Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
Lower extremity peripheral arterial disease (PAD) is the atherosclerotic obstruction of the lower extremity arteries that can lead to walking impairment, non-healing open wounds, gangrene or limb loss. It is estimated that PAD affects greater than 200 million people worldwide and is associated with advanced age, tobacco use, diabetes, hypertension, and hypercholesterolemia. Initial management of PAD involves risk factor modification and pharmacologic strategies, including the implementation of statin therapy. Statins, the most commonly used cholesterol lowering medications, also have beneficial pleiotropic (cholesterol independent) effects including improved patency rates from vascular reconstruction, decreased risk of stroke, myocardial infarction and improved survival. In this chapter, we will discuss the relevant clinical trials, prospective observation and retrospective studies that exemplify the effect of statins on PAD. We will then focus on statin’s cellular effects on endothelial and vascular smooth muscle cell function by examining effects on plaque progression, intimal hyperplasia, re-endothelialization, and angiogenesis/arteriogenesis.
Department of Surgery, Loyola University of Chicago, Maywood, IL, United States
Research Service, Edward Hines, Jr., VA Hospital, Hines, IL, United States
Kelly A. Langert
Research Service, Edward Hines, Jr., VA Hospital, Hines, IL, United States
Department of Molecular Pharmacology and Neuroscience, Loyola University of Chicago, Maywood, IL, United States
Kristopher Maier
Department of Surgery, Loyola University of Chicago, Maywood, IL, United States
Vivian Gahtan
Department of Surgery, Loyola University of Chicago, Maywood, IL, United States
Research Service, Edward Hines, Jr., VA Hospital, Hines, IL, United States
*Address all correspondence to: ashley.penton@luhs.org
1. Introduction
Peripheral arterial disease (PAD) is the atherosclerotic occlusion of the lower extremities that can lead to walking impairment, gangrene, limb loss and even death. An estimated 8–12 million Americans and greater than 200 million people worldwide are affected by PAD, with prevalence being approximately 10% in patients greater than 60–70 years old [1, 2, 3, 4]. To date, medical management includes behavioral modifications to mitigate risk factors (smoking cessation, control of hypertension, diabetes mellitus and hyperlipidemia) and treatment with medical therapies (statins, antiplatelet drugs, antihypertensives and glucose control). In addition, exercise programs are used to increase cardiovascular health and functional performance [1, 5]. Surgical or endovascular techniques are typically reserved for patients requiring arterial revascularization secondary to threatened limb viability [1].
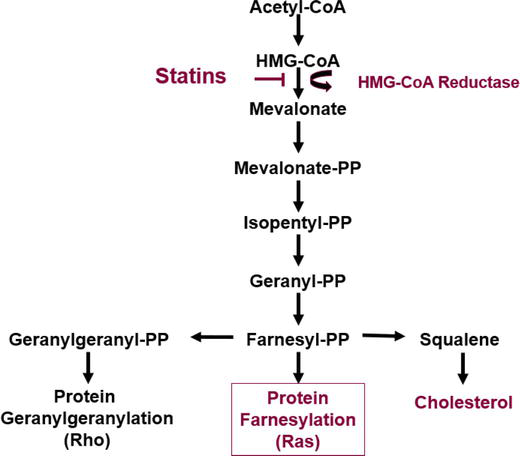
Statins, or 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitors, were initially implemented in the management of PAD for their lipid lowering mechanism. However, statins have demonstrated beneficial cholesterol independent, or “pleiotropic,” effects that reduce cardiovascular events, improve symptoms, graft patency, limb salvage and reduce surgical mortality [6, 7]. The pleiotropic effects are widely understood to be mediated by the ability of statins to inhibit HMG-CoA reductase, the rate-limiting enzyme of the cholesterol biosynthesis pathway (Figure 1) [8, 9]. Besides reducing cholesterol levels, inhibition of HMG-CoA reductase prevents the conversion of HMG-CoA to mevalonate, which limits the available pool of farnesylpyrophosphate (FPP) and geranylgeranylpyrophosphate (GGPP) isoprenoids. FPP and GGPP serve as important lipid posttranslational modifications of a variety of proteins, including small guanosine triphosphate (GTP) binding protein Ras, Rho and other Ras-like proteins, including Rab, Rac, Ral, and Rap [9]. Statins are believed to exert many of their pleiotropic effects through the downstream inhibition of FPP and GGPP, which disrupts the posttranslational modification and normal functioning of Ras and RhoA.
Figure 1.
Mevalonate/cholesterol synthesis pathway [8].
RhoA and Ras are GTP binding proteins that interact with downstream targets to elicit a variety of cellular responses [10]. In 1996, Rho-kinase (Rho-kinase-alpha/ROCK2 and Rho-kinase-B/ROCK1) was identified as the effector of RhoA [11]. Together, RhoA and Rho-kinase control multiple cell functions, including adhesion, proliferation, migration and calcium sensitivity of the contractile proteins [12, 13]. Specifically in vascular smooth muscle cells (VSMCs), Rho-kinase phosphorylates both myosin light chain (MLC) and myosin phosphatase, target subunit 1 (MYPT1). The phosphorylation of MYPT1 leads to inactivation of myosin phosphatase promoting MLC-induced vascular contraction and VSMC migration [12, 14]. RhoA/Rho-kinase is also implicated in the destabilization of nitric oxide synthase (eNOS), an important mediator of endothelial cell and smooth muscle cell function [12]. Ras, on the other hand, has been determined to regulate cell proliferation and survival through the activation of effector pathways, such as Raf and mitogen activated protein kinase (MAPK) [15]. MAPK has been implicated in VSMC proliferation and migration, which is relevant to the development of PAD.
This chapter will focus on the cholesterol independent effects of statins on PAD. The prospective randomized control trials, prospective observational and retrospective studies that examine the pleiotropic effects of statins on PAD will be reviewed. Then the cellular effects of statins on atherosclerosis, intimal hyperplasia (IH), re-endothelialization, angiogenesis and arteriogenesis through cholesterol independent pathways will be presented.
Since the late 1990s, clinical studies have shown statins (independent of lowering cholesterol) are associated with reduced major adverse cardiovascular events (MACE), having anti-inflammatory properties, improved PAD symptoms and having the potential to increase circulatory endothelial progenitor cells (EPCs). One of the first major prospective randomized clinical trials was the Heart Protection Study in 2002 (Table 1) [16]. This study randomized 20,536 adults in the United Kingdom with coronary artery disease (CAD), other occlusive arterial disease or diabetes to receive 40 mg simvastatin or matching placebo daily. They assessed mortality and fatal or nonfatal vascular events over a 5 year period. Irrespective of their cholesterol levels, simvastatin significantly reduced all-cause mortality, including cardiovascular and stroke related events, and nonfatal myocardial infarction (MI) or stroke. Another large prospective randomized controlled trial in 2008, the JUPITER (Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin) Trial (Table 1), randomized 17,802 healthy men and women without hyperlipidemia to receive 20 mg rosuvastatin daily or placebo [19]. They found rosuvastatin also significantly reduced MACE. Other prospective observational studies in the early 2000s found similar results [27, 28, 29]. In 2016, Ramos et al. retrospectively assessed 5480 statin users and non-statin users aged 35–85 with PAD (Ankle-Brachial Index (ABI) <0.90), but without history of cardiovascular disease [30]. This study found statin therapy in patients with PAD was associated with reduced MI, cardiac revascularization, ischemic stroke and all-cause mortality.
Treatment: 40 mg simvastatin daily for 5 years Control: Placebo
Mortality, fatal or nonfatal vascular events
Statin ↓ all-cause mortality, coronary death rate, other vascular death rate, nonfatal MI, nonfatal or fatal stroke, and coronary or noncoronary revascularization. These results applied to all patients - LDL below 116 mg/dl or total cholesterol below 193 mg/dl.
Treatment: 40 mg pravastatin daily for 5 years Control: Placebo
CRP at baseline, at 5 years. End point: death from CAD, or symptomatic nonfatal MI.
Pravastatin ↓ CRP levels at 5 years; other statins ↑ CRP levels. Changes persist after stratification by age, BMI, smoking status, blood pressure, baseline levels of total cholesterol, LDL, HDL and triglycerides.
Occurrence of MI, stroke, arterial revascularization, hospitalization for unstable angina, or death from CV causes.
Rosuvastatin ↓ LDL (50%) and CRP (37%). Rosuvastatin ↓ rates of MI, stroke, revascularization for unstable angina and for the combined end point of MI, stroke, or death from CV causes, and death from any cause. Trial stopped after 1.9 years.
Atorvastatin ↓ CRP at 1 month and 1 year. Nitrite levels (main component of oxidative stress, initial insult to endothelial dysfunction) ↓ at 1 month but not at 1 year.
prior MI or angina pectoris, total cholesterol 5.5–8.0 mmol/L
Treatment: Simvastatin 20 to 40 mg daily Control: Placebo
Auscultation of carotid and femoral arteries; symptoms of intermittent claudication and angina.
Follow up period of 5.4 years. Simvastatin ↓ new intermittent claudication (38%), ≥1 new bruits (30%), and new carotid bruits (48%) compared with placebo.
6 months and 1 year assessed treadmill exercise (2 miles/hr., 12.5% grade).
Simvastatin ↑ treadmill exercise time until onset of claudication from baseline by 54 seconds at 6 months (vs 9 seconds in placebo) and 95 seconds at 1 year (vs −10 seconds in placebo).
1 and 3 months assessed pain-free walking distance.
Both groups had ↑ pain free walking distance but no difference between the two groups, (at entry: 56 (53–108) m vs. 53 (53–106) m; after 3 months: 79 (53–108) m vs. 106 (66–159) m, for the treated and placebo group, respectively)
Peripheral blood collected at baseline and after 12 months of therapy. Assessed EPC numbers and miR-221/222
Atorvastatin ↑ miR-221/222 in CAD group compared to non-CAD group. miR-221/222 negatively correlated with EPC number in the CAD group. After 12 months, changes in lipid levels, atorvastatin > pravastatin. LLT with atorvastatin ↑ EPCs, ↓ miR-221/222. No change in either with pravastatin.
To clinically measure statins effect on systemic inflammation, studies have utilized plasma concentrations of C-reactive protein (CRP) as a marker. Men and women with elevated CRP levels are known to be at increased risk of future cardiovascular events [31]. Multiple prospective randomized controlled trials have shown statins decrease plasma CRP levels at 1 month, 6 months, 1 year, 2 years, and up to 5 years (Table 1) [17, 18, 19, 20]. Particularly, in patients with PAD, Bleda et al. demonstrated in sixty patients that 40 mg atorvastatin daily significantly decreased CRP levels at 1 year [20].
Studies have also indicated that statins improve leg function in patients with PAD. In 1998, the prospective randomized control trial, Scandinavian Simvastatin Survival Study, found that simvastatin (20-40 mg per day for 5 years) significantly reduced the incidence of new intermittent claudication by 38% as well as the incidence of carotid and femoral bruits (Table 1) [21]. In 2003, Aronow et al. showed simvastatin taken for 6 months and 1 year increased exercise treadmill time from baseline (Table 1) [22]. A prospective observational study performed with 392 men and women (age > 55) with ABI <0.90 and 249 men and women (age > 55) with an ABI between 0.90 and 1.50 showed statin users had better 6 minute walking distances and 4 meter walking velocities after 2 years [32]. While these studies demonstrated that statins improve leg function, a randomized controlled trial by Breger et al. showed no difference in pain-free walking distance (Table 1); however, these patients were assessed at a shorter time interval of 1 and 3 months, suggesting that the effects of oral statins on leg symptoms may be time dependent [23]. Of note, one novel study investigated intramuscular administration of pitavastatin loaded nanoparticles to patients with chronic limb threatening ischemia unsuitable for surgery (Table 1) [24]. They assessed patients at 26 weeks and found administration of nanoparticles was safe and improved leg symptoms in 5 of the 16 patients.
Although there are limited clinical studies on the effects of statins on EPCs, two prospective randomized clinical trials have examined patients with stable CAD and patients without CAD (Table 1) [25, 26]. These studies found atorvastatin increased peripheral circulatory quantity of EPCs in patients with CAD. Suggesting statins may influence repair after ischemic injury secondary to their contribution in mobilizing EPCs.
Clinically, statins reduce cholesterol levels, and in addition through their pleiotropic effects affect the following: reduce MACE, have anti-inflammatory properties independent of lowering cholesterol, improve PAD symptoms and have the potential to increase circulatory EPCs. Although American and European guidelines recommend statins as the first line lipid lowering agent for patients with a 10-year atherosclerotic cardiovascular disease (ASCVD) risk greater than 7.5%, there remains no specific guideline that determines whether high intensity (LDL reduction >50%), moderate intensity (LDL reduction 30–50%) or low intensity (LDL reduction <30%) statin is preferred for PAD specific disease [1, 33, 34, 35]. At present, American Heart Association guidelines recommend using estimated 10 year ASCVD risk to help guide statin intensity [35]. Recent studies have investigated whether statin intensity affects outcomes in patients with PAD. A meta-analysis by Sagris et al. found 39 studies with 275,670 patients with PAD, of which 49% were on statins [36]. Of this group, high intensity statins were associated with a 36% reduction of all-cause mortality in comparison to low intensity statins. In this study, statins were overall associated with reduction in all-cause mortality, cardiovascular mortality, MACE, risk for amputation, or loss of arterial patency. Other studies have shown similar benefits of high intensity statin therapy in PAD [37, 38]. Unfortunately, negative off-target effects, including serious muscle-related symptoms (myalgia, myositis, rhabdomyolysis), cognitive decline, hepatoxicity, new-onset diabetes, and peripheral neuropathy preclude the use of statins in some patients [39]. The population of patients intolerant of statin use is approximately 9.1% [40].
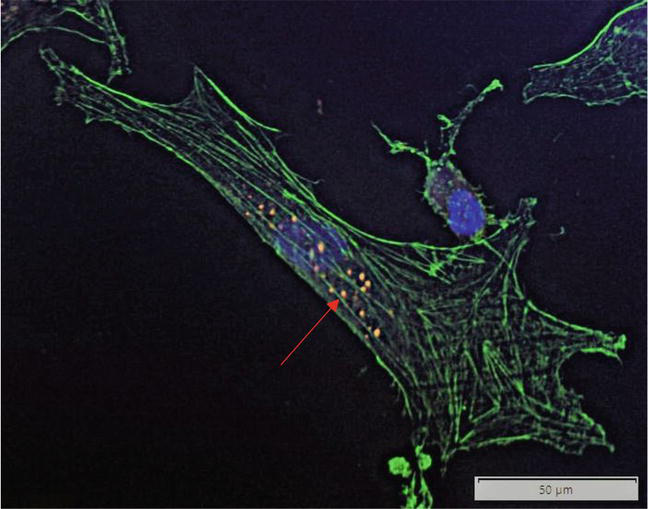
To harness the pleiotropic effects of statins while minimizing negative off-target effects, recent studies have focused on developing targeted statin therapies. These experimental drug delivery systems include statin-loaded hyaluronic acid tagged polysialic acid-polycaprolactone micelles, intramuscular or peri-sciatic injection of poly (lactic-co-glycolic acid) (PLGA) nanoparticles, and nanofiber eluting stents [24, 41, 42, 43, 44, 45]. We are currently investigating the effects of intraluminal administration of statin loaded chitosan-/PLGA nanoparticles on intimal hyperplasia in a rat carotid artery balloon injury model. Our goal is to optimize a delivery system that will facilitate local delivery of statins to vascular cells [46]. Preliminary data suggest that chitosan encapsulated nanoparticles are readily taken up by VSMCs (Figure 2). In the rest of this chapter, we will review the cellular effects of statins and incorporate novel drug delivery models that have been used to date.
Figure 2.
Chitosan-encapsulated PLGA nanoparticles (red, Alexa Fluor 488) readily taken up within VSMCs (green, Phalloidin).
PAD can lead to acute limb ischemia as a result of embolic or thrombotic disease secondary to underlying atherosclerosis. In order to understand the pleiotropic effects of statins, to understand the pathophysiology of atherosclerosis is paramount. Atherosclerosis is a chronic inflammatory, fibroproliferative disease process that is a result of prolonged endothelial damage [47, 48, 49]. A multitude of risk factors such as hyperlipidemia, hypertension, smoking, hemodynamic factors, immune reactions, or genetic factors can lead to endothelial damage. With endothelial damage comes endothelial dysfunction, which causes a leaky and defective endothelial lining, permitting plasma molecules and lipoproteins to extravasate into the subendothelial space [47]. When low-density lipoprotein (LDL) is retained in this space, it becomes oxidized. Oxidized-LDL is cytotoxic, pro-inflammatory, chemotactic and proatherogenic [47]. The milieu created causes the endothelial cells to express adhesion molecules that help recruit monocytes and T lymphocytes. Chemoattractants then stimulate the inflammatory cells to migrate into the subendothelial space where monocytes differentiate into macrophages and internalize oxidized LDL. Internalized oxidized LDL form foam cells, or lipid-loaded macrophages, a marker of early and late atherosclerosis [47]. Foam cells that undergo apoptosis and necrosis contribute to the formation of a soft and destabilizing lipid-rich core within the atherosclerotic plaque. As the disease process progresses, VSMCs change from a contractile to proliferative and secretory phenotype [48]. This induces VSMCs to migrate from the media to the intima where they proliferate and deposit a collagen-rich extracellular matrix (ECM), forming a thick fibrous cap to the lipid-filled lesion. As the lesion grows, perfusion by the vaso vasorum becomes limited, creating a hypoxic environment that can cause VSMC death and thinning of the fibrous cap [48]. The fibrous cap is further destabilized by macrophages secreting matrix metalloproteases (MMP) and tissue factor, placing the plaque at risk for rupture. Plaque rupture then leads to the risk of thrombus formation and occlusion, resulting in limb threatening ischemia.
As described, atherosclerosis is dependent on a complex interplay between the endothelial lining, inflammatory cells, lipoproteins and VSMCs. Of which, elevated plasma cholesterol levels have shown a clear association with increased atherosclerotic disease. Statins, therefore, were a promising medication to help minimize atherosclerotic burden by lowering plasma cholesterol levels. Particularly, statins are known to decrease LDL and remove existing LDL from the circulation; however, studies have demonstrated the overall benefits of statins are greater than anticipated due to their pleiotropic effects [29]. For example, in a cholesterol independent manner, statins stabilize atherosclerotic plaque, decrease endothelial dysfunction and reduce vascular inflammation.
3.1.1 Statin effect on plaque stability
Statins contribute to plaque stability by decreasing macrophage accumulation and inhibiting MMP and tissue factor production by cholesterol independent and dependent mechanisms [9, 14]. Previously, statins were thought to stabilize plaque through their cholesterol lowering mechanism, as lowering cholesterol via dietary mechanisms decreased macrophage and proteolytic enzyme accumulation [50]. This effect of statins was highlighted by Crisby et al’s study, which demonstrated atherosclerotic plaque (collected from carotid endarterectomy) in patients taking pravastatin for 3 months contained less lipid, higher interstitial collagen, less oxidized LDL, fewer macrophages, less MMP-2 and greater tissue inhibitor of MMP-1 in comparison to patients who received no statin therapy [51]. Later studies demonstrated statins work through additional mechanisms to stabilize plaques. For example, in Feig et al’s study, they demonstrated statins not only lower cholesterol to decrease macrophage accumulation, but, actively induce macrophage emigration through the chemokine receptor CCR7 [52]. Taken together, statins work through cholesterol dependent and independent mechanisms to reduce proteolytic enzyme activity, thereby reducing the risk of destabilizing the fibrous cap on atheromas.
3.1.2 Statin effect on endothelial dysfunction
One of the earliest components of atherosclerosis is endothelial dysfunction, which leads to the impaired synthesis, release and activity of endothelial-derived nitric oxide (NO) [9]. NO is a soluble gas synthesized by nitric oxide synthase (eNOS). NO is critical for vascular hemostasis due to its vasodilatory, anti-inflammatory and antioxidant properties [53]. Once endothelial dysfunction ensues, reactive oxygen species propagate its dysfunction. Statins have been documented to improve endothelial function by increasing NO production and reducing reactive oxygen species.
Statins minimize endothelial dysfunction by increasing eNOS and stabilizing eNOS mRNA, thereby increasing NO production [9, 54, 55]. Statins increase NO production through multiple mechanisms, including its interaction with RhoA, phosphatidylinositol 3-kinase (PI3k)/AKT and caveolin-1 [9, 56]. As previously discussed, RhoA downregulates eNOS and statins reduce RhoA through reducing production of mevalonate [57]. Therefore, statin mediated inhibition of the isoprenoid intermediate RhoA results in reduced inhibition of eNOS. In regards to PI3k/Akt, statins have been shown to enhance its phosphorylation, which increases Akt expression [58, 59]. Akt has been shown to enhance eNOS expression. Caveolin-1, on the other hand, is an inhibitory protein that binds eNOS, inhibiting the production of NO [56, 60]. Statins have been shown to decrease the expression of caveolin-1, thereby increasing eNOS production of NO [56]. Through these mechanisms, statins work to enhance eNOS which increases NO production and reduces endothelial dysfunction. Statins further reduce endothelial dysfunction through its antioxidant effects. Moon et al demonstrated that rosuvastatin administered to 99 patients who had experienced an atherosclerotic stroke resulted in a reduction of oxidative stress markers (specifically, malonidialdehyde and oxidized LDL) compared to patients without treatment [61]. The reduction of oxidative stress markers was thought to be secondary to statins ability to inhibit oxidant enzyme activity, such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and myeloperoxidase, while upregulating antioxidant enzymes, catalase and paraoxonase [62]. Further studies have demonstrated statins reduce reactive oxygen species through enhanced phosphorylation of PI3k/Akt and activation of the transcription factor Nfr2 [58]. Through these pleiotropic effects, statins help decrease reactive oxygen species and therefore reduce the progression of atherosclerosis.
3.1.3 Statin effect on vascular inflammation
Chronic inflammation is a large component of the initiation and progression of atherosclerosis. Statins appear to reduce vascular inflammation by reducing leukocyte adhesion and transendothelial migration [45, 63, 64]. Simvastatin in particular, has been shown to decrease leukocyte rolling and adhesion in vitro, and in vivo [65]. Other studies have shown that statins reduce the leukocyte-endothelial interaction by decreasing the RhoA-dependent expression of endothelial adhesion molecules including P- and E- selectins, intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 [66, 67]. Statins also hinder the recruitment of inflammatory cells into the vascular intima and across inflamed vascular endothelium by attenuating the expression of monocyte chemoattractant protein-1 (MCP-1) via a mechanism that may involve Cdc42 and RalA GTPases [63, 68, 69, 70]. Taken together, statin pleiotropic effects hinder the propagation of atherosclerosis, in part, by inhibiting leukocyte transendothelial migration.
In summary, statins stabilize atherosclerotic plaques by reducing macrophage and MMP accumulation, improve endothelial function by stabilizing and increasing NO via increased eNOS production, and diminish vascular inflammation by hindering leukocyte adhesion and transendothelial migration. The pleiotropic effect of statins on atherosclerosis is largely through inhibition of mevalonate-derived products, as well as through other pathways, including PI3k/Akt.
3.2 Intimal hyperplasia
When PAD is significant enough to warrant intervention, the management strategy can be through endovascular procedures (angioplasty, stenting) or surgically (endarterectomy or bypass); however, restenosis secondary to IH remains a significant challenge that affects patency rates after intervention [71, 72, 73]. IH is triggered by endothelial damage, which can be caused by balloon inflation, stent implantation or bypass graft placement [12, 73, 74, 75]. The endothelial damage that ensues stimulates the production of proinflammatory molecules and activation of circulatory monocytes that bind and penetrate the vascular wall, perpetuating a local inflammatory response. There is also a reduction of VSMC inhibitory factors, such as heparan sulphate, NO and natriuretic peptides. Simultaneously, there is production of plasminogen activators that contribute to the degradation of the ECM and activation of MMPs [74]. These factors together stimulate VSMC transition from a contractile state to a proliferative state through MAPKs [76]. Migration and proliferation of VSMCs from the medial and adventitial layer of the arterial wall into the intimal or subendothelial space then ensues. Intimal expansion subsequently occurs, secondary to VSMC accumulation and exuberant ECM synthesis stimulated by growth factors, such as TGF-beta and platelet derived growth factor [77].
One mechanism by which statins reduce IH is through the inhibition of the small GTPase RhoA [12]. As previously discussed, RhoA interacts with Rho-kinase to promote vascular contraction, VSMC migration and reduce eNOS gene expression, steps crucial in the pathophysiology of IH [78]. Therefore, by statins inhibiting RhoA, we would anticipate decreased VSMC migration, as well as the restoration of NO production. In 2005, Yamanouchi et al. demonstrated normocholesterolemic rats supplemented with oral pravastatin 10 mg/day exhibited reduced vein graft IH with suppressed cellular proliferation and increased cellular apoptosis [79]. In vitro, they demonstrated human umbilical vein endothelial cells (HUVECs) treated with pravastatin inhibited Rho-kinase activity and accelerated endothelial eNOS expression; however, there was no effect on Rho-kinase in VSMCs. Other in vivo studies have shown oral and intraperitoneal statins (pitavastatin, simvastatin, atorvastatin) reduce IH [80, 81, 82, 83, 84]. Studies using novel techniques have also assessed the effect of statins effect on IH [41, 85]. These techniques have been developed to locally deliver statins in one dose for the purposes of reducing systemic toxicity. For example, Helkin et al. assessed simvastatin loaded in hyaluronic acid tagged polysialic acid-polycaprolactone micelles delivered to rats intraluminally prior to carotid artery injury. This treatment demonstrated a 55% reduction of IH while oral simvastatin reduced IH by 25%. Other studies have incorporated statins into drug-eluting stents, with statin-eluting-stents decreasing IH in comparison to other stents [44, 86]. Particularly, Tsuki et al. developed a bioabsorbable statin incorporated polymeric nanoparticle-eluting stent [44]. They demonstrated in a pig coronary artery model, pitavastatin incorporated nanoparticle eluting stent attenuated in-stent stenosis, comparably to sirolimus-eluting stents, without delayed endothelial healing. Future studies are aimed at finding alternative means to deliver localized statins at the site of endothelial injury to minimize risks of systemic therapy.
Another cholesterol independent effect of statins is their inhibition of another small GTPase protein, Ras. Sakamoto et al. assessed the effect of fluvastatin on MAPKs, extracellular signal-regulated kinase 1 and 2 (ERK1/2) and p38MAPK phosphorylation in an organ-cultured rat tail artery [76]. ERK1/2 and p38MAPK are believed to be phosphorylated and activated by Ras, inducing the morphologic change of contractile VSMCs to the proliferative state [87]. They demonstrated that fluvastatin significantly decreased ERK1/2 and p38MAPK and restored VSMC contractility, suggesting that statins may inhibit proliferation via inhibition of MAPK phosphorylation. In 2020, Chu et al. evaluated the effect of atorvastatin on the phosphorylation of p38 MAPKs [81]. They found rats undergoing vein graft bypass had reduced IH and significantly decreased p38 MAPK phosphorylation (p < 0.05) when treated with oral atorvastatin. These studies indicate that the pleiotropic effects of statins hinder the progression of IH not only via endothelial cell dependent mechanisms but also via inhibition of VSMC migration.
In summary, statins decrease IH at least through two different pathways that are activated by RhoA and Ras. By inhibiting RhoA, statins inhibit Rho-kinase which decreases VSMC migration and NO production. By inhibiting Ras, statins inhibit the p38MAPK pathway, which decrease VSMC migration and proliferation.
3.3 Re-endothelialization
The leading pathophysiology of atherosclerosis and IH is endothelial damage. To accommodate for this, endothelial cells are continuously attempting to repair themselves by proliferating resident endothelial cells and circulating EPCs [88]. This concept of repair is termed re-endothelialization. Statins accelerate re-endothelialization by mobilizing, differentiating, and improving survival of EPCs [89, 90, 91]. A study by Walter et al demonstrated simvastatin administered to Sprague–Dawley rats accelerated re-endothelialization after carotid artery balloon injury and resulted in a dose dependent reduction of neointimal thickening [92]. In regards to re-endothelialization, simvastatin was shown to increase the rat’s circulating EPCs and induced adhesiveness at the site of endothelial injury by upregulating the integrin subunits alpha5, alpha(v), beta1 and beta5. By upregulating these integrin subunits, statins not only mobilize EPCs, but target them to the site of endothelial injury. Further studies demonstrated statins increase circulating EPCs through the phosphorylation of Akt, activating the PI3k/Akt signaling pathway [89]. These findings establish yet another pleiotropic mechanism by which statins specifically pre-empt vascular wall pathologies.
3.4 Angiogenesis and Arteriogenesis
Angiogenesis, in the adult, is the formation of new capillaries. It features sprouting of new endothelial cells from preexisting capillaries under the influence of angiogenic factors generated by a hypoxic environment [93, 94]. Arteriogenesis describes the remodeling of preexisting collateral arterioles. Typically, the collateral arterioles are high resistance and do not offer much blood flow to distal capillary beds. With proximal arterial occlusion, hemodynamic changes provoke arteriole remodeling, which encompasses proliferation of vascular cells and turnover of the vascular matrix [93, 94]. The complex interplay that ensues between vascular cells, adhesion molecules, chemokines and monocytes results in collateral arterioles with increased diameter and wall thickness, providing a natural bypass; however, this adaptation is not equivalent to direct arterial perfusion.
In patients with progressive PAD, endovascular or surgical revascularization is the preferred therapeutic strategy. However, not all patients are candidates due to the severity of disease or due to risk of intervention secondary to a patient’s severe comorbidities. In this population, patients rely on angiogenesis and arteriogenesis to promote perfusion to ischemic tissue. Unfortunately, endogenous angiogenesis and arteriogenesis are not enough to restore blood flow in the setting of critical limb ischemia. Therefore, a growing body of literature has investigated medical interventions including medical revascularization with the goal of increasing blood vessel growth and improving perfusion to ischemic extremities [93]. One of these medical interventions includes administration of statins.
3.4.1 Statin effect on angiogenesis
Statins have a dose-dependent biphasic effect on angiogenesis, with lower-doses acting in a pro-angiogenic manner and higher doses acting in an anti-angiogenic, pro-apoptotic manner [95, 96, 97, 98, 99]. The pro-angiogenic effects of statins appear to act through the intracellular signaling pathway, PI3 kinase/Akt (Table 2) [59, 101, 102, 103, 104]. One effect of enhancing the PI3 kinase/AKT signaling pathway is increasing capillary density. One study used both in vitro and in vivo models to demonstrate that statins enhance phosphorylation of Akt, increasing its substrate eNOS, which inhibited endothelial cell apoptosis and accelerated formation of endothelial cell tubules in a matrigel assay [59]. In vivo, statins promoted angiogenesis and blood flow recovery in a rabbit hindlimb ischemia model. One study suggested that the statin effect may be mediated by Notch1 signaling, a downstream effector of PI3Kinase/Akt (Table 2) [104]. Statins also influence angiogenesis through their effect on EPCs. EPCs have been demonstrated to be involved in neovascularization, thereby constituting post-natal vasculogenesis [95]. Statins have been shown to promote the survival, migration and differentiation of adult EPCs through Akt dependent mechanisms [97]. For example, in vitro low dose fluvastatin was shown to activate the Akt/eNOS pathway resulting in increased EPC migration and proliferation [101]. This same study showed in vivo enhanced incorporation of EPCs in ischemic tissue with increased capillary density.
Statin
Dose
Cell Type/Model
Biologic Effect
Reference
Simvastatin/ Rosuvastatin
1 μM and 10 μM
HUVECS/Ex vivo mouse aortic ring
In vitro: Statins↑ HUVEC tube formation at low dose. Ex vivo: Statins abolished new vessel formation, ↑ VE-cadherin and cell–cell adhesion.
In vitro: [Low]: 0.005–0.01 μM [High]: 0.05–1 μM In vivo: [Low]: 0.5 mg/kg/day [High]: 2.5 mg/kg/day
Human adult dermal microvascular ECs / WT and hypercholesterolemic C57BL/6 J mice 24 weeks
Statin ↑ EC proliferation, migration and differentiation at [low], ↓ at [high]. Antiangiogenic effects associated with ↓ EC release of VEGF, ↑ EC apoptosis. Effects reversed by GGPP. In vivo: Statin ↑ angiogenesis at [low] dose, ↓ at [high] dose in WT and hypercholesterolemic mice.
In vitro: Statin ↑ phosphorylation of Akt, inducing phosphorylation of eNOS, causing NO production; ↑ tubles in matrigel, ↓ apoptosis. In vivo: statins ↑ capillary formation.
In vitro: Statin activated endothelial y-secretase and Notch1 downstream of PI3K/akt (100 nmol, ↓ at 1microM) and ↑ ephrin B2 downstream (promoter of arteriogenesis). ↑ proangiogenic activity of ECs, ameliorated by Notch 1 inhibition. In vivo: Statin (3 mg/kg) ↑ blood flow recovery, capillary density, arterioles in WT mice but not in Notch1 deficient mice.
While lower doses of statins promote angiogenesis, high doses of statins have been shown to be anti-angiogenic and pro-apoptotic (Table 2). One mechanism by which high dose statins may decrease angiogenesis is through their inhibition of RhoA [97]. Weis et al. demonstrated in vitro that high doses of statins increased endothelial cell apoptosis, inhibited endothelial cell proliferation, migration, differentiation and decreased endothelial cell release of VEGF (Table 2) [100]. In vivo, mice given high dose statin had an angiostatic response after hindlimb ischemia. The in vitro anti-angiogenic effects were reversed with the administration of GGPP. Another mechanism by which statins may inhibit angiogenesis is through their increase of cell cycle inhibitors and downregulation of angiogenesis related genes (PAI-1, vitronectin, HoxD3, and Notch4) [105]. Interestingly, studies have indicated high doses of statin continue to stabilize eNOS mRNA, further demonstrating the duality of statins [106].
The biphasic angiogenic effect of statins is complex and may be due to the differing affinity for GGPP/RhoA and FPP/Ras inhibition. Zahedipour et al. have suggested that at low doses, statins preferentially inhibit cholesterol synthesis, which leaves products of GGPP/RhoA and FPP/Ras uninhibited [97]. By contrast, high dose statins significantly reduce all byproducts of the mevalonate pathway. The effect is reduction in cell proliferation and migration, consistent with the angiostatic environment high dose statins produce. Future studies will be needed to determine the optimal dosage and vehicle for statins to be used as a novel medical revascularization technique.
3.4.2 Statin effect on Arteriogenesis
In the past decade, studies have demonstrated that statins induce arteriogenesis. In particular, pitavastatin has been shown to increase angiogenesis and arteriogenesis in a murine hindlimb ischemia model [104]. Other studies have used novel drug carriers (PLGA nanoparticles) to administer pitavastatin into ischemic muscle of mice, rabbit and cynomolgus monkey [42, 43]. These studies demonstrated that intramuscular injection of polymeric nanoparticles delivering 0.5 mg/kg pitavastatin induced arteriogenesis and ameliorated exercise-induced ischemia. In vitro studies suggest that the effect of statins on arteriogenesis appears to be through Notch1 and Akt signaling. In the study by Kikuchi et al, they demonstrated pitavastatin activated Notch1 in HUVECs, which was inhibited by knockdown of Akt [104]. In vivo, they demonstrated that pitavastatin administered to wild-type mice undergoing hindlimb ischemia increased endothelial ephrin B2 (a downstream target of Notch1) and induced arteriogenesis. This effect was not apparent in Notch1 mutant mice, suggesting Notch1 facilitates the arteriogenic effects of statins. Similarly, Zacharek et al demonstrated rats subjected to middle cerebral artery occlusions treated with simvastatin demonstrated increased arteriogenesis [107]. Rats with increased arteriogenesis also demonstrated upregulation of Notch1 and its effector, notch intracellular domain. This effect was mitigated by inhibition of Notch signaling. These studies demonstrate statins effect of increased arteriogenesis is through activation of Notch1 and enhanced Akt signaling.
The prevalence of PAD is growing globally secondary to our aging population. The management of PAD is multi-disciplinary and begins with prompt risk factor management and initiation of medical therapies. Of these medical therapies, statins have been shown to be a great benefit given their cholesterol lowering abilities and pleiotropic effects. As described, cholesterol independent mechanisms of statins include stabilizing atherogenic plaques, reducing IH, increasing reendothelialization via mobilizing of EPCs, and promoting angiogenesis in a dose-dependent manner. The pleiotropic effects of statins are largely mediated through inhibition of mevalonate-derived products, such as RhoA and Ras GTPases, as well as through phosphorylation and activation of the PI3k/Akt pathway. The effects of statins on these pathways include increased NO production and endothelial stabilization, reduced vascular inflammation, reduced VSMC proliferation, increased EPC mobilization, decreased intimal hyperplasia, variable effects on angiogenesis and increased arteriogenesis. Future studies aim to develop techniques that will safely provide local delivery of statins to the area of PAD, in hopes of minimizing systemic toxicities and providing medical revascularization.
1.Society for Vascular Surgery Lower Extremity Guidelines Writing Group et al. Society for Vascular Surgery practice guidelines for atherosclerotic occlusive disease of the lower extremities: Management of asymptomatic disease and claudication. Journal of Vascular Surgery. 2015;61(3 Suppl):2S-41S
2.Criqui MH, Aboyans V. Epidemiology of peripheral artery disease. Circulation Research. 2015;116(9):1509-1526
3.Selvin E, Erlinger TP. Prevalence of and risk factors for peripheral arterial disease in the United States: Results from the National Health and nutrition examination survey, 1999-2000. Circulation. 2004;110(6):738-743
4.Nehler MR et al. Epidemiology of peripheral arterial disease and critical limb ischemia in an insured national population. Journal of Vascular Surgery. 2014;60(3):686-95 e2
5.Alnaeb ME et al. Statins and peripheral arterial disease: Potential mechanisms and clinical benefits. Annals of Vascular Surgery. 2006;20(5):696-705
6.Coppola G, Novo S. Statins and peripheral arterial disease: Effects on claudication, disease progression, and prevention of cardiovascular events. Archives of Medical Research. 2007;38(5):479-488
7.Daskalopoulou SS et al. Peripheral arterial disease: A missed opportunity to administer statins so as to reduce cardiac morbidity and mortality. Current Medicinal Chemistry. 2005;12(4):443-452
8.Sadowitz B, Maier KG, Gahtan V. Basic science review: Statin therapy--part I: The pleiotropic effects of statins in cardiovascular disease. Vascular and Endovascular Surgery. 2010;44(4):241-251
9.Liao JK, Laufs U. Pleiotropic effects of statins. Annual Review of Pharmacology and Toxicology. 2005;45:89-118
10.Satoh K, Fukumoto Y, Shimokawa H. Rho-kinase: Important new therapeutic target in cardiovascular diseases. American Journal of Physiology. Heart and Circulatory Physiology. 2011;301(2):H287-H296
11.Amano M et al. Phosphorylation and activation of myosin by rho-associated kinase (rho-kinase). The Journal of Biological Chemistry. 1996;271(34):20246-20249
12.Sugimoto M, Yamanouchi D, Komori K. Therapeutic approach against intimal hyperplasia of vein grafts through endothelial nitric oxide synthase/nitric oxide (eNOS/NO) and the rho/rho-kinase pathway. Surgery Today. 2009;39(6):459-465
13.Yao L et al. The role of RhoA/rho kinase pathway in endothelial dysfunction. Journal of Cardiovascular Disease Research. 2010;1(4):165-170
14.Fukata Y, Amano M, Kaibuchi K. Rho-rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends in Pharmacological Sciences. 2001;22(1):32-39
15.Nan X et al. Ras-GTP dimers activate the mitogen-activated protein kinase (MAPK) pathway. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(26):7996-8001
16.Heart Protection Study Collaborative. MRC/BHF heart protection study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet. 2002;360(9326):7-22
17.Ridker PM et al. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The cholesterol and recurrent events (CARE) investigators. Circulation. 1999;100(3):230-235
18.Albert MA et al. Effect of statin therapy on C-reactive protein levels: The pravastatin inflammation/CRP evaluation (PRINCE): A randomized trial and cohort study. JAMA. 2001;286(1):64-70
19.Ridker PM et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. The New England Journal of Medicine. 2008;359(21):2195-2207
20.Bleda S et al. Long-term pleiotropic effect of statins upon nitric oxide and C-reactive protein levels in patients with peripheral arterial disease. Heart Asia. 2011;3(1):130-134
21.Pedersen TR et al. Effect of simvastatin on ischemic signs and symptoms in the Scandinavian simvastatin survival study (4S). The American Journal of Cardiology. 1998;81(3):333-335
22.Aronow WS et al. Effect of simvastatin versus placebo on treadmill exercise time until the onset of intermittent claudication in older patients with peripheral arterial disease at six months and at one year after treatment. The American Journal of Cardiology. 2003;92(6):711-712
23.Bregar U et al. The influence of atorvastatin on walking performance in peripheral arterial disease. VASA. 2009;38(2):155-159
24.Matsumoto T et al. Pitavastatin-incorporated nanoparticles for chronic limb threatening ischemia: A phase I/IIa clinical trial. Journal of Atherosclerosis and Thrombosis. 2022;29(5):731-746
25.Minami Y et al. Effect of atorvastatin on microRNA 221/222 expression in endothelial progenitor cells obtained from patients with coronary artery disease. European Journal of Clinical Investigation. 2009;39(5):359-367
26.Vasa M et al. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation. 2001;103(24):2885-2890
27.Feringa HH et al. Cardioprotective medication is associated with improved survival in patients with peripheral arterial disease. Journal of the American College of Cardiology. 2006;47(6):1182-1187
28.Schillinger M et al. Statin therapy improves cardiovascular outcome of patients with peripheral artery disease. European Heart Journal. 2004;25(9):742-748
29.Gonzalez L, Helkin A, Gahtan V. Dyslipidemia part 2: Review of dyslipidemia treatment in patients with noncoronary vascular disease. Vascular and Endovascular Surgery. 2016;50(2):119-135
30.Ramos R et al. Statins for prevention of cardiovascular events in a low-risk population with low ankle brachial index. Journal of the American College of Cardiology. 2016;67(6):630-640
31.Ridker PM, Haughie P. Prospective studies of C-reactive protein as a risk factor for cardiovascular disease. Journal of Investigative Medicine. 1998;46(8):391-395
32.McDermott MM et al. Statin use and leg functioning in patients with and without lower-extremity peripheral arterial disease. Circulation. 2003;107(5):757-761
33.Frank U et al. ESVM guideline on peripheral arterial disease. VASA. 2019;48(Suppl 102):1-79
34.Gerhard-Herman MD et al. 2016 AHA/ACC guideline on the Management of Patients with Lower Extremity Peripheral Artery Disease: Executive summary: A report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation. 2017;135(12):e686-e725
35.Stone NJ et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association task force on practice guidelines. Circulation. 2014;129(25 Suppl 2):S1-S45
36.Sagris M et al. Statins and statin intensity in peripheral artery disease. VASA. 2022;51(4):198-211
37.Arya S et al. Association of Statin Dose with Amputation and Survival in patients with peripheral artery disease. Circulation. 2018;137(14):1435-1446
38.Sofat S et al. Effects of statin therapy and dose on cardiovascular and limb outcomes in peripheral arterial disease: A systematic review and meta-analysis. European Journal of Vascular and Endovascular Surgery. 2021;62(3):450-461
39.Bitzur R et al. Intolerance to statins: Mechanisms and management. Diabetes Care. 2013;36 Suppl 2(Suppl 2):S325-S330
40.Bytyci I et al. Prevalence of statin intolerance: A meta-analysis. European Heart Journal. 2022;43(34):3213-3223
41.Helkin A et al. Intraluminal delivery of simvastatin attenuates intimal hyperplasia after arterial injury. Vascular and Endovascular Surgery. 2019;53(5):379-386
42.Matsumoto T et al. Therapeutic Arteriogenesis/angiogenesis for peripheral arterial disease by nanoparticle-mediated delivery of Pitavastatin into vascular endothelial cells. Annals of Vascular Diseases. 2020;13(1):4-12
43.Oda S et al. Nanoparticle-mediated endothelial cell-selective delivery of pitavastatin induces functional collateral arteries (therapeutic arteriogenesis) in a rabbit model of chronic hind limb ischemia. Journal of Vascular Surgery. 2010;52(2):412-420
44.Tsukie N et al. Pitavastatin-incorporated nanoparticle-eluting stents attenuate in-stent stenosis without delayed endothelial healing effects in a porcine coronary artery model. Journal of Atherosclerosis and Thrombosis. 2013;20(1):32-45
45.Langert KA, Goshu B, Stubbs EB Jr. Attenuation of experimental autoimmune neuritis with locally administered lovastatin-encapsulating poly(lactic-co-glycolic) acid nanoparticles. Journal of Neurochemistry. 2017;140(2):334-346
46.Yang F et al. Chitosan/poly(lactic-co-glycolic)acid nanoparticle formulations with finely-tuned size distributions for enhanced Mucoadhesion. Pharmaceutics. 2022;14(1):95
47.Falk E. Pathogenesis of atherosclerosis. Journal of the American College of Cardiology. 2006;47(8 Suppl):C7-C12
48.Helkin A et al. Dyslipidemia part 1--review of lipid metabolism and vascular cell physiology. Vascular and Endovascular Surgery. 2016;50(2):107-118
49.Rafieian-Kopaei M et al. Atherosclerosis: Process, indicators, risk factors and new hopes. International Journal of Preventive Medicine. 2014;5(8):927-946
50.Libby P, Aikawa M. Mechanisms of plaque stabilization with statins. The American Journal of Cardiology. 2003;91(4A):4B-8B
51.Crisby M et al. Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases, and cell death in human carotid plaques: Implications for plaque stabilization. Circulation. 2001;103(7):926-933
52.Feig JE et al. Statins promote the regression of atherosclerosis via activation of the CCR7-dependent emigration pathway in macrophages. PLoS One. 2011;6(12):e28534
53.Cyr AR et al. Nitric oxide and endothelial dysfunction. Critical Care Clinics. 2020;36(2):307-321
54.Laufs U, Fata VL, Liao JK. Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. The Journal of Biological Chemistry. 1997;272(50):31725-31729
55.Suciu M. The role of nitric oxide (NO) and statins in endothelial dysfunction and atherosclerosis. Farmácia. 2009;57(2):1-10
56.Suh JW et al. HMG-CoA reductase inhibitor improves endothelial dysfunction in spontaneous hypertensive rats via down-regulation of caveolin-1 and activation of endothelial nitric oxide synthase. Journal of Korean Medical Science. 2010;25(1):16-23
57.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by rho GTPase. The Journal of Biological Chemistry. 1998;273(37):24266-24271
58.Chartoumpekis D et al. Simvastatin lowers reactive oxygen species level by Nrf2 activation via PI3K/Akt pathway. Biochemical and Biophysical Research Communications. 2010;396(2):463-466
59.Kureishi Y et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nature Medicine. 2000;6(9):1004-1010
60.Feron O et al. Hypercholesterolemia decreases nitric oxide production by promoting the interaction of caveolin and endothelial nitric oxide synthase. The Journal of Clinical Investigation. 1999;103(6):897-905
61.Moon GJ et al. Antioxidant effects of statins in patients with atherosclerotic cerebrovascular disease. Journal of Clinical Neurology. 2014;10(2):140-147
62.Davignon J, Jacob RF, Mason RP. The antioxidant effects of statins. Coronary Artery Disease. 2004;15(5):251-258
63.Langert KA, Von Zee CL, Stubbs EB Jr. Cdc42 GTPases facilitate TNF-alpha-mediated secretion of CCL2 from peripheral nerve microvascular endoneurial endothelial cells. Journal of the Peripheral Nervous System. 2013;18(3):199-208
64.Satny M, Hubacek JA, Vrablik M. Statins and inflammation. Current Atherosclerosis Reports. 2021;23(12):80
65.Obama R et al. Direct inhibition by a statin of TNFalpha-induced leukocyte recruitment in rat pial venules - in vivo confocal microscopic study. Pathophysiology. 2004;11(2):121-128
66.Honjo M et al. Statin inhibits leukocyte-endothelial interaction and prevents neuronal death induced by ischemia-reperfusion injury in the rat retina. Archives of Ophthalmology. 2002;120(12):1707-1713
67.Wojciak-Stothard B, Williams L, Ridley AJ. Monocyte adhesion and spreading on human endothelial cells is dependent on rho-regulated receptor clustering. The Journal of Cell Biology. 1999;145(6):1293-1307
68.Diomede L et al. In vivo anti-inflammatory effect of statins is mediated by nonsterol mevalonate products. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21(8):1327-1332
69.Langert KA, Pervan CL, Stubbs EB Jr. Novel role of Cdc42 and RalA GTPases in TNF-alpha mediated secretion of CCL2. Small GTPases. 2014;5:e29260
70.Romano M et al. Inhibition of monocyte chemotactic protein-1 synthesis by statins. Laboratory Investigation. 2000;80(7):1095-1100
71.Collins TC, Beyth RJ. Process of care and outcomes in peripheral arterial disease. The American Journal of the Medical Sciences. 2003;325(3):125-134
72.Desai SS et al. Outcomes after endovascular repair of arterial trauma. Journal of Vascular Surgery. 2014;60(5):1309-1314
73.Schillinger M, Minar E. Restenosis after percutaneous angioplasty: The role of vascular inflammation. Vascular Health and Risk Management. 2005;1(1):73-78
74.Lemson MS et al. Intimal hyperplasia in vascular grafts. European Journal of Vascular and Endovascular Surgery. 2000;19(4):336-350
75.Serrano CV Jr et al. Coronary angioplasty results in leukocyte and platelet activation with adhesion molecule expression. Evidence of inflammatory responses in coronary angioplasty. Journal of the American College of Cardiology. 1997;29(6):1276-1283
76.Sakamoto K et al. Fluvastatin prevents vascular hyperplasia by inhibiting phenotype modulation and proliferation through extracellular signal-regulated kinase 1 and 2 and p38 mitogen-activated protein kinase inactivation in organ-cultured artery. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(2):327-333
77.Wilcox JN et al. Perivascular responses after angioplasty which may contribute to postangioplasty restenosis: A role for circulating myofibroblast precursors? Annals of the New York Academy of Sciences. 2001;947:68-90 discussion 90-2
78.Ming XF et al. Rho GTPase/rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Molecular and Cellular Biology. 2002;22(24):8467-8477
79.Yamanouchi D et al. Hydrophilic statin suppresses vein graft intimal hyperplasia via endothelial cell-tropic rho-kinase inhibition. Journal of Vascular Surgery. 2005;42(4):757-764
80.Aydin U et al. Effects of atorvastatin on vascular intimal hyperplasia: An experimental rodent model. Angiology. 2009;60(3):370-377
81.Chu T et al. Atorvastatin reduces accumulation of vascular smooth muscle cells to inhibit intimal hyperplasia via p38 MAPK pathway inhibition in a rat model of vein graft. Arquivos Brasileiros de Cardiologia. 2020;115(4):630-636
82.Desai P et al. Fluvastatin inhibits intimal hyperplasia in wild-type but not Thbs1-null mice. The Journal of Surgical Research. 2017;210:1-7
83.Fujita H et al. Pitavastatin inhibits intimal hyperplasia in rabbit vein graft. The Journal of Surgical Research. 2008;148(2):238-243
84.Yucel S et al. Reduced intimal hyperplasia in rabbits via medical therapy after carotid venous bypass. Texas Heart Institute Journal. 2009;36(5):387-392
85.Zhang L et al. Local delivery of pravastatin inhibits intimal formation in a mouse vein graft model. The Canadian Journal of Cardiology. 2012;28(6):750-757
86.Miyauchi K et al. Effectiveness of statin-eluting stent on early inflammatory response and neointimal thickness in a porcine coronary model. Circulation Journal. 2008;72(5):832-838
87.Hawes BE et al. Distinct pathways of Gi- and Gq-mediated mitogen-activated protein kinase activation. The Journal of Biological Chemistry. 1995;270(29):17148-17153
88.Versari D, Lerman LO, Lerman A. The importance of reendothelialization after arterial injury. Current Pharmaceutical Design. 2007;13(17):1811-1824
89.Dimmeler S et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. The Journal of Clinical Investigation. 2001;108(3):391-397
90.Llevadot J et al. HMG-CoA reductase inhibitor mobilizes bone marrow--derived endothelial progenitor cells. The Journal of Clinical Investigation. 2001;108(3):399-405
91.Walter DH, Dimmeler S, Zeiher AM. Effects of statins on endothelium and endothelial progenitor cell recruitment. Seminars in Vascular Medicine. 2004;4(4):385-393
92.Walter DH et al. Statin therapy accelerates reendothelialization: A novel effect involving mobilization and incorporation of bone marrow-derived endothelial progenitor cells. Circulation. 2002;105(25):3017-3024
93.Annex BH, Cooke JP. New directions in therapeutic angiogenesis and Arteriogenesis in peripheral arterial disease. Circulation Research. 2021;128(12):1944-1957
94.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nature Medicine. 2000;6(4):389-395
95.Ii M, Losordo DW. Statins and the endothelium. Vascular Pharmacology. 2007;46(1):1-9
96.Skaletz-Rorowski A, Walsh K. Statin therapy and angiogenesis. Current Opinion in Lipidology. 2003;14(6):599-603
97.Zahedipour F et al. Pleiotropic properties of statins via angiogenesis modulation in cardiovascular disease. Drug Discovery Today. 2022;27(10):103325
98.Muqri F et al. Thrombospondin-5 and fluvastatin promote angiogenesis and are protective against endothelial cell apoptosis. Journal of Cellular Biochemistry. 2020;121(10):4154-4165
99.Khaidakov M et al. Statins and angiogenesis: Is it about connections? Biochemical and Biophysical Research Communications. 2009;387(3):543-547
100.Weis M et al. Statins have biphasic effects on angiogenesis. Circulation. 2002;105(6):739-745
101.Shao H et al. Statin and stromal cell-derived factor-1 additively promote angiogenesis by enhancement of progenitor cells incorporation into new vessels. Stem Cells. 2008;26(5):1376-1384
102.Zhou J et al. Rosuvastatin enhances angiogenesis via eNOS-dependent mobilization of endothelial progenitor cells. PLoS One. 2013;8(5):e63126
103.Xiao Y et al. Transgelin 2 participates in lovastatin-induced anti-angiogenic effects in endothelial cells through a phosphorylated myosin light chain-related mechanism. PLoS One. 2012;7(10):e46510
104.Kikuchi R et al. Pitavastatin-induced angiogenesis and arteriogenesis is mediated by Notch1 in a murine hindlimb ischemia model without induction of VEGF. Laboratory Investigation. 2011;91(5):691-703
105.Vincent L et al. Cerivastatin, an inhibitor of 3-hydroxy-3-methylglutaryl coenzyme a reductase, inhibits endothelial cell proliferation induced by angiogenic factors in vitro and angiogenesis in in vivo models. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22(4):623-629
106.Urbich C et al. Double-edged role of statins in angiogenesis signaling. Circulation Research. 2002;90(6):737-744
107.Zacharek A et al. Simvastatin increases notch signaling activity and promotes arteriogenesis after stroke. Stroke. 2009;40(1):254-260
Written By
Ashley Penton, Kelly A. Langert, Kristopher Maier and Vivian Gahtan
Submitted: 25 January 2023Reviewed: 31 January 2023Published: 02 March 2023
 Open access peer-reviewed chapter
Open access peer-reviewed chapter