 Open access peer-reviewed chapter
Open access peer-reviewed chapter
Abstract
The chronic inflammatory state is a common condition in obesity. It has become a health problem with pandemic proportions that, in some developing countries, jointly to overweight, affects more than 50% of their population. As a part of the scenario, we observe how a significant proportion of people with overweight or obesity have raised the acute inflammatory response markers. This situation shows us how this chronic condition can trigger aggressive inflammatory phenomena in critically ill patients with other clinical conditions, occasioning torpid clinical evolution, ominous results, and low-rate survival. This chapter pretends to describe the influence of a low-grade inflammatory state on the clinical outcome of patients who develop a systemic inflammatory response.
Keywords
- chronic diseases
- low-rate inflammation
- obesity
- cancer
- systemic inflammatory response
1. Introduction
The term inflammation has been known for over a thousand years, becoming the common pathway for different diseases [1]. In this way, inflammation becomes one of the essential physiological mechanisms of systemic regulation, a process where cellular and molecular events occur, decreasing injury progression, and restoring tissue homeostasis [2].
Homeostatic compensations, which are responses of organisms to damage, illness, or significant environmental challenges, can represent trade-offs required to preserve essential bodily processes but, over time, lead to further abnormalities in body function [3]. The immune system provides a mechanism for the body to discern between healthy foreign cells and substances and to phagocytose or produce sensitized lymphocytes, specialized proteins, or antibodies to eradicate the invader [3, 4]. When tissue suffers damage, the affected components yield several chemical substances, which significantly alter the proximity of healthy tissues. Therefore, inflammation refers to a complete range of tissue changes [3].
A natural conserved process in inflammation, regardless of being systemic or local, is defined by the activation of immune and non-immune cells that defend the host by removing cell damage, dropped, or altered host molecules (pathogens or endogenous), as well as toxins, while promoting tissue repair and recovery [5].
Evidence indicates how changes in metabolic and inflammatory processes affect several chemical compounds in the body [6]. The presence of inflammation plays a significant role as an indicator of complications or development of diseases such as overweight, obesity, diabetes mellitus, cardiovascular diseases, and cancer [7]. The excess fatty tissue produces a state of low-grade inflammation, and the presence of inflammatory markers establishes several links with pathways in some organs that can drive obesity-related inflammation [7].
Obesity worldwide is defined as excess adipose tissue [3]. The measure of body mass index (BMI), which is determined as follows, is a proxy indicator for body fat composition calculated by dividing the body weight in kilograms (kg) by the height squared (m2); obesity is defined as BMI ≥ 30 kg/m2 while overweight considered as BMI ≥25.0 kg/m2 [3, 8, 9]; however, nowadays, it is recognized that BMI is a good screening tool, but it does not permit knowing the actual estimation of the fat mass and its impacts, such as the source of inflammatory mediators and endocrine organ; and ideally must be complemented by the body composition analysis, through different tools [DEXA (dual-energy X-ray absorptiometry), IMR (magnetic resonance imaging), BIA (bioelectrical impedance analysis), and themselves could be the objective of another discussion panel].
Nowadays, obesity has a 39% worldwide prevalence registered in 2021, existing strong evidence to support its close relationship to chronic low-grade systemic inflammation. The overweight obesity trendline considers the phenomenon one of the most important preventable risk factors for developing various chronic diseases [10], and it is linked to low-grade inflammatory phenomena.
During the COVID-19 pandemic, chronic low-grade inflammation was a common condition in patients with a severe hyperinflammatory syndrome, now named multisystemic inflammatory syndrome, defined by the centers for disease control and prevention (CDC) as a serious condition characterized by the presence of fever in a period over 24 hours, high levels of inflammatory markers and physical and biochemical evidence of multiorgan damage in a critically ill subject [11].
This chapter supports that this persistent medical condition can induce aggressive inflammatory processes (i.e., cytokine storm) in critically ill patients with diverse clinical disorders, leading to a sluggish clinical course, troubling outcomes, and a poor survival rate. This chapter explains how a low-grade inflammatory state impacts the patient’s prognosis and offers an overview of the most severe disorders affecting the public’s health worldwide.
2. Obesity and inflammatory phenomena impaction on intermediate metabolism
Overweight-obesity phenomenon is an altered metabolic condition and affects people at the pandemic global level; its origin is explained by a disproportionated relationship between energy intake and metabolic necessities and energy expenditure, and the result is the excess of adipose tissue (fat mass) that triggers a chronic low-grade inflammatory state.
A biological reaction to cellular damage, “inflammation,” is characterized by interactions that predict disruption of immune homeostasis [12].
The inflammatory mechanisms are the expression of a nonspecific inflammatory response to any disruptor of the homeostasis in a biological organism and imply the activation of humoral and cellular mediators, such as the infiltrating macrophages located in the middle of the adipose tissue in a complex immunologic net of phenomena and triggering insulin resistance obesity-related [12].
As part of these complex inflammatory phenomena in the adipose tissue, various macrophage phenotypes lead to those cells’ pro-inflammatory, anti-inflammatory, and even pleiotropic functions.
The immune system cell interacts with hypertrophic adipocytes through crosstalk, with chemotactic mediators that conduct the macrophage movements in infiltrating tissues, and consequently, the secretion of pro-inflammatory substances (TNFα, IL-6, IL-1 beta, and MCP-1, among others) and a chronic inflammatory state [13]. Monocyte–macrophages, described in 1882, are part of the innate immune system and represent one of the most primitive and unspecific host defense mechanisms in front of foreign pathogens, allergens, toxins, and even xenobiotic substances [12]. One of their most important roles is clearing cells and tissue damage, free chemotactic mediators, activating and solving sterile inflammatory events, and being the initial step for wound healing, tissue regeneration, and scar formation [13].
The molecules functioning, such as modulators for inflammation, depends on a broad group based on pattern recognition receptor, including toll-like receptors (TLRs), C-type lectin receptors (CLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene I (RIG-I)-like helicase receptors (RLR’s), and scavenger receptors, among others [14]. Monocyte–macrophage identify more than 1000 molecular patterns, damage-associated and pathogen-associated (DAMP’s and PAMP’s), acting as immune mediators with a pleiotropic and dynamic profile of secreting products (cytokines, chemokines, and growth factors) [15].
The role of a chronic sterile inflammatory phenomenon has been better understood over the years and has been elucidated in areas such as allergy, autoimmune phenomena, and cancer, and a prominent clue position the interactive role of macrophages-adipose tissue during the insulin resistance obesity-induced and their satellite mechanisms [16, 17, 18, 19, 20, 21].
Adipose tissue, functionally classified as white and brown adipose tissue (WAT and BAT, respectively), has a variety of metabolic actions. WAT goes 5 to 50% of total body weight; it is in subcutaneous, visceral epicardial, and around blood vessels; on the other hand, BAT has an inversely proportional relationship with age [22, 23]. The beige adipose tissue plays a heterogeneous role in glucose and lipid metabolism, helping to improve the intermediate metabolism [23].
The homeostasis maintaining the adipose tissue is considered an endocrine organ, with chemotactic capabilities to produce endocrine signals from WAT [24]. In obese people, its functionally turns anomalous and expresses an inflammatory phenotype linked to systemic inflammation, metabolic syndrome, and insulin resistance [22]; in the whole process, TNFα is fundamental to the development of an inflammatory phenotype, besides TLR4 that is a crucial mediator molecule expressed in adipocytes and macrophages, involved in the induction of insulin resistance through fatty acids [25, 26] (Figure 1).
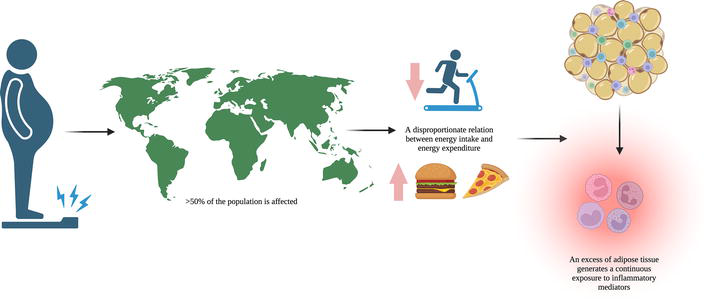
Figure 1.
Obesity and chronic inflammation. Obesity is one of the most prevalent diseases, affecting more than 50% of the population. This illness is explained by a disproportionated relation between energy intake and energy expenditure, which results in excess adipose tissue (considered an endocrine organ with chemotactic capabilities to produce endocrine signals from white adipose tissue) and puts patients into continuous exposure to inflammatory mediators, ending in a chronic inflammatory state that has diverse repercussions.
3. Inflammatory conditions on chronic diseases and their relationship with obesity
Chronic low-grade inflammation is joint in numerous chronic pathologies, some of which coincidentally have obesity according to the BMI of patients. Still, we know that it is not necessarily the better indicator of the quantity of fatty tissue in our body, and it is a good reason because currently is increasing the use of diverse tools to evaluate body composition in areas with a high prevalence of obesity.
The newest tools to determine the amount of fatty tissue overall and percentage involve all of them than discriminate lean mass, water, and fat mass such as IMR, computed tomography, and BIA. In the clinical ambulatory set, BIA has an advantage concerning IMR and CT and is a cost-effective tool that gives us information about subrogate measurements, such as phase angle, and has a predictive role in the patient outcome.
Fat mass is a source of lipids, an endocrine and metabolically active organ. Lipids forming the fat mass are involved in the innate immune response, specifically in triggering inflammatory mediators associated with the humoral immune unspecific response.
As we mentioned above, humoral inflammatory mediators are closely related to pleiotropic macrophage functions involved in various chronic conditions (coronary artery disease, diabetes type 2 obesity-related, or systemic unspecific response to damage in critically ill patients’ ability to overcome or over-response with an acute inflammatory response [27].
Inflammatory processes are highly ontogenically conserved response physiological mechanisms. It has passed in an evolutive way, probably from an innate response to microbial inversion to an unspecific response to tissular damage in front of any damage mechanism [27].
It is one of the most ancient pathological processes recorded during human history; even classical Greeks
The loss of control of the inflammation leads to damage in the systemic innate immune response, taking it to an imbalance between pro-, and anti-inflammatory molecular mediators, involving lipids, lysoglycerophospholipids, endocannabinoids, and their anatomical sources in the body. They all present in chronic disease development, such as cancer, diabetes mellitus, atherosclerosis, autoimmune diseases, and neurodegenerative processes, among other chronic inflammatory pathologies, which disrupt the tissular and systemic homeostasis [28].
The acute inflammatory response is triggered by innate immune cells such as macrophages, mast cells, and dendritic cells, involved in pathogen recognition through PAMPs or cell damage with impact on damage-associated molecular patterns (DAMPs). The complete cycle primarily induces local vasodilatation and cell recruitment, the release of complement proteins to stop microbes or molecules that have exceeded the epithelial or other natural barriers [28].
At this stage, the milieu has inflammatory characteristics with massive quantities of pro-inflammatory cytokines and omega-six-like pro-inflammatory lipid molecules as arachidonic acid-derived eicosanoids (prostaglandins and leukotrienes), all together potentiating the acute inflammatory immune response [29].
If the damage mechanisms stop the immune system and vascular endothelium put in on a lipid mediator class switch to break the eicosanoids and other inflammatory mediators, re-toward the metabolism to produce omega-three polyunsaturated fatty acids that initiate and lead the inflammatory phenomena to be solved. If the inflammation is extensive, it can be self-maintenance and promote the development of fibrosis and loss of tissular and organic functioning [30].
The acute inflammatory processes can be perpetuated secondary to the persistence of the damage source or
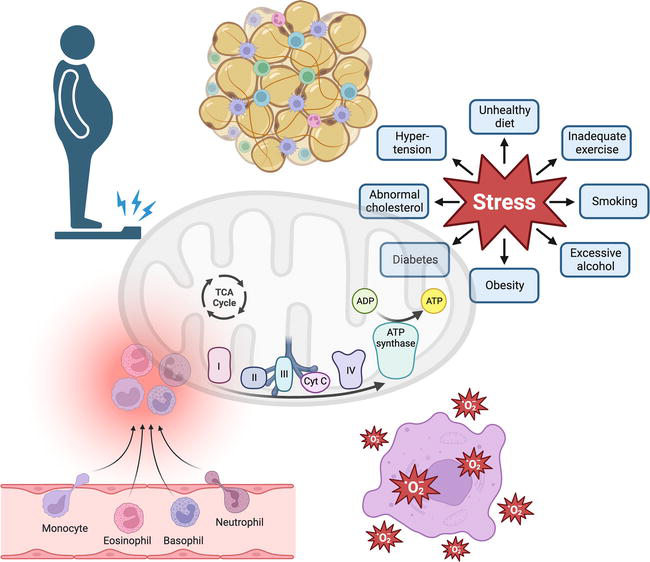
Figure 2.
Chronic low-grade inflammation and chronic diseases. Chronic low-grade inflammation is joint in numerous chronic pathologies, some of which coincidentally have obesity, where the immune system cell plays a fundamental role in interacting with hypertrophic adipocytes through crosstalk, with chemotactic mediators that conduct the macrophage movements in infiltrating tissues, and consequently, the secretion of pro-inflammatory substances (TNFα, IL-6, IL-1 beta, MCP-1, among others).
3.1 Inflammation and diabetes
Diabetes is one of the more prevalent chronic diseases around the world, and type 2 is usually associated with the complex overweight-obesity and insulin resistance; all of them involved in an imbalance between energy intake and expenditure, with the accumulation of fat mass and the consequent chronic low-grade inflammatory state, with macrophage activation through chemotactic signals from high-lipid content adipocytes, and consequently, elevated levels of pro-inflammatory cytokines that perpetuate the insulin resistance in obesity. It predisposes to the development of diabetes mellitus type 2 [13, 33].
The effects of the adipose tissue are endothelial dysfunction and the subsequent development of cardiovascular diseases. As a shred of evidence, in this milieu, a complex cross-talking between endocrine, immune (chronic inflammatory state), and neuronal networks is developing, relating to the loss of insulin sensitivity in target cells (adipocytes, hepatocytes, and myocytes immune cells, among others). This condition alters the insulin ability to control glucose levels and lipid homeostasis toward hyperglycemia, hyperlipidemia, and clinical data of diabetes type 2 [34, 35].
In the initial steps, the alterations are partially compensated through an increased insulin release and secretion from pancreatic beta-cells, leading to the exhaust of the beta-cells, reduced expression of insulin receptors, and altered metabolic feedback for the insulin [36, 37], perpetuating the chronic inflammatory state in the liver, skeletal muscle mass, endocrine pancreatic islets, and brain.
In the concrete case of obesity and inflammation related to metabolic diseases, the senescence of cells joins the aging to chronic disorders. Then aging enhances the burden of cellular senescence in almost all tissues with metabolic activity during the pathophysiological process of obesity in adulthood, diabetes mellitus type 2, and nonalcoholic fatty liver related to obesity, independently from the aging alone [38].
The senescence has a profound effect on the cells and toward the tissues to be dysfunctional and show inflammation stigmas in progenitor and mature cells, well-differentiated, or non-proliferating cells [38].
The insulin resistance-hyperinsulinemia pathophysiologic way, triggering senescence tissue in human adipose and liver cells, and the senescence perpetuate the insulin resistance in a vicious circle that profoundly affects people with these metabolic issues in age, body mass index, and hyperinsulinemia degree independent mechanism [39].
All the above-described phenomena reflect the relevance of the inflammatory mechanisms in obesity, insulin resistance, and diabetes and their profound effect on the health-disease interplay in humans (Figure 3).
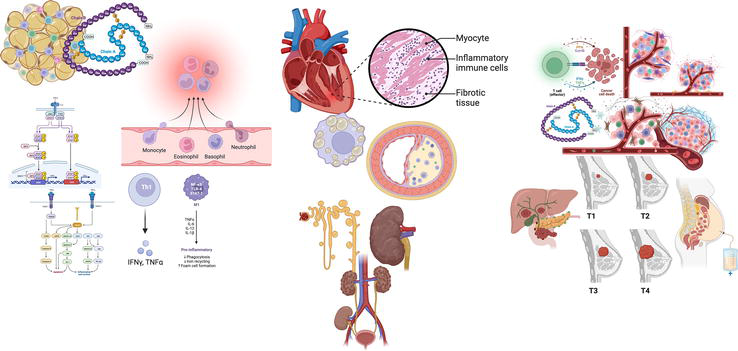
Figure 3.
Loss of control of inflammatory-anti-inflammatory balance and chronic diseases. The loss of control of inflammation leads to damage in the systemic innate immune response, taking it to an imbalance between pro, and anti-inflammatory molecular mediators, generating metabolic disturbances linked to low-grade chronic inflammation, promoting obesity-related hyperglycemia, insulin signaling, and high activity of glycolytic pathway and oxidative stress, reactions which end up triggering a low-grade chronic inflammatory state and being a central factor for many other diseases, as diabetes, cardiovascular diseases, chronic kidney disease, and even some types of cancer.
3.2 Inflammation and cardiovascular diseases (hypertension, ischemic cardiopathy, and stroke)
In cardiovascular diseases, atherosclerosis is essential and involves molecular mechanisms that result reflected in pathophysiologic issues joined to an inflammatory response. Recent publications describe the pro-inflammatory cellular stress, the role of the endothelium, and its functioning releasing molecules with autocrine, paracrine, and endocrine actions that sequentially trigger and perpetuate the inflammatory low-grade phenomena in cardiovascular disease and stroke, which are characteristics of the immune response that lead to inflammation and turn an atheroma plaque stable to unstable [40].
Various mechanisms induce inflammatory processes in atherosclerosis, including endothelium aging, metabolism alterations, and autoimmune and infectious damage. Along the described mechanisms, atherosclerosis triggers life-threat pathophysiological conditions such as cardiogenic shock, myocardial infarction, and stroke, all of them with acute systemic hyperinflammation that potentiate other phenomena such as procoagulant cascades, coinciding both kinds of inflammatory processes acute and chronic [41].
Atherosclerosis represents an inflammatory disease with a chronicity trend, affecting the elastic and muscular line in the arterial vessels, implicating the formation of atheroma plaques (rich in cholesterol) that cause obstruction and stenosis in the stable plaque and thrombosis in unstable plaque due to the activation of complement and coagulation cascades [40].
Regarding atherosclerosis pathophysiology, the inflammation that characterizes the disease is joined to aging but is not necessarily the only factor; it could be related to obesity, insulin resistance with cellular senescence, and genetic predisposition (familial dyslipidemias), among others [42]. The inflammatory phenomenon can be considered as nonclassical inflammation (Figure 3).
3.3 Chronic kidney disease and inflammation
Chronic kidney disease has become one of the most common illnesses worldwide, especially in low- and middle-income countries, reaching over 800 million people globally with this diagnosis (10% of the overall population) and emerging as one of the leading causes of mortality [43].
Persistent low-grade inflammation is an elemental factor in this disease’s development, making the kidneys one of the most highly susceptible organs to damage from pro-inflammatory cytokines and oxidative stress [44]. The renal system controls a significant portion of blood circulation, with the kidneys as the primary organs for maintaining the corticomedullary osmotic gradient for fluid absorption and urine concentration [45].
In the face of stressors such as energy deprivation, hormones, and various vasoactive molecules (including prostaglandins and nitric oxide) are produced to maintain homeostasis. However, these regulatory molecules are disrupted when inflammation results in kidney damage [46].
Renal tubules also contain many inflammatory markers responsible for renal insults and damage. Nevertheless, in remark to dysregulation states, such as diabetes, complications of acute kidney injury, and other uncontrolled inflammatory responses, these inflammatory mediators stop being regulated, which contributes to maladaptive response, higher energy demands, and a significant risk to ischemia, leading to oxidative damage as well, secondary to increased reactive oxygen species and decreased nitric oxide due to increased homocysteine levels [47].
Other factors trigger the emergence of a constant inflammation cascade that involves the patients with this diagnosis, which, in addition to the previous mechanism, occurs as a result of the treatment that has been given, for example, in dialysis, where factors such as impurities, microorganism presence, and even genetic and epigenetic influence, make the patient prone to an increase in the production of pro-inflammatory cytokines and oxidative stress, contributing to chronic inflammatory status [48].
CKD, then, is linked to an inflammatory state, and some authors describe the role of low-grade systemic inflammation in the development of CKD with alterations in laboratory inflammatory markers frequently coincidental with metabolic syndrome, nonalcoholic fatty liver disease, diabetes type 2, or cardiovascular disease [45, 48, 49]. As in other pathological conditions, the inflammatory phenomenon initiates with pro-inflammatory cytokines release, with an increased blood flow and chemotaxis mediating leukocyte infiltration [50]. The inflammatory low-grade state persists and is perpetuated by phenomena such as poor diet, alterations in gut microbiota, pregnancy in women, childhood infections, and cell stress in a previous phase of the disease establishment [51].
In CKD, substantial settings enhance and perpetuate the inflammatory state, infections, malnourishment, oxidative stress, dyslipidemia, overload volume, dialysis, and reduced clearance of inflammatory mediators [44].
The kidneys are essential to maintaining homeostasis, along they are involved with clearing cytokines and foreign antigens during renal clearance, removing pro-inflammatory cytokines and PAMPs as part of the kidney mechanisms that control the inflammation [51]. The tissue kidney interaction with renal dendritic cells and macrophages subtype M2 generates immune tolerance phenomena.
The own renal mechanism limits the inflammatory processes. It helps to immune tolerance, causing high susceptibility to damage mediated by inflammatory molecules in the kidney, which receives approximately 25% of the cardiac output in a minute, turning over to the kidney propensity to damage by inflammation and chronic renal damage, exacerbated by endothelial compromise by ischemic phenomena [52, 53] (Figure 3).
3.4 Cancer and inflammation
Adipose tissue is dysfunctional in obesity and even in overweight. The crosstalk between microenvironmental conditions, adipocytes, and immune system cells turns the typical phenotype of the cells to a malignant phenotype characterized by immortal cells with loss of mechanisms that control the proliferation in an altered context of endocrine signals that promote cancer development through autocrine, paracrine, and endocrine cell signaling [11].
In 2016, after analyzing over 1000 epidemiological reports, the IARC Working Group published a viewpoint [54]. Enough evidence exists to conclude that obesity is related to cancer development in at least thirteen anatomical locations, with a broad spectrum of pathophysiologic alterations in association with overweight-obesity and their involvement from sex hormone metabolism to intermediate metabolism disturbances (e.g., insulin resistance), adipocytokine expression with an exacerbated pro-inflammatory-oxidative status [55, 56, 57].
As part of the metabolic alterations in obesity, adipocytes have subcellular and metabolic disturbances that include endoplasmic reticulum and mitochondrial dysfunction, with an enhanced level of reactive oxygen species as a direct consequence of an overload of nutrients, conditions that generate a microenvironment favoring the inflammation, metabolic disturbance (dyslipidemia, hyperglycemia, and oxidative stress) inducing DNA damage and genomic instability, linked to low-grade chronic inflammation, joined to obesity-related hyperglycemia, enhanced insulin signaling, high activity of glycolytic pathway, and oxidative stress [57, 58, 59, 60, 61, 62, 63, 64, 65].
All previously mentioned changes influence the relationship between cell–cell and cell-extracellular matrix in tumors located in the vicinity of areas with adipose tissue [65], with a variety of common pathways with mechanisms involved with chronic low-grade inflammatory phenomena and all together promote angiogenesis, invasion, metalloproteinase activation, reactive oxygen species release mediated through molecular ways such as phosphoinositide 3-kinase (PI3K), vascular endothelial growth factor (VEGF), and signaling interplay such as PI3K/Akt [66, 67, 68, 69, 70, 71, 72].
To sum up, in cancer, one of the most critical elements to condition the tissue malignant transformation is obesity and its influence on the chronic or sustained low-grade inflammation with metabolic disruption, elevated tissue levels of pro-inflammatory cytokines, C reactive protein, and others that reinforce the pro-inflammatory context generating a deleterious effect on the nutritional state of cancer patients increasing the malnourishment related to tumor-cachexia and significant inflammatory mediators levels [73].
Then cancer occurs by chronic inflammatory pathway activation and a failure to resolve them [74]. In this scenario, the substantial effect of the molecular mechanisms that induce obesity generates a relationship in carcinogenesis [74]. The elevated levels of pro-inflammatory molecules make a microenvironment disrupt cell processes [75]. Inflammation in obese produces a dysregulated innate immunity activation of cytokines in adipocytokine metabolism, potentially enhancing angiogenesis and tumor progression [74]. The mechanisms identify with the prolonged inflammation that contributes to the proliferation of cancer with angiogenesis, tissue remodeling, and DNA damage [75]. A mechanism identifier is in the regulation of p53 (tumor suppressor gene). The evidence shows that chronic levels of inflammation in tumorigenicity can result from the inhibition of this gene with the inhibited cell apoptosis induced in cell proliferation [75]. The presence of increased levels in inflammation parameters (interleukin IL-1, IL-6, and C–reactive protein) and oxidative stress (glutathione peroxidase and superoxide dismutase) contributes to the cell damage that promotes the development of types of cancer such as breast, colorectal, esophageal, and liver cancer [76].
Cancer has a close relationship with obesity, and obesity exerts a profound influence on insulin resistance, arterial hypertension, dyslipidemia, and other disturbances in the intermediate metabolism. During the rapid increase of the obesity pandemic, cancer cases rose too, and then began the explosion of papers describing the influence of obesity and low-grade inflammation in the genesis, maintenance, and progression of various malignant diseases [77]. The interplay between adipose tissue, immune system cells, and molecules and the development of malignant cells reflect a homeostatic disruption in the balance between the anti-inflammatory and pro-inflammatory milieu [77].
In the case of cancer, macrophages play a crucial role in development and progression. Still, they are not the only involved cells B lymphocytes interact too in sustained inflammatory conditions [78]; besides pro-inflammatory mediators such as adipocytokines, that join to dyslipidemia, hyperglycemia, and hypercholesterolemia worsening the health biological behavior of the macrophages and potentiate their detrimental effects [78] (Figures 3 and 4).
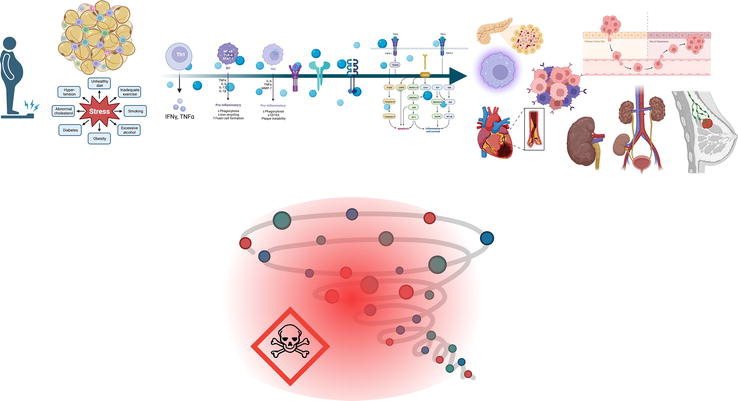
Figure 4.
Resumé low-grade chronic inflammation associated with chronic diseases and Acute Hyperinflammation. As part of the metabolic alterations in obesity, adipocytes have subcellular and metabolic disturbances that include endoplasmic reticulum and mitochondrial dysfunction, with an enhanced level of reactive oxygen species as a direct consequence of an overload of nutrients, conditions that generate a microenvironment favoring the inflammation, metabolic disturbance (dyslipidemia, hyperglycemia, and oxidative stress) inducing DNA damage, and genomic instability.
4. Influence of chronic inflammatory conditions on the clinical outcomes in acute situations (acute systemic inflammatory response)
The presence of a chronic state with low-grade systemic inflammation plays an essential role in the etiology and pathogenesis of multiple diseases [79]. The evidence demonstrates a link between chronic inflammation and acute exacerbation in various illnesses [80]. Inflammatory cytokines such as IL-6 and tumor necrosis factor-alpha (TNF-alpha), created after persistent inflammation and oxidative stress, start a vicious cycle [80].
Despite the previously mentioned mechanisms, the chronic low-grade inflammatory state in aging diseases belongs to phenomena named nonclassical inflammation (meta-inflammation, quasi-inflammation, or para-inflammation) [28, 81, 82, 83, 84], and it is part of an ontogenic mechanism evolutive preserved to the maintenance of homeostasis (allostasis; understanding as they achieve the stability through a changing milieu), and it involves the vascular endothelium, immune system, and lipid metabolism primarily in response to different challenges induced by the internal or external conditions [84].
In the tissues, the pro-inflammatory mechanisms include a feedback loop to synthesize and release mediators, such as cytokines, even in diseases not considered inflammatory states per se (cancer, neurologic, and psychiatric disorders) [85, 86, 87, 88].
The term canonical inflammation has recently re-emerged. It is characterized by a focal reaction in the microvascular bed and triggers an exudative response with leukocyte migration by chemotaxis to the local damaged tissues. The pathophysiological mechanisms involve endothelial response at the level of postcapillary venules, causing endothelitis, a phenomenon observed only in vertebrates with lymphatic microcapillaries net and microcirculatory units in various organs. It generated the classical signs of inflammation (rubor (redness), tumor, heat, pain, and loss of function) [89, 90]. On the other hand, invertebrates develop nonclassical local inflammation, characterized by phagocyte accumulation in damaged tissue, without any microvascular compromise [89].
In any inflammatory process, the goal of the local inflammation is to isolate and eliminate any damage-related damage. Still, in the low-grade inflammatory response, the four local signs of inflammation are not involved in the sustained response [87, 89].
The low-grade inflammation is known as para-inflammation, and it is considered a nonclassical type of inflammation with long-term effects of the factors damage-related, in the absence of signs of an inflammatory focus, delocalization of the process, not enough mechanical barriers, interconnected with the tissular aging, metabolic mediators related to damage and systemic changes in the endothelium (endotheliosis; all of them observed in metabolic syndrome, morbid obesity, DM type 2, and aging-related sarcopenia) [91, 92].
Based on the inflammatory mechanisms previously named, recently has been delineated the hyperinflammation or high-grade systemic inflammatory state, tied to an exacerbated systemic effect of inflammation, triggers mediators leading to systemic endothelitis, resulting in life-threatening endothelial disorders, joined to leukocytes activation inside blood vessels, with subsequent activation of hemostatic mechanisms, complements factors and kallikrein-kinin systems.
In patients who have a basis of low-grade chronic inflammatory state related to obesity, metabolic alterations, and aging, among others, a new damage condition can release a cytokine storm, enhancing the latent microcirculatory issues, perpetuating the damage that could be limited in an acute inflammatory state, turning it severe and transforming in a life treating condition, such as in severe COVID-19 and the cytokine storm observed in obese and diabetic patients.
Therefore, the confluence of the local and systemic chronic-low-level inflammation and acute inflammatory conditions can lead to the perfect storm where the chronic conditions inflammation-related will worsen the inflammatory response in current damage and turn over the phenomena to hyperinflammation (Figure 4).
5. Conclusions
Chronic low-grade inflammatory state derived from obesity is a present condition in a wide variety of chronic and aging-related diseases, such as diabetes type 2, cardiovascular diseases and stroke, chronic kidney disease, and cancer, among the most important. During the past four years, we observed a “perfect storm” of the coincidental phenomena involving chronic low-grade inflammation in chronic diseases and emerging acute diseases with an exacerbating inflammatory response, clearly represented by severe COVID-19 cases [93], triggering a multisystemic inflammatory syndrome worsening the prognosis in critically ill patients. Throughout the process, we understood how the chronic low-grade inflammatory states allied to aging-related diseases potentiate the systemic acute response triggering a massive release of inflammatory mediators, generating the now-named “cytokine storm” and putting in this scenario the fundamental importance of regulating and modifying the people behavior to turn to a healthier lifestyle, reducing the overweight-obesity rates around the world through a better diet and increasing the rates of physical activity, to improve their health conditions and health-related quality of life through the changes in modifiable risk factors.
References
- 1.
Medzhitov R. Inflammation 2010: New adventures of an old flame. Cell. 2010; 140 (6):771-776 - 2.
Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2018; 9 (6):7204-7218 - 3.
Hall JE, Hall ME. Tratado de fisiología médica. 14th ed. Barcelona: Elsevier; 2021 - 4.
Lemke G. How macrophages deal with death. Nature Reviews. Immunology. 2019; 19 (9):539-549 - 5.
Netea MG, Balkwill F, Chonchol M, Cominelli F, Donath MY, Giamarellos-Bourboulis EJ, et al. A guiding map for inflammation. Nature Immunology. 2017; 18 (8):826-831 - 6.
Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014; 156 (1-2):20-44 - 7.
Lempesis IG, Georgakopoulou VE. Physiopathological mechanisms related to inflammation in obesity and type 2 diabetes mellitus. World Journal of Experimental Medicine. 2023; 13 (3):7-16 - 8.
Szukiewicz D. Molecular mechanisms for the vicious cycle between insulin resistance and the inflammatory response in obesity. IJMS. 2023; 24 (12):9818 - 9.
Cuciureanu M, Caratașu CC, Gabrielian L, Frăsinariu OE, Checheriță LE, Trandafir LM, et al. 360-degree perspectives on obesity. Medicina. 2023; 59 (6):1119 - 10.
Sanhueza S, Simón L, Cifuentes M, Quest AFG. The adipocyte–macrophage relationship in cancer: A potential target for antioxidant therapy. Antioxidants. 2023; 12 (1):126 - 11.
Hamad Saied M, van der Griend L, van Straalen JW, Wulffraat NM, Vastert S, Jansen MHA. The protective effect of COVID-19 vaccines on developing multisystem inflammatory syndrome in children (MIS-C): A systematic literature review and meta-analysis. Pediatric Rheumatology Online Journal. 2023; 21 (1):80 - 12.
Minihane AM, Vinoy S, Russell WR, Baka A, Roche HM, Tuohy KM, et al. Low-grade inflammation, diet composition and health: Current research evidence and its translation. The British Journal of Nutrition. 2015; 114 (7):999-1012 - 13.
Guria S, Hoory A, Das S, Chattopadhyay D, Mukherjee S. Adipose tissue macrophages and their role in obesity-associated insulin resistance: An overview of the complex dynamics at play. Bioscience Reports. 2023; 43 (3):BSR20220200 - 14.
Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunological Reviews. 2009; 227 (1):221-233 - 15.
Zhang X, Mosser D. Macrophage activation by endogenous danger signals. The Journal of Pathology. 2008; 214 (2):161-178 - 16.
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. The Journal of Clinical Investigation. 2012; 122 (3):787-795 - 17.
Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, et al. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007; 447 (7140):92-96 - 18.
Ma WT, Gao F, Gu K, Chen DK. The role of monocytes and macrophages in autoimmune diseases: A comprehensive review. Frontiers in Immunology. 2019; 10 :1140 - 19.
Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011; 475 (7355):222-225 - 20.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. The Journal of Clinical Investigation. 2003; 112 (12):1796-1808 - 21.
Xu H, Barnes GT, Yang Q , Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. The Journal of Clinical Investigation. 2003; 112 (12):1821-1830 - 22.
Chait A, Den Hartigh LJ. Adipose tissue distribution, inflammation and its metabolic consequences, including diabetes and cardiovascular disease. Frontier in Cardiovascular Medicine. 2020; 7 :22 - 23.
Li Y, Yun K, Mu R. A review on the biology and properties of adipose tissue macrophages involved in adipose tissue physiological and pathophysiological processes. Lipids in Health and Disease. 2020; 19 (1):164 - 24.
Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. The Journal of Clinical Endocrinology & Metabolism. 2004; 89 (6):2548-2556 - 25.
Nisoli E, Briscini L, Giordano A, Tonello C, Wiesbrock SM, Uysal KT, et al. Tumor necrosis factor α mediates apoptosis of brown adipocytes and defective brown adipocyte function in obesity. Proceedings of the National Academic Science USA. 2000; 97 (14):8033-8038 - 26.
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid–induced insulin resistance. The Journal of Clinical Investigation. 2006; 116 (11):3015-3025 - 27.
Radzioch D, Giera M, De Sanctis JB. Editorial: Quo Vadis lipid mediators – Lipid mediators implication in inflammation and chronic inflammatory diseases. Frontiers in Immunology. 2021; 12 :699276 - 28.
Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008; 454 (7203):428-435 - 29.
Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nature Reviews. Immunology. 2015; 15 (8):511-523 - 30.
Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: Signals in resolution. Nature Immunology. 2001; 2 (7):612-619 - 31.
Fullerton JN, Gilroy DW. Resolution of inflammation: A new therapeutic frontier. Nature Reviews. Drug Discovery. 2016; 15 (8):551-567 - 32.
Aoki T, Narumiya S. Prostaglandins and chronic inflammation. Trends in Pharmacological Sciences. 2012; 33 (6):304-311 - 33.
Harford KA, Reynolds CM, McGillicuddy FC, Roche HM. Fats, inflammation and insulin resistance: Insights to the role of macrophage and T-cell accumulation in adipose tissue. The Proceedings of the Nutrition Society. 2011; 70 (4):408-417 - 34.
Engin AB. Adipocyte-macrophage cross-talk in obesity. In: Engin AB, Engin A, editors. Obesity and Lipotoxicity. Cham: Springer International Publishing; 2017. pp. 327-343. Available from: http://link.springer.com/10.1007/978-3-319-48382-5_14 - 35.
Asterholm IW, Halberg N, Scherer PE. Mouse models of lipodystrophy: Key reagents for the understanding of the metabolic syndrome. Drug Discovery Today: Disease Models. 2007; 4 (1):17-24 - 36.
Mukhuty A, Fouzder C, Mukherjee S, Malick C, Mukhopadhyay S, Bhattacharya S, et al. Palmitate induced Fetuin-A secretion from pancreatic β-cells adversely affects its function and elicits inflammation. Biochemical and Biophysical Research Communications. 2017; 491 (4):1118-1124 - 37.
Jia Q , Morgan-Bathke ME, Jensen MD. Adipose tissue macrophage burden, systemic inflammation, and insulin resistance. American Journal of Physiology-Endocrinology and Metabolism. 2020; 319 (2):E254-E264 - 38.
Gustafson B, Nerstedt A, Spinelli R, Beguinot F, Smith U. Type 2 diabetes, independent of obesity and age, is characterized by senescent and dysfunctional mature human adipose cells. Diabetes. 2022; 71 (11):2372-2383 - 39.
Spinelli R, Baboota RK, Gogg S, Beguinot F, Blüher M, Nerstedt A, et al. Increased cell senescence in human metabolic disorders. Journal of Clinical Investigation. 2023; 133 (12):e169922 - 40.
Gusev E, Sarapultsev A. Atherosclerosis and inflammation: Insights from the theory of general pathological processes. IJMS. 2023; 24 (9):7910 - 41.
Xiang Q , Tian F, Xu J, Du X, Zhang S, Liu L. New insight into dyslipidemia-induced cellular senescence in atherosclerosis. Biological Reviews. 2022; 97 (5):1844-1867 - 42.
Santos RD, Gidding SS, Hegele RA, Cuchel MA, Barter PJ, Watts GF, et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: A consensus statement from the international atherosclerosis society severe familial hypercholesterolemia panel. The Lancet Diabetes & Endocrinology. 2016; 4 (10):850-861 - 43.
Kovesdy CP. Epidemiology of chronic kidney disease: An update 2022. Kidney International Supplements. 2022; 12 (1):7-11 - 44.
Kadatane SP, Satariano M, Massey M, Mongan K, Raina R. The role of inflammation in CKD. Cell. 2023; 12 (12):1581 - 45.
Mihai S, Codrici E, Popescu ID, Enciu AM, Albulescu L, Necula LG, et al. Inflammation-related mechanisms in chronic kidney disease prediction, progression, and outcome. Journal of Immunology Research. 2018; 2018 :1-16 - 46.
Damkjær M, Vafaee M, Møller ML, Braad PE, Petersen H, Høilund-Carlsen PF, et al. Renal cortical and medullary blood flow responses to altered NO availability in humans. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2010; 299 (6):R1449-R1455 - 47.
Liu BC, Tang TT, Lv LL, Lan HY. Renal tubule injury: A driving force toward chronic kidney disease. Kidney International. 2018; 93 (3):568-579 - 48.
Akchurin OM, Kaskel F. Update on inflammation in chronic kidney disease. Blood Purification. 2015; 39 (1-3):84-92 - 49.
United States Renal Data System. 2022 USRDS Annual Data Report: Epidemiology of Kidney Disease in the United States. National Institutes of Health; 2022 - 50.
Pahwa R, Goyal A, Jialal I. Chronic Inflammation. Treasure Island, FL: StatPearls Publishing; 2023. Available from: http://www.ncbi.nlm.nih.gov/books/NBK493173/ - 51.
Kurts C, Panzer U, Anders HJ, Rees AJ. The immune system and kidney disease: Basic concepts and clinical implications. Nature Reviews. Immunology. 2013; 13 (10):738-753 - 52.
Verma SK, Molitoris BA. Renal endothelial injury and microvascular dysfunction in acute kidney injury. Seminars in Nephrology. 2015; 35 (1):96-107 - 53.
Bouglé A, Duranteau J. Pathophysiology of sepsis-induced acute kidney injury: The role of global renal blood flow and renal vascular resistance. In: Kellum JA, Ronco C, Vincent JL, editors. Contributions to Nephrology. Karger AG; 2011. pp. 89-97. Available from: https://www.karger.com/Article/FullText/329243 - 54.
Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K. Body fatness and cancer – Viewpoint of the IARC working group. The New England Journal of Medicine. 2016; 375 (8):794-798 - 55.
Frisch CM, Zimmermann K, Zilleßen P, Pfeifer A, Racké K, Mayer P. Non-small cell lung cancer cell survival crucially depends on functional insulin receptors. Endocrine-Related Cancer. 2015; 22 (4):609-621 - 56.
Słabuszewska-Jóźwiak A, Lukaszuk A, Janicka-Kośnik M, Wdowiak A, Jakiel G. Role of leptin and adiponectin in endometrial Cancer. IJMS. 2022; 23 (10):5307 - 57.
Miethe C, Zamora M, Torres L, Raign KG, Groll CJ, Price RS. Characterizing the differential physiological effects of adipocytokines visfatin and resistin in liver cancer cells. Hormone Molecular Biology and Clinical Investigation. 2019; 2019 (2):38. DOI: 10.1515/hmbci-2018-0068/html - 58.
Cunarro J, Casado S, Lugilde J, Tovar S. Hypothalamic mitochondrial dysfunction as a target in obesity and metabolic disease. Frontiers in Endocrinology. 2018; 9 :283 - 59.
Assumpção JAF, Pasquarelli-do-Nascimento G, Duarte MSV, Bonamino MH, Magalhães KG. The ambiguous role of obesity in oncology by promoting cancer but boosting antitumor immunotherapy. Journal of Biomedical Science. 2022; 29 (1):12 - 60.
Hauck AK, Huang Y, Hertzel AV, Bernlohr DA. Adipose oxidative stress and protein carbonylation. Journal of Biological Chemistry. 2019; 294 (4):1083-1088 - 61.
Jackisch L, Murphy AM, Kumar S, Randeva H, Tripathi G, McTernan PG. Tunicamycin-induced endoplasmic reticulum stress mediates mitochondrial dysfunction in human adipocytes. The Journal of Clinical Endocrinology & Metabolism. 2020; 105 (9):2905-2918 - 62.
Nishikawa M, Hashida M, Takakura Y. Catalase delivery for inhibiting ROS-mediated tissue injury and tumor metastasis. Advanced Drug Delivery Reviews. 2009; 61 (4):319-326 - 63.
Mondal S, Adhikari N, Banerjee S, Amin SA, Jha T. Matrix metalloproteinase-9 (MMP-9) and its inhibitors in cancer: A minireview. European Journal of Medicinal Chemistry. 2020; 194 :112260 - 64.
Usman M, Volpi EV. DNA damage in obesity: Initiator, promoter and predictor of cancer. Mutation Research/Reviews in Mutation Research. 2018; 778 :23-37 - 65.
Quail DF, Dannenberg AJ. The obese adipose tissue microenvironment in cancer development and progression. Nature Reviews. Endocrinology. 2019; 15 (3):139-154 - 66.
Lu Y, Zhou XY, Zhou CL, Liu J, Yong T, Fan Y, et al. Insulin receptor tyrosine kinase substrate (IRTKS) promotes the tumorigenesis of pancreatic cancer via PI3K/AKT signaling. Human Cell. 2022; 35 (6):1885-1899 - 67.
Hu DX, Sun QF, Xu L, Lu HD, Zhang F, Li ZM, et al. Knockdown of DEAD-box 51 inhibits tumor growth of esophageal squamous cell carcinoma via the PI3K/AKT pathway. WJG. 2022;28 (4):464-478 - 68.
Gu JW, Young E, Patterson SG, Makey KL, Wells J, Huang M, et al. Postmenopausal obesity promotes tumor angiogenesis and breast cancer progression in mice. Cancer Biology & Therapy. 2011; 11 (10):910-917 - 69.
Seo BR, Bhardwaj P, Choi S, Gonzalez J, Andresen Eguiluz RC, Wang K, et al. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Science Translational Medicine. 2015; 2015 (7):301. DOI: 10.1126/scitranslmed.3010467 - 70.
Gonzalez-Perez R, Lanier V, Newman G. Leptin’s pro-Angiogenic signature in breast cancer. Cancers. 2013; 5 (4):1140-1162 - 71.
Miethe C, Torres L, Zamora M, Price RS. Inhibition of PI3K/Akt and ERK signaling decreases visfatin-induced invasion in liver cancer cells. Hormone Molecular Biology and Clinical Investigation. 2021; 42 (4):357-366 - 72.
Ramos-Andrade I, Moraes J, Brandão-Costa RM, Vargas Da Silva S, De Souza A, Da Silva C, et al. Obese adipose tissue extracellular vesicles raise breast cancer cell malignancy. Endocrine-Related Cancer. 2020; 27 (10):571-582 - 73.
Dibaba DT, Judd SE, Gilchrist SC, Cushman M, Pisu M, Safford M, et al. Association between obesity and biomarkers of inflammation and metabolism with cancer mortality in a prospective cohort study. Metabolism. 2019; 94 :69-76 - 74.
Lathigara D, Kaushal D, Wilson RB. Molecular mechanisms of Western diet-induced obesity and obesity-related carcinogenesis—A narrative review. Metabolites. 2023; 13 (5):675 - 75.
Nigam M, Mishra AP, Deb VK, Dimri DB, Tiwari V, Bungau SG, et al. Evaluation of the association of chronic inflammation and cancer: Insights and implications. Biomedicine & Pharmacotherapy. 2023; 164 :115015 - 76.
Savic Vujovic K, Zivkovic A, Dozic I, Cirkovic A, Medic B, Srebro D, et al. Oxidative stress and inflammation biomarkers in postoperative pain modulation in surgically treated patients with laryngeal Cancer—Pilot study. Cell. 2023; 12 (10):1391 - 77.
Singh A, Mayengbam SS, Yaduvanshi H, Wani MR, Bhat MK. Obesity programs macrophages to support cancer progression. Cancer Research. 2022; 82 (23):4303-4312 - 78.
Oleinika K, Slisere B, Catalán D, Rosser EC. B cell contribution to immunometabolic dysfunction and impaired immune responses in obesity. Clinical and Experimental Immunology. 2022; 210 (3):263-272 - 79.
Sharebiani H, Keramat S, Chavoshan A, Fazeli B, Stanek A. The influence of antioxidants on oxidative stress-induced vascular aging in obesity. Antioxidants. 2023; 12 (6):1295 - 80.
Luft VC, Schmidt MI, Pankow JS, Couper D, Ballantyne CM, Young JH, et al. Chronic inflammation role in the obesity-diabetes association: A case-cohort study. Diabetology and Metabolic Syndrome. 2013; 5 (1):31 - 81.
Russo S, Kwiatkowski M, Govorukhina N, Bischoff R, Melgert BN. Meta-inflammation and metabolic reprogramming of macrophages in diabetes and obesity: The importance of metabolites. Frontiers in Immunology. 2021; 12 :746151 - 82.
Wang G, Shen G, Jiang X, Chen Z, Yin T. Assessment of para-inflammation in a wound healing model. Experimental Therapy Medicine. 2020; 2020 :15. Available from:http://www.spandidos-publications.com/10.3892/etm.2020.8666 - 83.
Guarner V, Rubio-Ruiz ME. Low-grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. In: Yashin AI, Jazwinski SM, editors. Interdisciplinary Topics in Gerontology and Geriatrics. S. Karger AG; 2015. pp. 99-106. Available from: https://www.karger.com/Article/FullText/364934 - 84.
Fülöp T, Larbi A, Witkowski JM. Human Inflammaging. Gerontology. 2019; 65 (5):495-504 - 85.
Lan T, Chen L, Wei X. Inflammatory cytokines in cancer: Comprehensive understanding and clinical Progress in gene therapy. Cell. 2021; 10 (1):100 - 86.
Upthegrove R, Khandaker GM. Cytokines, oxidative stress and cellular markers of inflammation in schizophrenia. In: Khandaker GM, Meyer U, Jones PB, editors. Neuroinflammation and Schizophrenia. Cham: Springer International Publishing; 2019. pp. 49-66. DOI: 10.1007/7854_2018_88 - 87.
Gusev E, Zhuravleva Y. Inflammation: A new look at an old problem. IJMS. 2022; 23 (9):4596 - 88.
Gusev EY, Zotova NV. Cellular stress and general pathological processes. CPD. 2019; 25 (3):251-297 - 89.
Gusev EY, Zhuravleva YA, Zotova NV. Correlation of the evolution of immunity and inflammation in vertebrates. Biology Bulletin Reviews. 2019; 9 (4):358-372 - 90.
Rather LJ. Disturbance of function (functio laesa): The legendary fifth cardinal sign of inflammation, added by Galen to the four cardinal signs of Celsus. Bulletin of the New York Academy of Medicine. 1971; 47 (3):303-322 - 91.
Tylutka A, Morawin B, Walas Ł, Michałek M, Gwara A, Zembron-Lacny A. Assessment of metabolic syndrome predictors in relation to inflammation and visceral fat tissue in older adults. Scientific Reports. 2023; 13 (1):89 - 92.
Antuña E, Cachán-Vega C, Bermejo-Millo JC, Potes Y, Caballero B, Vega-Naredo I, et al. Inflammaging: Implications in sarcopenia. IJMS. 2022; 23 (23):15039 - 93.
Martínez-Colón GJ, Ratnasiri K, Chen H, Jiang S, Zanley E, Rustagi A, et al. SARS-CoV-2 infection drives an inflammatory response in human adipose tissue through infection of adipocytes and macrophages. Science Translational Medicine. 2022; 14 (674):eabm9151