciences. 2023;24(3):1-14. DOI: 10.3390/ijms24032963
15.Bhattacharyya S, Midwood KS, Varga J. Tenascin-C in fibrosis in multiple organs. Translational implications. Seminars in Cell & Developmental Biology. 2022;128:130-136. DOI: 10.1016/j.semcdb.2022.03.019
16.Marzeda AM, Midwood KS. Internal affairs: Tenascin-C as clinically relevant, endogenous driver of innate immunity. Journal of Histochemistry and Cytochemistry. 2018;66(4):289-304. DOI: 10.1369/0022155418757443
17.Lemanska-Perek A, Adamik B. Fibronectin and its soluble EDA-FN isoforms as biomarkers for inflammation and sepsis. Advances in Clinical and Experimental Medicine. 2019;28(11):1561-1567. DOI: 10.17219/acem/104531
18.Dhanesa N, Chorawala MR, Jain M, Bhalla A, Thedens D, Nayak M, et al. Fn-EDA (fibronectin containing extra domain A) in the plasma, but not endothelial cells, exacerbates stroke outcome by promoting thrombo-inflammation. Stroke. 2019;50:1201-1209. DOI: 10.1161/STROKEAHA.118.023697
19.Xiong J, Zhao Y, Lin Y, Chen Y, Weng Q, Shi C, et al. Identification and characterization of innate lymphoid cells generated from pluripotent stem cells. Cell Reports. 2022;41(5):1-23. DOI: 10.1016/j.celrep.2022.111569
20.Borgia F, Pomi F, Alessandrello C, Vaccaro H, Gangemi S. Potential role of innate lymphoid cells in the pathogenesis and treatment of skin diseases. Journal of Clinical Medicine. 2023;12(3043):1-21. DOI: 10.3390/jcm12083043
21.Jin W, Zheng Y, Zhu P. T cell abnormalities in systemic sclerosis. Autoimmunity Reviews. 2022;21(11):1-10. DOI: 10.1016/j.autrev.2022.103185
22.Al-Adwi Y, Westra J, van Goor H, Burgess JK, Denton CP, Mulder DJ. Macrophages as determinants and regulators of fibrosis in systemic sclerosis. Rheumatology. 2022;2022:1-23. DOI: 10.1093/rheumatology/keac410
23.Sakkas LI, Bogdanos DP. The role of T cells in systemic sclerosis: An update. Immuno. 2022;2(3):534-547. DOI: 10.3390/immuno2030034
24.Gensous N, Charrier M, Duluc D, Contin-Bordes C, Truchetet ME, Lazaro E, et al. T follicular helper cells in autoimmune disorders. Frontiers in Immunology. 2018;2018:9. DOI: 10.3389/fimmu.2018.01637
25.Wei X, Niu X. T follicular helper cells in autoimmune diseases. Journal of Autoimmunity. 2023;134:102976. DOI: 10.1016/j.jaut.2022.102976
26.Beurier P, Ricard L, Eshagh D, Malard F, Siblany L, Fain O, et al. Tfh cells in systemic sclerosis. Journal of Translational Medicine. 2021;19(374):1-9. DOI: 10.1186/s12967-021-03049-0
27.Ricard L, Jachiet V, Malard F, Ye Y, Stocker N, Riviere S, et al. Circulating follicular helper T cells are increased in systemic sclerosis and promote plasmablast differentiation through the IL-21 pathway which can be inhibited by ruxolitinib. Annals of the Rheumatic Disease. 2019;78(4):539-550. DOI: 10.1136/annrheumdis-2018-214382
28.Beesley CF, Goldman NR, Taher TE, Denton CP, Abraham DJ, Mageed RA, et al. Dysregulated B cell function and disease pathogenesis in systemic sclerosis. Frontiers in Immunology. 2023;2023:13
29.De Luca G, Tomelleri A, Dagna L, Matucci-Cerinic M. The target on B cells in systemic sclerosis. “A midsummer dream” to extinguish inflammation and prevent early disease progression to fibrosis. Clinical Rheumatics. 2021;40:2529-2533. DOI: 10.1007/s10067-021-05733-4
30.Liem S, Neppelenbroek S, Fehres CM, Wortel C, Toes RE, Hulzinga T, et al. Autoreactive B cell responses targeting nuclear antigen in systemic sclerosis. Seminars in Arthritis and Rheumatism. 2023;58:152136. DOI: 10.1016/j.semarthrit.2022.152136
31.Choreno-Parra JA, Cervantes-Rosete D, Jimenez-Alvarez LA, Ramirez-Martinez G, Marquez-Garcia JE, Cruz-Lagunas A, et al. Dendritic cells drive probiotic inflammation and aberrant T cell polarization in systemic sclerosis. Rheumatology. 2022;2022:1-12. DOI: 10.1093/rheumatology/keac489
32.Moudgil KD, Venkatesha SH. The anti-inflammatory and immunomodulatory activities of natural products to control autoimmune inflammation. International Journal of Molecular Sciences. 2023;24(95):1-32. DOI: 10.3390/ijms24010095
33.Fioretto BS, Rosa I, Matucci-Cerinic M, Romano E, Manetti M. Current trends in vascular biomarkers for systemic sclerosis: A narrative review. International Journal of Molecular Sciences. 2023;24(4097):1-33. DOI: 10.3390/ijms24044097
34.Deuteralou K, Kitas G, Garyfallos A, Dimitoulas T. Novel insights into the role of inflammasomes in autoimmune and metabolic rhemuatic diseases. Rheumatology International. 2018;2018:1-10. DOI: 10.1007/s00296-018-4074-5
35.Lin C, Jiang Z, Cao L, Zou H, Zhu X. Role of NLRP3 inflammasome in systemic sclerosis. Arthritis Research & Therapy. 2022;24(196):1-10. DOI: 10.1186/s13075-022-02889-5
36.Henderson J, O’Reilly S. Inflammasome lights up in systemic sclerosis. Arthritis Research & Therapy. 2017;19(20s):1-2. DOI: 10.1186/s13075-017-1420-z
37.Henderson J, Bhattacharyya S, Varga J, O’Reilly. Targetting TLRs and the inflammasome in systemic sclerosis. Journal of Pharmaceutical Therapy. 2018;2018:1-39. DOI: 10.1016/j.pharmthera.2018.08.003
38.Cavalli G, Colafrancesco S, Emmi G, Imazio M, Lopalco G, Maggio MC, et al. Interleukin-1 alpha: A comprehensive review of IL-1 alpha in the pathogenesis ad treatment of autoimmune and inflammatory diseases. Autoimmunity Reviews. 2021;20(102763):1-15. DOI: 10.1016/j.autrev.2021.102763
39.Xu D, Mu R, Wei X. The role of IL-1 family cytokines in the pathogenesis of SSc. Frontiers in Immunology. 2025;2019(10):1-8. DOI: 10.3389/fimmu.2019.02025
40.Artlett CM. The IL-1 family of cytokines. Do they have a role in scleroderma fibrosis? Immunology Letters. 2018;195:30-37. DOI: 10.1016/j.imlet.2017.11.012
41.Sedimbi SK, Hagglof T, Karlsson MCI. IL-18 in inflammatory and autoimmune disease. Cellular and Molecular Life Sciences. 2013;13(919973):1-14. DOI: 10.3389/fimmu.2022.919973
42.Lin E, Vincent FB, Sahhar J, Ngian GS, Kandane-Rathnayake R, Mende R, et al. Analysis of serum interleukin-1 alpha, IL-1B and IL-18 in patients with systemic sclerosis. Clinical Translational Immunology. 2019;8(e1045):1-11. DOI: 10.1002%2Fcti2.1045
43.Di Carmine S, Scott MM, McLean MH, McSorley HS. The role of IL-33 in organ fibrosis. Discovery Immunology. 2022;1(1):1-11. DOI: 10.1093/discim/kyac006
44.Li L, Zhu H, Zuo X. Interleukin-33 in systemic sclerosis: Expression and pathogenesis. Frontiers in Immunology. 2018;9(2663):1-7. DOI: 10.3389/fimmu.2018.02663
45.Mortafa M, ELShourgbagy EW, Elsaged RA. The role of IL-33 in severity of SSc. Egyptian Journal of Medical Microbiology. 2022;31(2):1-5. DOI: 10.21608/ejmm.2022.228611
46.Steele H, Cheng J, Willicut A, Dell G, Breckenridge J, Culberson E, et al. TNF super family control of tissue remodelling and fibrosis. Frontiers in Immunology. 2023;14(1219907):1-24. DOI: 10.3389/fimmu.2023.1219907
47.John SR. IL-6 family cytokines. Cold Spring Harbor Perspectives in Biology. 2018;10(a028415):1-17. DOI: 10.1101/cshperspect.a028415
48.Metcalfe RD, Putoczki TL, Griffin MD. Structural understanding of IL-6 family cytokine signaling and targeted therapy: Focus on IL-11. Frontiers in Immunology. 2020;11(1424):1-25. DOI: 10.3389/fimmu.2020.01424
49.Zheng B, Keen KJ, Fritzler MJ, Ryerson CJ, Wilcox P, Whalen BA, et al. Circulating cytokines levels in SSc related interstitial lung disease and idiopathic pulmonary fibrosis. Scientific Reports. 2023;13(6647):1-7. DOI: 10.1038/s41598-023-31232-4
50.Kawaguchi Y. Contribution of IL-6 to the pathogenesis of systemic sclerosis. Journal of Scleroderma Related Disorders. 2017;2(Suppl2):S6-S12. DOI: 10.5301/jsrd.5000258
51.Brembilla NC, Senra L, Boehncke WH. The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Frontiers in Immunology. 2018;9(1682):1-13. DOI: 10.3389/fimmu.2018.01682
52.Wei L, Abraham D, Ong V. The Yin and yang of IL-17 in systemic sclerosis. Frontiers in Immunology. 2022;13(885609):1-7. DOI: 10.3389/fimmu.2022.885609
Written By
Dwitya Elvira and Raveinal Masri
Submitted: 08 May 2023Reviewed: 01 June 2023Published: 09 October 2023