
Abstract
Fibrosis in systemic sclerosis (SSc or scleroderma) is characterized by an abundance of chromosome segregation defects and chromosome instability (CIN) that lead to overactivation of autoimmunity and inflammation. This chapter will emphasize the most recent findings on the involvement of centromere and telomere dysfunction in scleroderma. We will discuss how centromere and telomere dysfunction contribute to CIN, fibrosis, and cellular autoimmunity in scleroderma. We will also summarize how chromosome segregation defects in the form of aneuploidy and micronuclei formation activate the Cyclic GMP–AMP synthase (cGAS) Stimulator of interferon genes (STING) pathway of cellular immunity. Activation of this pathway induces production of inflammatory cytokines IFNβ and IL6. Finally, we will summarize the most recent therapies to block the cGAS-STING pathway and treat fibrosis.
Keywords
- centromeres
- chromosome instability
- telomeres
- fibrosis
- cGAS-STING pathway
- micronuclei
- IFNβ
- IL6
1. Introduction
1.1 Pathogenesis and diagnosis of scleroderma fibrosis
Scleroderma results from increased production and accumulation of collagen and other extracellular matrix (ECM) proteins in body tissues culminating in fibrosis, a thickening of the connective tissue [1]. Fibroblasts, the resident cells of connective tissue are key players of fibrosis in SSc [2]. After tissue damage and immune cell activation, fibroblasts in the ECM differentiate into secretory myofibroblasts in a tissue microenvironment rich in fibroblast growth factor (FGF), transforming growth factor beta (TGFβ), interferon beta (IFNβ) and interleukins 1 and 6 [1, 2]. Buildup of myofibroblasts is responsible for the disproportionate production and accumulation of collagen and ECM proteins in SSc fibrosis [1, 2, 3, 4]. Excessive buildup of collagen in the skin, gastrointestinal tract, lungs, kidneys, heart, or other tissues together with microvascular damage and cell-mediated autoimmunity lead to organ dysfunction.
Scleroderma is subclassified into limited cutaneous (lcSSc) or diffuse cutaneous (dcSSc) based on the degree of hardening and thickening of the skin and organ involvement. LcSSc usually affects the skin of the face and lower limbs, while dcSSc affects large areas of the skin and major organs. Although both types of SSc are very incapacitating, dcSSc patients have higher morbidity and mortality due to increased inflammation and fibrosis of internal organs, especially interstitial lung disease (ILD) and heart fibrosis [5, 6, 7, 8]. The cause of SSc pathogenesis is yet to be uncovered, although a combination of factors, including immune system dysregulation [8], genetic and epigenetic factors such as mutations in HLA genes [9, 10, 11], and reactive oxidizing species (ROS) [12] play a major role. Exposure to environmental factors such as silica, bleomycin, aromatic and chlorinated solvents, ketones, and trichloroethylene have also been linked to the condition in some individuals [2, 3, 4, 8, 13, 14, 15, 16].
The diagnosis of SSc relies on clinical evaluation and detection of antibodies that recognize the nucleus of the patient’s own cells, the so-called antinuclear antibodies (ANAs) [17, 18]. ANAs recognize nuclear components and are detected in as many as 95% of SSc patients [19]. Interestingly, some of these antibodies localize to centromere regions, the middle portion of chromosomes. Anticentromere antibodies (ACAs) were first identified in the lcSSc CREST variant (CREST: calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) [20] and served as crucial reagents to study centromere biology. ACAs are found in up to 43% of lcSSc (mostly all cases of CREST), recognize many centromere proteins (CENPA, CENPB, CENPC, and so on, to CENPT), and are associated with a generally more favorable prognosis [20]. However, ACA-positivity is associated with the development of pulmonary arterial hypertension [18, 19, 20]. CENPB autoantibodies are present in almost all ACA positive lcSSc patients [21]. CENPB autoantibodies can also be found in Sjogren syndrome and neoplasia [21, 22]. CENPA autoantibodies appear to be more specific for SSc patients at risk of pulmonary vascular disease. Autoantibodies that recognize CENPC alone are associated with Sjogren syndrome and CENPF autoantibodies are associated with cancers [22].
ANAs specific for SSc recognize topoisomerase I, an enzyme that breaks single stranded DNA, relaxes supercoiled DNA, and facilitates chromosome condensation [18]. Antitopoisomerase-I antibodies (ATAs; anti-Scl-70) are mostly found in dcSSc patients (prevalence 20–40%), and correlate with poor prognosis, lung fibrosis, and disease progression [17, 18]. Anti-RNA polymerase III autoantibodies are detected in dcSSc patients (prevalence 10–25%), and serve as markers of rapid disease development and renal crisis [17, 18].
Recent studies have also identified ANAs that localize to telomeres, the protective cap of chromosomes, in 9% of SSc patients [23, 24]. These antibodies recognize the shelterin telomeric repeat binding factor 1 (TERF1) protein and are linked to severe lung disease [23]. Despite their clinical value for specific diagnosis and prognosis of SSc, the etiology of ACAs, ATAs, and anti-telomere antibodies in SSc patients is unknown.
1.2 Centromeres
Centromeres are structural units that modulate the proper division of chromosomes. Destabilization of centromere function results in chromosome instability (CIN), a hallmark of birth defects, cancers, and fibrosis [25, 26, 27, 28]. Centromeres have two vital functions; (i) recruit centromere proteins to form the kinetochore, and (ii) keep sister chromatids together before chromosome segregation [29, 30]. Defects in either of these vital functions results in structural and numerical CIN, presented in the forms of lagging chromosomes, dicentric chromosomes, aneuploidy, and micronuclei (Figure 1) [31, 32].
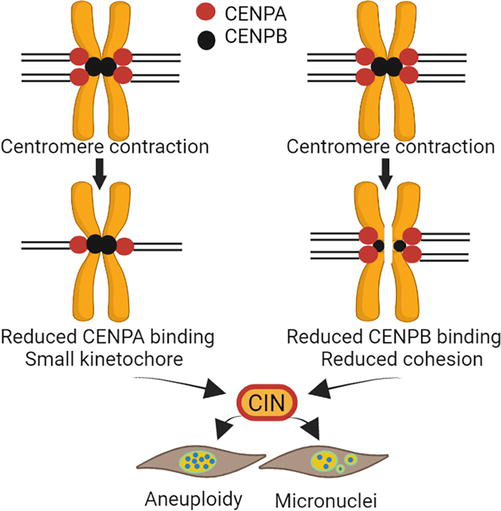
Figure 1.
Centromere function. The H3 CENPA deposits to centromere DNA to initiate the assembly of the kinetochore, which is necessary for microtubule attachment and chromosome segregation. CENPB binds to arrays in the centromere sequence and interacts with CENPA and other centromere proteins to facilitate centromere function. CENPB also binds pericentromere arrays to facilitate chromatid adhesion. Destabilizing these processes, such as centromere sequence deletion or contraction, could reduce CENPA or CENPB binding, leading to CIN. This figure was created with
Centromere DNA sequences contain arrays of high order repeats (HORs), which can extend several megabases in length (Figure 2) [33]. The size of centromere arrays varies among individuals and disease processes. HORs are composed of 171 bp α-satellite units organized in a head-to-tail fashion [33]. During the evolution of hominids, centromeric α-repeats in each chromosome became homogeneous. Today, the α-repeats in each centromere array have 98–100% similarity, yet they have only ~75% similarity to the α-repeats of other centromeres arrays [34, 35]. The centromere core is composed of 1 or 2 arrays [36], while the periphery of the centromeres, the pericentromere, is more diverse, with shorter arrays or monomers of alpha and other repeat types, as well as transposon-like elements [36, 37, 38]. Centromeres have been difficult to study because of their repetitive nature, but the genomic assembly of human centromeres has recently been achieved [39, 40, 41]. Even so, the human population has a large variation in centromere size and content [41, 42, 43].
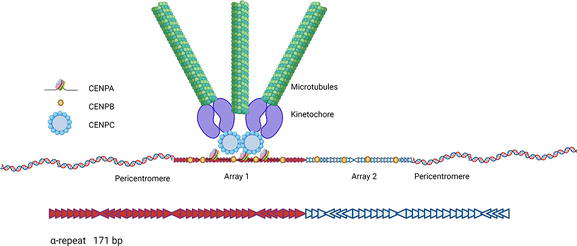
Figure 2.
Organization of human centromeres. Centromere sequences are composed of α-repeats, ~171 bp units, organized in a head-to-tail fashion (arrows). Centromeres sequences are composed of one or two arrays of HORs. Pericentromere sequence contains α- and other satellites. The H3 histone CENPA deposits to the largest array of the core to start the assembly of the kinetochore, which is necessary for microtubule attachment and chromosome replication. H3K9me3 remains abundant at the pericentromeres. CENPB binding to centromere sequences and its interaction with CENPA and CENPC stabilize the formation of the kinetochore. CENPB also binds pericentromere arrays to facilitate chromatid cohesion. This figure was created with
The function of the centromere is modulated epigenetically by CENPA, a H3-histone variant that forms nucleosomes with H2A, H2B, and H4 at active centromere arrays or elsewhere and defines the location of kinetochore formation (Figure 2) [29]. CENPB, a protein that binds to specific DNA sequences in centromeres (CENPB boxes), enhances this centromere chromatin interaction. CENPB interaction with H3K9me3 helps maintain sister chromatid cohesion [30, 44, 45]. Despite the prevalence of ACAs in SSc, the role that centromere sequences and proteins play in the pathogenesis of SSc has remained unexplored.
1.3 Telomeres
Telomeres are specialized chromatin structures that protect chromosome ends. Telomere sequences are regions of TTAGGG repeats that shrink in dividing cells during cell replication [46, 47, 48, 49]. The main functions of the telomeres are to (i) protect the terminal regions of the chromosomes from progressive degradation, and (ii) prevent chromosome fusion by ensuring that the DNA repair systems do not mistakenly recognize the very ends of the DNA as DSBs. Excessive telomere shortening or telomere uncapping can produce telomere fusions, which results in CIN, mostly in the forms of dicentric, lagging chromosomes, and micronuclei (Figure 3). Telomerase holoenzyme helps maintain telomere length by synthesizing telomeric DNA [46]. Shelterin complex proteins bind and protect telomeres from degradation and allow access of telomerase and helicase to telomere DNA [46, 47, 48, 49]. Although telomerase activity is reduced in somatic cells, it is upregulated in cells that undergo rapid expansion, such as the cells involved in ILD and fibrosis [47, 48]. Thus, it is a distinct possibility that telomerase plays a crucial role in SSc fibrosis.
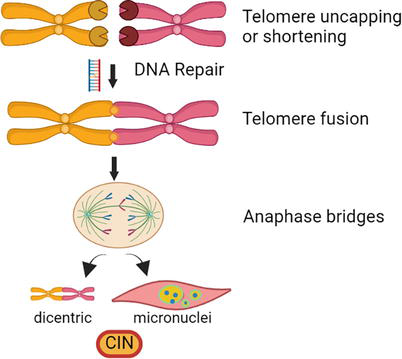
Figure 3.
Telomere function. The telomeric chromatin structure protects chromosome ends from recombination, repair, and fusion. Telomerase holoenzyme counteracts telomere DNA degradation during replication by adding DNA to telomeres, thus preserving telomeres for future cell division. Deletion or degradation of telomeres produces CIN. Recombination and subsequent repair of degraded telomeres can fuse chromosomes, leading to dicentric and/or micronuclei formation. This figure was created with
1.4 Chromosome instability and activation of cGAS-STING/type I interferon pathway
cGAS is an enzyme that senses cytosolic double-stranded DNA from pathogens or damaged self-DNA, such as micronuclei, as part of the innate immune response (Figure 4) [50, 51, 52, 53]. Upon binding to cytosolic DNA, cGAS produces cGAMP, which interacts with STING to trigger downstream activation of the type I interferon pathway [52, 53]. STING recruits TBK1 to phosphorylate the transcription factor IRF3, which translocates to the nucleus and induce production of type I interferons including IFNβ [53]. Activation of STING also triggers RELA (p65), a component of the NFKB that induces IFNβ and pro-inflammatory cytokines, such as IL6 (Figure 4) [49]. cGAS-STING pathway activation is impeded by BANF, which competes with cGAS binding to intracellular DNA, and TREX1, which associates with cGAS and degrades the DNA [54, 55, 56]. Fibroblasts from cGAS KO mice do not produce IFNβ following DNA transfection [56]. Aberrant detection of cytosolic DNA has been implicated in cancers and autoimmune diseases [57]. It has also been suggested that cGAS-STING is overactivated in autoimmune diseases such as systemic lupus erythematosus (SLE) [58, 59], and interferons are known to be increased in SSc [60, 61]. Additionally, there are recent findings showing that the cGAS-STING pathway independently triggers fibrosis [62, 63]. As we show below, lesion fibroblasts from SSc patients show increased activation of the cGAS-STING/type I Interferon pathway [64].
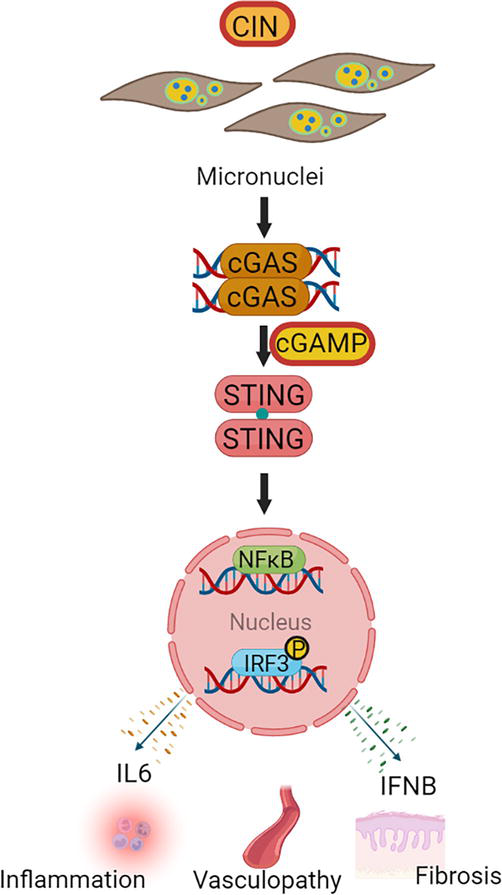
Figure 4.
Function of cGAS-STING pathway. cGAS senses cytosolic DNA from viruses as well as damaged DNA or micronuclei from CIN. Binding of cGAS to cytosolic DNA produces cGAMP, a molecule that interacts with STING to activate the type I interferon pathway. STING activates the expression of NF-κB and phosphorylates IRF3 to induce the production of IL6 and IFNβ, cytokines that mediate inflammation, vasculopathy, and fibrosis.
2. Chromosome abnormalities in scleroderma cells
Several investigators have noted chromosomal abnormalities in SSc [28, 64, 65, 66, 67, 68, 69, 70], including numerical and structural CIN. These findings included micronuclei formation, lagging chromosomes, aneuploidy and polyploidy, chromatid aberrations with gaps and breaks, acentric chromosome fragments, rearrangements, dicentric, and ring forms. These aberrations were specific to lymphocytes, lesion fibroblasts, and bone marrow cells. These cytogenetic abnormalities were more common in advanced forms of SSc, but not in CREST or Raynaud phenomena. Our studies showed increased aneuploidy in forearm lesion skin fibroblasts from dcSSc but not lcSSc patients, which is in agreement with these findings [64]. However, we did not see chromosome abnormalities in lymphocytes or macrophages from the same patients and, the reason for these inconsistencies remains to be clarified. Abnormal nuclear morphology and micronuclei formation were the most common CIN structural abnormalities found in involved skin fibroblasts from SSc patients, suggesting that lagging chromosomes and chromosome fragments failed to be incorporated into the nucleus and were enveloped separately with the nuclear membrane [64, 69, 70]. The clastogenic agent bleomycin further increased micronuclei formation in SSc patient fibroblasts from affected and unaffected skin, but not in skin fibroblasts from healthy controls [69]. The increased chromosomal breakage in dcSSc was not associated with cancer development or chromosome breakage syndromes [67], although it appears to also be common in SSc family members [65].
3. Centromere defects in scleroderma
Abnormal centromere assembly, localization, and function may lead to errors in chromosome segregation and production of centromere antibodies. Recent studies in our laboratory demonstrated that centromere DNA sequences are shorter at several chromosomes in SSc patients, particularly in lesion dermal fibroblasts [64]. Although changes in the length of centromeres among human populations have been reported [41, 42, 43], such dramatic centromere deletion in several chromosomes is pervasive in SSc. Centromere deletions appear to dominate the landscape of several chromosomes in dcSSc, yet centromere arrays in chromosomes 1 and 2 were consistently found deleted in SSc patients [64]. It is unclear why these centromere arrays were particularly deleted, and it is not known whether the deletions contribute to SSc pathology. Further studies indicated these centromere changes were not the result of culture conditions or the previous therapy received by the patients. It remains to be explored whether changes at centromere sequences directly affect the deposition and strength of the kinetochore during chromosome segregation. However, centromere deletions in SSc were found to correlate with abnormal CENPA expression and deposition, which lead to CIN. In fact, SSc fibroblasts show a lack CENPA deposition in micronuclei with CENPB boxes, suggesting that centromere DNA sequences influence centromere function and chromosome segregation as shown in Figure 1.
A second important observation was understanding of how centromere changes in SSc patients lead to the production of ACAs [64]. Our studies identified specific centromere defects in lcSSc patients that produce ACAs. Nuclear proteins CENPA and CENPB leaked and colocalized into the cytoplasm and cell membrane of lcSSc dermal fibroblasts. The integrity of the nuclear membrane appears to be one critical factor that contributes to the leaking of centromere proteins, although it is expected that other factors play a role in the specificity of centromere protein misplacement. It remains to be explored what factors, other than rupture of the nuclear membrane, lead to leaking of CENP proteins only in lcSSc patients that produce ACAs. CENPB in the cytoplasm of lcSSc lesion fibroblasts co-localizes with MHC class II molecules DRB1 and DRB5, which suggests a likely mechanism for ACA production in lcSSc [64]. It is possible that the size and complexity in structure of CENPB and perhaps other CENP proteins are more immunogenic. Whether lesion fibroblasts are the sole drivers of aberrant autoimmunity in lcSSc or whether other cell types also act as antigen-presenting cells is unknown.
4. Telomere defects in scleroderma
Studies aimed at understanding the role of telomeres in SSc have found that, similar to centromere deletions, telomere attrition is detected in SSc patients. These recent findings were evident in a subset of SSc patients and telomere shortening was also found to be more prominent in patients with ILD [71, 72]. Whether such telomere deletions also exist in involved skin fibroblasts remains to be studied. Other studies revealed that shorter telomeres in lymphocytes of SSc patients correlated with the production of autoantibodies targeting telomere-associated proteins (shelterin) in a subset of patients. Further, the production of telomere antibodies also correlated with ILD [23, 24]. The recent discovery of telomere autoantibodies in SSc patients may not have been possible before due to the spatiotemporal expression of these proteins and tissue specificity [73]. In contrast, ACAs were discovered in CREST patients even before we understood the structure of centromeres. It is possible that telomere attrition leads to an excess of shelterin proteins that leak to the cytoplasm and act as antigens in a subset of SSc patients. Taken together, these studies suggest that telomere dysfunction affects chromosome segregation and may promote inflammation and fibrosis in SSc as indicated in Figure 4.
5. Chromosome instability and cGAS-STING activation in scleroderma and fibrosis
Production of micronuclei and release of DNA into the cytoplasm can activate the cGAS-STING pathway of inflammation and fibrosis [50, 51, 52, 53]. Previous reports have shown that SSc fibroblasts produce increased amounts of IL6 and IFNβ, the final molecules activated in the cGAS-STING/type I interferon pathway, although they can be activated by other cellular and immune pathways [74, 75]. Although IL6 and IFNβ may be promising targets to treat SSc, the molecular mechanisms of how these proteins are activated was only recently discovered. The production of IL6 in serum of patients and dermal fibroblasts appears to be specific to SSc clinical phenotypes of dcSSc, such as ILD, pulmonary arterial hypertension, gastrointestinal involvement, and cardiac involvement [76, 77]. On the other hand, increased production of IFNβ and activation of IFN-regulated genes has been observed in the serum and involved skin tissue of patients with SSc [75]. Furthermore, the IFNβ score correlated with markers of disease severity [75]. We observed that increased amounts of micronuclei in SSc dermal fibroblasts strongly correlated with the expression of
Although inhibitors of the cGAS-STING pathway have been only examined in autoimmunity in SLE, blocking the final cytokines of the pathway have been explored in SSc. Blockage of IL6 has shown to be promising for reversing the production of TGFβ, a cytokine involved in dermal fibrosis [78]. Further, the efficacy of IL6 inhibitors, particularly Tocilizumab in clinical trials, finishing phase 3, has been satisfactory in preserving lung function, although the primary skin fibrosis endpoint was not met [79, 80]. IFNβ inhibitors per se have been suggested but never studied in SSc [81]. We believe that by blocking the pathway early would prevent the release of both IL6 and IFNβ, attacking the fibrotic and inflammatory component of the disease with a synergistic effect.
We confirmed that cGAS activation was responsible for the IFNβ production in SSc skin cells, treating lesion fibroblasts with G150, a cGAS-specific inhibitor [63]. G150 treatment strongly reduced the cGAS-induced enzymatic (2′3’ cGAMP) activity and the production of IFNβ, although the levels of IL6 were not tested. Future research needs to address if cGAS or STING inhibition blocks both cytokines, but also reduces fibrosis of the skin and other organs. Even though current cGAS-STING pathway inhibitors show promising efficacy, they present toxicity to the cells and may harm the body in the long term [82, 83]. Recently, novel CGAS-STING inhibitors have been discovered showing different degrees of specificity for cell types and phenotypes [82, 83]. Therefore, the specificity of new molecules for treating SSc subtypes and phenotypes deserves to be addressed thoroughly for optimal efficacy and safety.
6. Conclusions
We find that telomere and centromere defects at the genetic and epigenetic levels are prevalent in SSc patients and may contribute to chromosome missegregation. Centromere and telomere sequence deletions appear to be important factors that contribute to chromosome segregation defects, and hence, lead to CIN. Such centromere and telomere deletions may have stemmed from ROS or perhaps from exposure to environmental factors that damage the DNA and/or interfere with DNA repair pathways. This review brings to light a new target pathway in SSc, the cGAS-STING/IFNβ pathway, which can contribute to fibrosis. Targeting the cGAS-STING/ IFNβ pathway with newly-developed molecules could prove to be a viable route to treat SSc fibrosis.
Acknowledgments
We are grateful to Drs. David Markovitz, Mark Kaplan, Dinesh Khanna, and Harmit Malik for discussions. R.C-G. was supported by The Hormel Institute, the Rheumatology Research Foundation, an NIH/NCI grant R21-CA259630, and the National Scleroderma Foundation. R.C.-G. is the recipient of the Mark Flapan Award and the Marta Marx Fund for the Eradication of Scleroderma Award from the National Scleroderma Foundation.
Acronyms and abbreviations
Cyclic GMP–AMP synthase | |
Stimulator of interferon genes | |
Systemic Sclerosis | |
limited cutaneous systemic sclerosis | |
diffuse cutaneous systemic sclerosis | |
reactive oxidizing species | |
extracellular matrix | |
fibroblast growth factor | |
transforming growth factor β | |
interferon beta | |
interleukin 1 | |
interleukin 6 | |
antinuclear antibodies | |
anticentromere antibodies | |
calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia | |
antitopoisomerase-I antibodies | |
double-stranded breaks | |
telomeric repeat binding factor 1 | |
centromere protein | |
chromosome instability | |
high order repeats | |
TANK-binding kinase 1 | |
REL-associated protein | |
Nuclear factor kappa B | |
Interferon regulatory factor 3 | |
BAF Nuclear Assembly Factor 1 | |
Three Prime Repair Exonuclease 1 | |
Systemic lupus erythematosus |
References
- 1.
Gilbane AJ, Denton CP, Holmes AM. Scleroderma pathogenesis: A pivotal role for fibroblasts as effector cells. Arthritis Research & Therapy. 2013; 15 :215. DOI: 10.1186/ar4230 - 2.
Katsumoto TR, Whitfield ML, Connolly MK. The pathogenesis of systemic sclerosis. Annual Review of Pathology. 2011; 6 :509-537. DOI: 10.1146/annurev-pathol-011110-130312 - 3.
Bhattacharyya S, Wei J, Varga J. Understanding fibrosis in systemic sclerosis: Shifting paradigms, emerging opportunities. Nature Reviews Rheumatology. 2011; 8 :42-54. DOI: 10.1038/nrrheum.2011.149 - 4.
Varga J, Abraham D. Systemic sclerosis: A prototypic multisystem fibrotic disorder. The Journal of Clinical Investigation. 2007; 117 :557-567. DOI: 10.1172/JCI31139 - 5.
Barnes J, Mayes MD. Epidemiology of systemic sclerosis: Incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Current Opinion in Rheumatology. 2012; 24 :165-170. DOI: 10.1097/BOR.0b013e32834ff2e8 - 6.
Elhai M, Meune C, Boubaya M, Avouac J, Hachulla E, Balbir-Gurman A, et al. Mapping and predicting mortality from systemic sclerosis. Annals of the Rheumatic Diseases. 2017; 76 :1897-1905. DOI: 10.1136/annrheumdis-2017-211448 - 7.
Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. The New England Journal of Medicine. 2009; 360 :1989-2003. DOI: 10.1056/NEJMra0806188 - 8.
Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017; 390 :1685-1699. DOI: 10.1016/S0140-6736(17)30933-9 - 9.
Feghali-Bostwick C, Medsger TA Jr, Wright TM. Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis and Rheumatism. 2003; 48 :1956-1963. DOI: 10.1002/art.11173 - 10.
Tsou PS, Sawalha AH. Unfolding the pathogenesis of scleroderma through genomics and epigenomics. Journal of Autoimmunity. 2017; 83 :73-94. DOI: 10.1016/j.jaut.2017.05.004 - 11.
Fioretto BS, Rosa I, Romano E, Wang Y, Guiducci S, Zhang G, et al. The contribution of epigenetics to the pathogenesis and gender dimorphism of systemic sclerosis: A comprehensive overview. Therapeutic Advance Musculoskeletal Disease. 2020; 12 :1759720X20918456. DOI: 10.1177/1759720X20918456 - 12.
Vona R, Giovannetti A, Gambardella L, Malorni W, Pietraforte D, Straface E. Oxidative stress in the pathogenesis of systemic scleroderma: An overview. Journal of Cellular and Molecular Medicine. 2018; 22 (7):3308-3314. DOI: 10.1111/jcmm.13630 - 13.
Nietert PJ, Silver RM. Systemic sclerosis: Environmental and occupational risk factors. Current Opinion in Rheumatology. 2000; 12 :520-526. DOI: 10.1097/00002281-200011000-00008 - 14.
Shivakumar DS, Kamath NS, Naik A. Silica associated systemic sclerosis: An occupational health hazard. BML Case Reports. 2023; 16 :e253952. DOI: 10.1136/bcr-2022-253952 - 15.
Muntyanu A, Milan R, Rahme E, LaChance A, Ouchene L, Cormier M, Litvinov IV, Hudson M, Baron M, Netchiporouk E; Canadian Scleroderma Research Group. Exposure to silica and systemic sclerosis: A retrospective cohort study based on the Canadian scleroderma research group. Frontiers in Medicine (Lausanne). 2022;9:984907. DOI: 10.3389/fmed.2022.984907 - 16.
Huang J, Puente H, Wareing NE, Wu M, Mayes MD, Karmouty- Quintana H, et al. STAT6 suppression prevents bleomycin-induced dermal fibrosis. The FASEB Journal. 2023; 37 :e22761. DOI: 10.1096/fj.202200994R - 17.
Affandi AJ, Radstake TR, Marut W. Update on biomarkers in systemic sclerosis: Tools for diagnosis and treatment. Seminars in Immunopathology. 2015; 37 :475-487. DOI: 10.1007/s00281-015-0506-4 - 18.
Kuwana M. Circulating anti-nuclear antibodies in systemic sclerosis: Utility in diagnosis and disease subsetting. Journal of Nippon Medical School. 2017; 84 :56-63. DOI: 10.1272/jnms.84.56 - 19.
Salazar GA, Assassi S, Wigley F, Hummers L, Varga J, Hinchcliff M, et al. Antinuclear antibody-negative systemic sclerosis. Seminars in Arthritis and Rheumatism. 2015; 44 :680-686. DOI: 10.1016/j.semarthrit.2014.11.006 - 20.
Song G, Hu C, Zhu H, Wang L, Zhang F, Li Y, et al. New centromere autoantigens identified in systemic sclerosis using centromere protein microarrays. The Journal of Rheumatology. 2013; 40 :461-468. DOI: 10.3899/jrheum.120264 - 21.
Prasad RM, Bellacosa A, Yen TJ. Clinical and molecular features of anti-CENP-B autoantibodies. Journal of Molecular Pathology. 2021; 2 :281-295. DOI: 10.3390/jmp2040024 - 22.
Kajio N, Takeshita M, Suzuki K, Kaneda Y, Yamane H, Ikeura K, et al. Anti-centromere antibodies target centromere-kinetochore macrocomplex: A comprehensive autoantigen profiling. Annals of the Rheumatic Diseases. 2021; 80 :651-659. DOI: 10.1136/annrheumdis-2020-218881 - 23.
Adler BL, Boin F, Wolters PJ, Bingham CO, Shah AA, Greider C, et al. Autoantibodies targeting telomere-associated proteins in systemic sclerosis. Annals of the Rheumatic Diseases. 2021; 80 :912-919. DOI: 10.1136/annrheumdis-2020-218918 - 24.
Vulsteke JB, Smith V, Bonroy C, Derua R, Blockmans D, De Haes P, et al. Identification of new telomere- and telomerase-associated autoantigens in systemic sclerosis. Journal of Autoimmunity. 2023; 135 :102988. DOI: 10.1016/j.jaut.2022.102988 - 25.
Verdaasdonk JS, Bloom K. Centromeres: Unique chromatin structures that drive chromosome segregation. Nature Reviews. Molecular Cell Biology. 2011; 12 :320-332. DOI: 10.1038/nrm3107 - 26.
Yuen KW, Montpetit B, Hieter P. The kinetochore and cancer: what's the connection? Current Opinion in Cell Biology. 2005; 17 :576-582. DOI: 10.1016/j.ceb.2005.09.012 - 27.
Teh MT, Tilakaratne WM, Chaplin T, Young BD, Ariyawardana A, Pitiyage G, et al. Fingerprinting genomic instability in oral submucous fibrosis. Journal of Oral Pathology & Medicine. 2008; 37 :430-436. DOI: 10.1111/j.1600-0714.2008.00643.x - 28.
Emerit I. Chromosomal breakage in systemic sclerosis and related disorders. Dermatologica. 1976; 153 :145-156. DOI: 10.1159/000251109 - 29.
De Rop V, Padeganeh A, Maddox PS. CENP-A: The key player behind centromere identity, propagation, and kinetochore assembly. Chromosoma. 2012; 121 :527-538. DOI: 10.1007/s00412-012-0386-5 - 30.
Fachinetti D, Han JS, McMahon MA, Ly P, Abdullah A, Wong AJ, et al. DNA sequence-specific binding of CENP-B enhances the Fidelity of human centromere function. Developmental Cell. 2015; 33 :314-327. DOI: 10.1016/j.devcel.2015.03.020 - 31.
Krupina K, Goginashvili A, Cleveland DW. Causes and consequences of micronuclei. Current Opinion in Cell Biology. 2021; 70 :91-99. DOI: 10.1016/j.ceb.2021.01.004 - 32.
Fenech M, Knasmueller S, Bolognesi C, Holland N, Bonassi S, Kirsch-Volders M. Micronuclei as biomarkers of DNA damage, aneuploidy, inducers of chromosomal hypermutation and as sources of pro-inflammatory DNA in humans. Mutation Research, Reviews in Mutation Research. 2020; 786 :108342. DOI: 10.1016/j.mrrev.2020.108342 - 33.
Liehr T. Microscopic and submicroscopic copy number variations (CNVs) in genetics and counseling. In: Benign & Pathological Chromosomal Imbalances. 1st ed. Amsterdam: Academic Press, Elsevier; 2013. p. 220. DOI: 10.1016/c2012-0-00243-8 - 34.
Koga A, Hirai Y, Terada S, Jahan I, Baicharoen S, Arsaithamkul V, et al. Evolutionary origin of higher-order repeat structure in alpha-satellite DNA of primate centromeres. DNA Research. 2014; 21 :407-415. DOI: 10.1093/dnares/dsu005 - 35.
Roizès G. Human centromeric alphoid domains are periodically homogenized so that they vary substantially between homologues. Mechanism and implications for centromere functioning. Nucleic Acids Research. 2006; 34 :1912-1924. DOI: 10.1093/nar/gkl137 - 36.
Ng TM, Waples WG, Lavoie BD, Biggins S. Pericentromeric sister chromatid cohesion promotes kinetochore biorientation. Molecular Biology of the Cell. 2009; 20 :3818-3827. DOI: 10.1091/mbc.e09-04-0330 - 37.
Zahn J, Kaplan MH, Fischer S, Dai M, Meng F, Saha AK, et al. Expansion of a novel endogenous retrovirus throughout the pericentromeres of modern humans. Genome Biology. 2015; 16 :74. DOI: 10.1186/s13059-015-0641-1 - 38.
Contreras-Galindo R, Kaplan MH, He S, Contreras-Galindo AC, Gonzalez-Hernandez MJ, Kappes F, et al. HIV infection reveals widespread expansion of novel centromeric human endogenous retroviruses. Genome Research. 2013; 23 :1505-1513. DOI: 10.1101/gr.144303.112 - 39.
Jain M, Olsen HE, Turner DJ, Stoddart D, Bulazel KV, Paten B, et al. Linear assembly of a human centromere on the Y chromosome. Nature Biotechnology. 2018; 36 :321-323. DOI: 10.1038/nbt.4109 - 40.
Miga KH, Koren S, Rhie A, Vollger MR, Gershman A, Bzikadze A, et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature. 2020; 585 :79-84. DOI: 10.1038/s41586-020-2547-7 - 41.
Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, et al. The complete sequence of a human genome. Science. 2022; 376 :44-53. DOI: 10.1126/science.abj6987 - 42.
Altemose N, Logsdon GA, Bzikadze AV, Sidhwani P, Langley SA, Caldas GV, et al. Complete genomic and epigenetic maps of human centromeres. Science. 2022; 376 :eabl4178. DOI: 10.1126/science.abl4178 - 43.
Contreras-Galindo R, Fischer S, Saha AK, Lundy JD, Cervantes PW, Mourad M, et al. Rapid molecular assays to study human centromere genomics. Genome Research. 2017; 27 :2040-2049. DOI: 10.1101/gr.219709.116 - 44.
Rosandić M, Paar V, Basar I, Gluncić M, Pavin N, Pilas I. CENP-B box and pJalpha sequence distribution in human alpha satellite higher-order repeats (HOR). Chromosome Research. 2006; 14 :735-753. DOI: 10.1007/s10577-006-1078-x - 45.
Ohzeki J, Nakano M, Okada T, Masumoto H. CENP-B box is required for de novo centromere chromatin assembly on human alphoid DNA. The Journal of Cell Biology. 2002; 159 :765-775. DOI: 10.1083/jcb.200207112 - 46.
Shay JW, Wright WE. Telomeres and telomerase: Three decades of progress. Nature Reviews. Genetics. 2019; 20 :299-309. DOI: 10.1038/s41576-019-0099-1 - 47.
Liu T, Ullenbruch M, Young Choi Y, Yu H, Ding L, Xaubet A, et al. Telomerase and telomere length in pulmonary fibrosis. American Journal of Respiratory Cell and Molecular Biology. 2013; 49 :260-268. DOI: 10.1165/rcmb.2012-0514OC - 48.
Arish N, Petukhov D, Wallach-Dayan SB. The role of telomerase and telomeres in interstitial lung diseases: From molecules to clinical implications. International Journal of Molecular Sciences. 2019; 20 :2996. DOI: 10.3390/ijms20122996 - 49.
Srinivas N, Rachakonda S, Kumar R. Telomeres and telomere length: A general overview. Cancers (Basel). 2020; 12 :558. DOI: 10.3390/cancers12030558 - 50.
Bai J, Liu F. Nuclear cGAS: Sequestration and beyond. Protein & Cell. 2022; 13 :90-101. DOI: 10.1007/s13238-021-00869-0 - 51.
Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017; 548 :461-465. DOI: 10.1038/nature23449 - 52.
Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013; 339 :786-791. DOI: 10.1126/science.1232458 - 53.
Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nature Reviews. Immunology. 2021; 21 :548-569. DOI: 10.1038/s41577-021-00524-z - 54.
Guey B, Wischnewski M, Decout A, Makasheva K, Kaynak M, Sakar MS, et al. BAF restricts cGAS on nuclear DNA to prevent innate immune activation. Science. 2020; 369 :823-828. DOI: 10.1126/science.aaw6421 - 55.
Hemphill WO, Simpson SR, Liu M, Salsbury FR Jr, Hollis T, Grayson JM, et al. TREX1 as a novel immunotherapeutic target. Frontiers in Immunology. 2021; 12 :660184. DOI: 10.3389/fimmu.2021.660184 - 56.
Fu Y, Fang Y, Lin Z, Yang L, Zheng L, Hu H, et al. Inhibition of cGAS-mediated interferon response facilitates transgene expression. iScience. 2020; 23 :101026. DOI: 10.1016/j.isci.2020.101026 - 57.
Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. The Journal of Experimental Medicine. 2018; 215 :1287-1299. DOI: 10.1084/jem.20180139 - 58.
Thim-Uam A, Prabakaran T, Tansakul M, Makjaroen J, Wongkongkathep P, Chantaravisoot N, et al. STING mediates lupus via the activation of conventional dendritic cell maturation and plasmacytoid dendritic cell differentiation. iScience. 2020; 23 :101530. DOI: 10.1016/j.isci.2020.101530 - 59.
Murayama G, Chiba A, Kuga T, Makiyama A, Yamaji K, Tamura N, et al. Inhibition of mTOR suppresses IFNα production and the STING pathway in monocytes from systemic lupus erythematosus patients. Rheumatology (Oxford, England). 2020; 59 :2992-3002. DOI: 10.1093/rheumatology/keaa060 - 60.
Fernandez-Ruiz R, Niewold TB. Type I interferons in autoimmunity. The Journal of Investigative Dermatology. 2022; 142 (3 Pt B):793-803. DOI: 10.1016/j.jid.2021.11.031 - 61.
Vlachogiannis NI, Tual-Chalot S, Zormpas E, Bonini F, Ntouros PA, Pappa M, et al. Adenosine-to-inosine RNA editing contributes to type I interferon responses in systemic sclerosis. Journal of Autoimmunity. 2021; 125 :102755. DOI: 10.1016/j.jaut.2021.102755 - 62.
Yong H, Wang S, Song F. Activation of cGAS/STING pathway upon TDP-43-mediated mitochondrial injury may be involved in the pathogenesis of liver fibrosis. Liver International. 2021; 41 :1969-1971. DOI: 10.1111/liv.14895 - 63.
Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, et al. Mitochondrial damage and activation of the STING pathway Lead to renal inflammation and fibrosis. Cell Metabolism. 2019; 30 :784-799.e5. DOI: 10.1016/j.cmet.2019.08.003 - 64.
Paul S, Kaplan MH, Khanna D, McCourt PM, Saha AK, Tsou PS, et al. Centromere defects, chromosome instability, and cGAS-STING activation in systemic sclerosis. Nature Communications. 2022; 13 :7074. DOI: 10.1038/s41467-022-34775-8 - 65.
Emerit I, Housset E, Feingold J. Chromosomal breakage and scleroderma: Studies in family members. The Journal of Laboratory and Clinical Medicine. 1976; 88 :81-86 - 66.
Wolff DJ, Needleman BW, Wasserman SS, Schwartz S. Spontaneous and clastogen induced chromosomal breakage in scleroderma. The Journal of Rheumatology. 1991; 18 :837-840 - 67.
Pan SF, Rodnan GP, Deutsch M, Wald N. Chromosomal abnormalities in progressive systemic sclerosis (scleroderma) with consideration of radiation effects. The Journal of Laboratory and Clinical Medicine. 1975; 86 :300-308 - 68.
Jabs EW, Tuck-Muller CM, Anhalt GJ, Earnshaw W, Wise RA, Wigley F. Cytogenetic survey in systemic sclerosis: Correlation of aneuploidy with the presence of anticentromere antibodies. Cytogenetics and Cell Genetics. 1993; 63 :169-175. DOI: 10.1159/000133527 - 69.
Martins EP, Fuzzi HT, Kayser C, Alarcon RT, Rocha MG, Chauffaille ML, et al. Increased chromosome damage in systemic sclerosis skin fibroblasts. Scandinavian Journal of Rheumatology. 2010; 39 :398-401. DOI: 10.3109/03009741003685640 - 70.
Migliore L, Bevilacqua C, Scarpato R. Cytogenetic study and FISH analysis in lymphocytes of systemic lupus erythematosus (SLE) and systemic sclerosis (SS) patients. Mutagenesis. 1999; 14 :227-231. DOI: 10.1093/mutage/14.2.227 - 71.
Usategui A, Municio C, Arias-Salgado EG, Martín M, Fernández-Varas B, Del Rey MJ, et al. Evidence of telomere attrition and a potential role for DNA damage in systemic sclerosis. Immunity & Ageing. 2022; 19 :7. DOI: 10.1186/s12979-022-00263-2 - 72.
Lakota K, Hanumanthu VS, Agrawal R, Carns M, Armanios M, Varga J. Short lymphocyte, but not granulocyte, telomere length in a subset of patients with systemic sclerosis. Annals of the Rheumatic Diseases. 2019; 78 :1142-1144. DOI: 10.1136/annrheumdis-2018-214499 - 73.
Wagner KD, Ying Y, Leong W, Jiang J, Hu X, Chen Y, et al. The differential spatiotemporal expression pattern of shelterin genes throughout lifespan. Aging. 2017; 9 :1219-1232. DOI: 10.18632/aging.101223 - 74.
Khan K, Xu S, Nihtyanova S, Derrett-Smith E, Abraham D, Denton CP, et al. Clinical and pathological significance of interleukin 6 overexpression in systemic sclerosis. Annals of the Rheumatic Diseases. 2012; 71 :1235-1242. DOI: 10.1136/annrheumdis-2011-200955 - 75.
Wu M, Assassi S. The role of type 1 interferon in systemic sclerosis. Frontiers in Immunology. 2013; 4 :266. DOI: 10.3389/fimmu.2013.00266 - 76.
Lin X, Ding M, Chen T, Min S, Wang D, Jiang G. Peripheral blood IL-6 levels in systemic sclerosis patients: Correlation between IL-6 levels and clinical phenotypes. Journal of Cosmetic Dermatology. 2022; 21 :6086-6091. DOI: 10.1111/jocd.15133 - 77.
O’Reilly S, Cant R, Ciechomska M, van Laar JM. Interleukin-6: A new therapeutic target in systemic sclerosis? Clinical Translational Immunology. 2013; 2 (4):e4. DOI: 10.1038/cti.2013.2 - 78.
Denton CP, Ong VH, Xu S, Chen-Harris H, Modrusan Z, Lafyatis R, et al. Therapeutic interleukin-6 blockade reverses transforming growth factor-beta pathway activation in dermal fibroblasts: Insights from the faSScinate clinical trial in systemic sclerosis. Annals of the Rheumatic Diseases. 2018; 77 :1362-1371. DOI: 10.1136/annrheumdis-2018-213031 - 79.
Bohdziewicz A, Pawlik KK, Maciejewska M, Sikora M, Alda-Malicka R, Czuwara J, et al. Future treatment options in systemic sclerosis-potential targets and ongoing clinical trials. Journal of Clinical Medicine. 2022; 11 :1310. DOI: 10.3390/jcm11051310 - 80.
Khanna D, Lin CJF, Furst DE, Goldin J, Kim G, Kuwana M, et al. Tocilizumab in systemic sclerosis: A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. Respiratory Medicine. 2020; 8 :963-974. DOI: 10.1016/S2213-2600(20)30318-0 - 81.
Kakkar V, Assassi S, Allanore Y, Kuwana M, Denton CP, Khanna D, et al. Type 1 interferon activation in systemic sclerosis: A biomarker, a target or the culprit. Current Opinion in Rheumatology. 2022; 34 :357-364. DOI: 10.1097/BOR.0000000000000907 - 82.
Guerini D. STING agonists/antagonists: Their potential as therapeutics and future developments. Cell. 2022; 11 :1159. DOI: 10.3390/cells11071159 - 83.
Li Q , Tian S, Liang J, Fan J, Lai J, Chen Q. Therapeutic development by targeting the cGAS-STING pathway in autoimmune disease and cancer. Frontiers in Pharmacology. 2021; 12 :779425. DOI: 10.3389/fphar.2021.779425