 Open access peer-reviewed chapter
Open access peer-reviewed chapter
Abstract
The bone marrow (BM) is an integral part of the immune system that communicates with other immune tissues via the bloodstream but does not have lymphatic vessels. It is the primary site of lymphopoiesis, where B cells and early T-cell progenitors develop, from late fetal life onwards, and a secondary lymphoid organ for B lymphocytes. At the same time, it regulates the function and dynamics of the immune system in a steady state and disease conditions. Activating and inhibitory signals from various marrow elements regulate the traffic of lymphocyte subtypes (B, T, and NK), including direct cell contact and released factors from stromal cells. This chapter is a review of the life cycle and dynamics of lymphoid cells in health and representative immune-associated disorders. Understanding the central bone marrow’s role may clarify the pathologic changes and open potential therapeutic channels in some disorders.
Keywords
- lymphopoiesis
- bone marrow niche
- lymphocyte function regulation
- bone marrow lymphocyte types
- bone marrow immune role in disease
1. Introduction
Both hematopoietic and lymphoid systems arise in the bone marrow from the late fetus and prevail throughout life with a finite control by lineage-specific and broadly acting biological factors and a homeostatic signaling network, allowing sensitive responsiveness to fluctuating body needs [1]. The complex functional structure of the bone marrow exhibits age and context adaptation [2].
The bone marrow (BM) is a central immune organ that does not have lymphatic vessels but communicates with other lymphoid tissues via the bloodstream. It is also a secondary lymphoid organ for mature B cells. It differs from other immune organs in lacking a fixed lymphocyte organization but contains interstitially scattered cells within its parenchyma, sometimes, forming small aggregates, especially in the elderly. In the bone marrow, regulatory cells and short- and long-acting signals orchestrate the immune system and critically underlie the pathogenesis of many reactive and neoplastic conditions [1]. This chapter reviews the bone marrow structure and function as a part of the immune system, including the following three sections:
Bone marrow lymphopoiesis: ontogeny and maturation
The milieu and kinetics of bone marrow lymphoid elements
Dynamics of BM lymphoid cells in health and disease
2. Bone marrow lymphopoiesis: ontogeny and maturation
The hematopoietic marrow extends throughout the medullary bone cavities of the body from the late fetus along the life span, gradually contracting toward the axial skeleton with age. In these locations, interaction with a complex connective tissue stroma with cellular and extracellular matrix components is critical for supporting and regulating proliferation, maturation, maintenance, senescence, and final destruction of hematopoietic elements.
Stromal elements comprise bone, vasculature, and a network of mesenchymal and reticular cells, critical for bone marrow functioning. Other elements include sympathetic and parasympathetic innervation, adipocytes, resident macrophages [3], neutrophils [4], megakaryocytes [5], T lymphocytes, and dendritic cells (DCs). The extracellular matrix of macromolecules, including fibronectin, vitronectin, collagens (types: I, II, IV, and VI), and proteoglycans, forms an integral part of the hematopoietic stem cell (HSC) microenvironment. Specific HSC niches refer to the arrays of stromal cells, specific locations, soluble molecules, signaling cascade, and gradients, together with the shear stress, temperature, and oxygen tension, which determine the stem cell behavior at any given time [6]. The niche dynamics promote specific HSC properties such as; quiescence, self-renewal, and proliferation. In contrast to other organs, the bone marrow has no clear demarcation by distinct cell density or stromal cell distribution.
Both hematopoietic and lymphoid progenitors have a common origin from pluripotent CD34+ stem cells (multipotent progenitors (MPPs)) in the bone marrow. Lymphoid lineages originate from a common progenitor with the central control of the Ikaros gene; a zinc finger gene encoding DNA-binding protein transcription factor, which plays crucial functions in hematopoiesis and regulation of immune cell development. Along with other transcription factors, it also regulates the expression of other genes influencing the phenotypic characteristics of lymphocytes, including immunoglobulin heavy and light chain gene rearrangement, and cluster of differentiation 3 (CD3) complex antigen receptors.
After an initial commitment, T-cell precursors migrate to the thymus, while B-cell progenitors complete their primary maturation in the bone marrow. Figure 1 outlines the central and peripheral compartments, and stages of lymphopoiesis [7]. Receptor gene recombinations occur along B- and T-lymphocyte differentiation before emigrating to the peripheral lymphoid tissues or the peripheral blood [1].
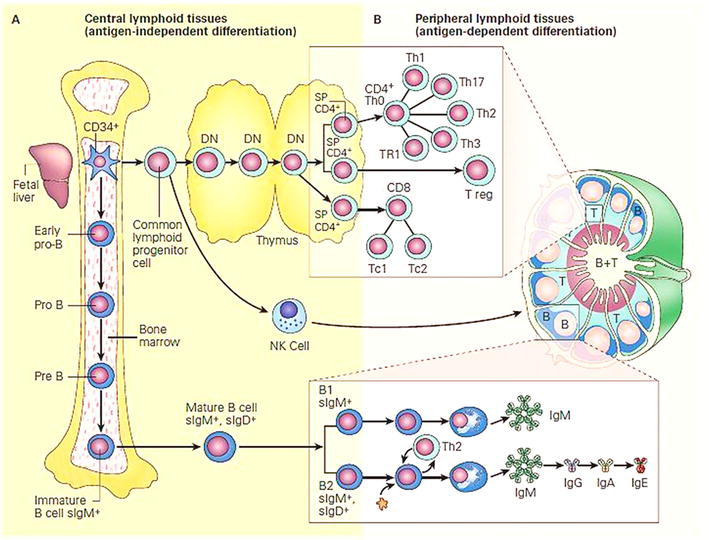
Figure 1.
Schematic representation of the central and peripheral compartments of lymphoid differentiation: Panel a: Development of the immune system from stem cells in bone marrow, and differentiating in central lymphoid tissues (bone marrow and thymus) is independent of antigen contact. Panel B: Migration of cells into peripheral lymphoid tissues (lymph nodes, spleen, and mucosa-associated lymphoid tissues) is antigen-dependent [
2.1 B-cell maturation
2.1.1 Primary B-cell maturation
Primary B-cell maturation proceeds through four main stages; pro-B, pre-B, immature B cells, and mature B cells [8], with an essential requirement for stromal interaction by direct cell contact and released growth factors [9]. Direct contact of pro-B cells with stromal cells occurs via V and LA-4/M1, and then c-kit receptor/stromal cells-stem-cell factor interaction initiates B-cell division and interleukin-7 (IL-7) receptor expression. Eventually, the downregulation of adhesion molecules releases the B cells.
The recombination of heavy chain variable gene region segments marks the pre-B cells [10]. The next stage of immature B cells expresses membrane immunoglobulin M (IgM).
B cells recognizing self-antigens undergo apoptosis and the surviving cells differentiate along two subpopulations: B1 cells expressing CD19+, CD5+, and sIgM+, and B2 cells expressing CD19+, sIgM+, and sIgD+. Both subtypes migrate into the peripheral lymphoid organs [10].
2.1.2 Secondary B-cell differentiation
In peripheral lymphoid organs, B-cell differentiation is antigen-driven with regulation by sequentially acting transcription factors that induce initial upregulation of PAX5, IRF8, and BACH2 genes, followed by IRF4, XBP1, and BLIMP1 genes [11]. In parallel with these changes, B cells express stage-specific markers, including cluster of differentiation 27 (CD27), cluster of differentiation 38 (CD38), and cluster of differentiation 138 (CD138), with simultaneous downregulation of the B-cell markers such as cluster of differentiation 19 (CD19) and cluster of differentiation 20 (CD20).
In the germinal centers, signals from the helper T cells activate B cells that proliferate, undergo somatic hypermutation, and finally generate memory B cells and long-lived plasma cells [11].
Although B-cell development is conceptually unidirectional, recent evidence suggests a possible phenotypic “reflex” from immature to pre-B cell subsets, probably through receptor editing, though the existence of multiple differentiation pathways is also possible [12].
2.1.3 The plasma cell stage
The maintenance of plasma cell survival in the bone marrow depends on stromal cell signals, and cytokines, including megakaryocyte proliferation-inducing ligand-APRIL and interleukin-6 (IL-6) [13], eosinophils APRIL and IL-6, [14], and monocytes APRIL [15], while the granulocyte colony-stimulating factor (G-CSF) mobilizes plasma cells [16].
2.2 T-cell maturation
After a short bone marrow phase, cytokines and major histocompatibility complex class I or II (MHC-I or II) on thymus epithelium promote the differentiation of T precursor cells into T-central (Tc), regulatory T cells (Treg), and T helper (Th) subpopulations, while those with self-specific T-cell receptor (TCR) undergo apoptosis [17].
Bone marrow T cells contribute to immune homeostasis where IL-7, interleukin-15 (IL-15), and tumor necrosis family (TNF) family members activate CD8+ T cells. Priming of naive CD4+ and CD8+ T cells may occur in response to bloodborne antigens presented by bone marrow dendritic cells (DCs) and other myeloid elements. Survival of memory CD8+ and CD4+ T cells in the bone marrow requires IL-7 and IL-15, and Major Histocompatibility Complex (MCH) and IL-7, respectively [17].
At least two memory T-cell niches sustain the viability and functionality of CD4+ T cells [17], similar to the hematopoietic stem cell niches [18]. CD45RA memory T cells include CCR7+ (central memory cells) and CCR7 effector memory cells [19], while noncirculating cells constitute “Tissue-resident memory T cells (Trm cells)” [18].
2.3 Natural killer cells
Natural killer (NK) cells develop from bone marrow CD34+ stem cells and undergo similar maturation stages to other lymphoid cells, but lack antigen-specific receptors. They express receptors for activating cellular killing and other receptors recognizing self-MHC alleles that inhibit the killing of normal cells. NK cells have the appearance of large granular lymphocytes with neither T- nor B-cell antigens and do not express cluster of differentiation 16 (CD16) and cluster of differentiation 56 (CD56) (an NK-specific adhesion molecule), helping their identification in the peripheral circulation [1].
2.4 Regulation of bone marrow lymphopoiesis
In the bone marrow, lymphopoiesis shares a common perivascular niche with myelopoiesis with several niche factors regulating their maturation and functions, including adipocytes [20], regulatory T cells (Tregs) [21], monocytic cells, mesenchymal cells [22], and nerves [23]. External signals also contribute to lymphocyte regulation such as sex steroids. This arrangement allows a versatile hemopoietic response under stress conditions, related to competitive and differential requirements for cytokines such as C-X-C motif chemokine ligand 12 (CXCL12) and stem cell factor (SCF) [24].
Bone marrow stromal cells maintain the survival and function of B lymphocytes, long-lived plasma cells, and memory T cells. They also release inhibitory factors such as transforming growth factor-β (TGF-β) and hepatocyte growth factor (HGF). In steady states, resident, low-migrating mature cluster of differentiation 19 (CD19)-negative plasma cells are the major B-cell component in the bone marrow. During an immune response, CD19+ plasma cells predominate [25]. Reciprocally, activated T cells secrete cytokines that promote the terminal differentiation of myeloid precursors. Unlike hemopoietic cells, plasma cells do not have fixed anatomical niches in the bone marrow [26], but receive supporting signals from many cells, including, CXCL12-producing mesenchymal cells [27], granulocytes, megakaryocytes, and myeloid cells [13]. Additional factors influencing plasma cell kinetics include megakaryocyte proliferation-inducing ligand APRIL and IL-6 [14], eosinophils APRIL and IL-6 [13], and monocytes APRIL [15]. On the other hand, G-CSF mobilizes plasma cells [16].
3. The milieu and kinetics of BM lymphoid elements
The bone marrow hosts a mixture of mature lymphocytes in specific niches, which promotes their long-term survival, including B, T, and NK cells [28]. It harbors about 12% of all lymphoid cells in the human body at any given time. Most T cells are recirculating from peripheral lymphoid tissues including memory CD4+ and CD8+ cells. Plasma cells similarly migrate into the bone marrow where they reside by losing response to CXCL12 and C-X-C motif chemokine ligand 9 (CXCL9) signals [25].
In normal adults, bone marrow lymphocytes range between 10 and 20% of cellularity; the majority are small cells, resembling those in the peripheral blood, and scatter interstitially with occasionally small aggregates, especially in the elderly. They comprise a mixture of B, T, and NK cells with their subtypes. T cells are the predominant lymphocytes in the bone marrow with a T-to-B lymphocyte ratio of 4 to 1. The CD4+ to CD8+ ratio is 1:2; the reverse of that in the peripheral blood.
3.1 The bone marrow lymphocyte mix
3.1.1 Bone marrow, B lymphocytes
The bone marrow is the site for B lymphopoiesis throughout life. Early stages of B-cell maturation prevail during fetal life, whereas mature B cells predominate in adults [26]. In infants and young children, the marrow has a higher percentage of lymphocytes (up to 50% of cellularity), including a proportion of larger lymphocytes with immature morphology (hematogones) that represent lymphoid progenitors and decrease with age [27]. The bone marrow has a higher proportion of IgM- than IgG (immunoglobulin G)-bearing B cells, which is the reverse of the peripheral blood [28]. They also express lower levels of peanut agglutinin (PNA) relative to germinal centers or circulating memory B cells [26].
In the adult bone marrow, most B cells have prior activation in germinal centers of peripheral lymphoid tissues, evidenced by somatic hypermutation, isotypic diversification, and antigen selection. Those expressing IgM lack cluster of differentiation 10 (CD10), lower cluster of differentiation 24 (CD24), and co-expression of immunoglobulin D (IgD), which differentiate them from the
3.1.2 Bone marrow, plasma cells
Plasma cells are terminal, nonproliferative B cells that develop through T-cell-dependent or -independent pathways [29]. A continuum of plasma cells’ maturation stages ranging from the less mature, proliferative short-lived (CD138+ B220+ or plasmablasts), to the mature long-lived PCs (LLPCs) are present. The latter make up more than 50% of total bone marrow plasma cells [29].
Factors maintaining survival of plasma cells in the bone marrow include membrane-bound ligands like cluster of differentiation 80 (CD80) and secreted proteins such as APRIL, interleukin-6 (IL-6), B-cell activating factor (BAFF), and CXCL12 (or stromal cell-derived factor-1 (SDF-1)) [30]. In Giemsa-stained marrow, plasma cells are scattered singly or in small groups representing about 2% of cellularity in normal adults. In biopsy sections, they appear close to capillaries [27]. They have a unique migration pattern with alternating high motility and low-rate migration or arrest, closely linked to their survival [29]. APRIL-secreting cells dynamically coalesce and recruit migrating plasma cells into clusters and release signals that promote the overall motility of plasma cells [31], and enhance their fitness and survival [29]. Other factors enhancing plasma cell motility include CXCL12 and its receptor C-X-C motif chemokine receptor 4 (CXCR4), fibronectin, and intercellular adhesion molecule 1 (ICAM-1) ligand [32]. While very late antigen-4 (VLA-4) and vascular cell adhesion molecule-1 (VCAM-1) binding promote cell arrest and tight adhesion, blocking either pathway leads to egress of plasma cells [29].
3.1.3 Bone marrow T lymphocytes
Mature T cells in the bone marrow are in constant exchange with the blood and make up a part of the total body recirculating lymphocyte pool that also includes the thoracic duct, spleen, and lymph nodes [28]. In the absence of ongoing immune responses or inflammations, homing to the bone marrow is a “default” pathway for the maintenance of recirculating memory T cells.
Regulation of bone marrow T-cell niches differs between steady-state conditions, immune response, and different disease states. Recirculating T lymphocytes compete with resident T cells for the same niche locations in the bone marrow [17]. The proliferation rates of cluster of differentiation 4 (CD4) and cluster of differentiation 8 (CD8) T cells are higher in the bone marrow than in the spleen and lymph nodes. T cells in the bone marrow have high expression of CXCR4, C-C chemokine receptor type 5 (CCR5), C-X-C motif chemokine receptor 6 (CXCR6), and C-X3-C motif chemokine receptor 1 (CX3CR1), but not of C-X-C motif chemokine receptor 3 (CXCR3). They respond to inflammatory chemokines, including C-C motif chemokine ligand 3 (CCL3), C-C motif chemokine ligand 4 (CCL4), C-C motif chemokine ligand 5 (CCL5), C-X-C motif chemokine ligand 16 (CXCL16), and C-X3-C motif chemokine ligand 1 (CX3CL1).
The recirculation of CD8 T cells involves rolling in bone marrow microvessels through L-, P-, and E-selectins. They stick to endothelial cells by the lymphocyte integrin α4β1, activated by SDF-1 (CXCL12) and the endothelial adhesion molecule VCAM-1. Modulating the expression of T-cell receptor CXCR4 by antigen, interleukin (IL)-2, and tumor necrosis factor (TNF) promotes the directed motility of T cells [17]. Granulocyte colony-stimulating factor (G-CSF) mobilizes regulatory T cells from the bone marrow.
3.2 T lymphocytes networking with other bone marrow elements
In the bone marrow, T lymphocytes contribute to the homeostasis of hematopoiesis and bone. T cells act mainly on the proliferative/differentiation phase of myelopoiesis rather than the stem cell maintenance and the bone remodeling system [18]. Many of the molecular signals in the immune response also contribute to bone homeostasis. Examples of osteoclast activators include interleukin-17 (IL-17) and receptor activator of nuclear factor κΒ ligand (RANK-L), expressed by activated CD4 and CD8 T cells. On the other hand, interferon-ɣ (IFN-ɣ) and interleukin-4 (IL-4) released by T cells inhibit bone resorption [17].
4. Dynamics of bone marrow lymphoid cells in health and disease
Bone marrow lymphocytes have central roles in many physiological and pathological processes. Memory B cells contribute to long-term antibody production, inflammations, tissue repair, and bone metabolism. T cells interact with other marrow elements and modulate their functions such as mesenchymal stromal cells, osteoclasts, osteoblasts, and hematopoietic precursors [17].
The lymphocyte traffic, which maintains homeostasis of the immune system, requires spatial and functional versatility. The cellular redistribution of mitochondria is an important factor in regulating these mechanisms permitting the motility of emigrating cells [33].
4.1 Aging
Aging alters basic cellular mechanisms leading to genomic instability, telomere shortening, epigenetic dysregulation, and cellular senescence, all contributing to a deranged hematopoiesis and immune system and increasing inflammatory diseases in the elderly. Common effects include reduced B cells and their subsets and impairment of antibody responses. The reduced generative capacity and altered microenvironment reduce HSC self-renewal and their preferential differentiation toward myeloid cells, B-1 and B-2 subsets, and regulatory B cells (Bregs), naive T cell, production with an increase in regulatory T cells (Tregs) [34].
Inflammatory changes in aging convert myeloid cells into pro-inflammatory 4-1BB ligand (4-1BBL)-expressing cells, leading to the activation of CD8+ T cells secreting antitumor granzyme B. The reduction of B-cell subsets suppresses antibody affinity and diversity and impairs antibody responses, with the expansion of age-related B cells (ABCs) contributing to inflammation via pro-inflammatory T-cell activation and cytokine release [34]. The downregulation of XBP-1 and B-lymphocyte-induced maturation protein-1 (Blimp-1) transcription factors, and upregulation of the plasma cell-inhibiting factor PAX-5, reduce B1 and spontaneously IgM-secreting B-1 cells and their diversity in the elderly.
On the other hand, the reduced E47 messenger RNA (mRNA) stability and activation-induced deoxycytidine deaminase (AID) transcription inhibit B-2 cell functions and isotype switching [34]. ABCs uniquely express cluster of differentiation 11b (CD11b), cluster of differentiation 11c (CD11c), and T-bet, and respond to innate activation stimuli, such as toll-like receptor 7 (TLR7) signals. They also secrete autoreactive antibodies and anti-inflammatory cytokine interleukin-10 (IL-10), reflecting their primary immunoregulatory function. They have distinct subsets that vary with extrinsic factors, such as age and antigen load. A major subset is a T-bet expressing TH1 cells that expand in response to innate stimuli, such as TLR7 and toll-like receptor 9 (TLR9) ligands. T-bet expression by B cells promotes immunoglobulin G2a (IgG2a) antibody isotype switching. A smaller subset is T-bet negative expressing C-X-C Motif Chemokine Receptor 5 (CXCR5) and it causes higher levels of immunoglobulin G1 (IgG1). Double-negative memory B cells may represent an intermediate stage from ABC to plasma cells. The age-related adipose-resident B cells (AABs) express pro-inflammatory markers [34].
4.2 Modulation of hematopoiesis in inflammatory conditions and infections
Significant alterations in leukocyte production occur in many inflammatory conditions, with an increase in granulopoiesis at the expense of lymphopoiesis. The mechanisms underlying such alterations are mostly due to modulation of the bone marrow microenvironment by stress signals and reduction of growth and retention factors, particularly stem cell factor and CXCL12, which preferentially inhibit lymphopoiesis [24]. However, overstressing hematopoietic stem and progenitor cells (HSPCs) in chronic inflammation leads to cell (DNA) damage and bone marrow failure. Furthermore, toxic insults to bone marrow stroma, e.g., chemotherapy may result in clonal hematopoiesis and potentially malignant transformation.
Figure 2 represents the inflammatory bone marrow microenvironment and its consequences [35]. An inflammatory microenvironment changes mesenchymal stem cells (MSCs) into an inflammatory, secretory phenotype, e.g., nestin+, Gli1+, and leptin-receptor+ (LepR) cells with the release of pro-inflammatory signals that alter the HSC niche and the erythroid precursors. Consequently, alterations in HSC function and output, including myeloid cell expansion, innate immune cells’/myeloid-derived suppressor cells’ (MDSCs’) recruitment, increased platelets’ release by megakaryocytes and decreased the development of lymphoid cells. The impaired erythroid differentiation accounts partially for the associated anemia and the macrophage inflammatory phenotype further contributes to microenvironment inflammation. Adipocytes also increase with the release of inflammatory signals [35].
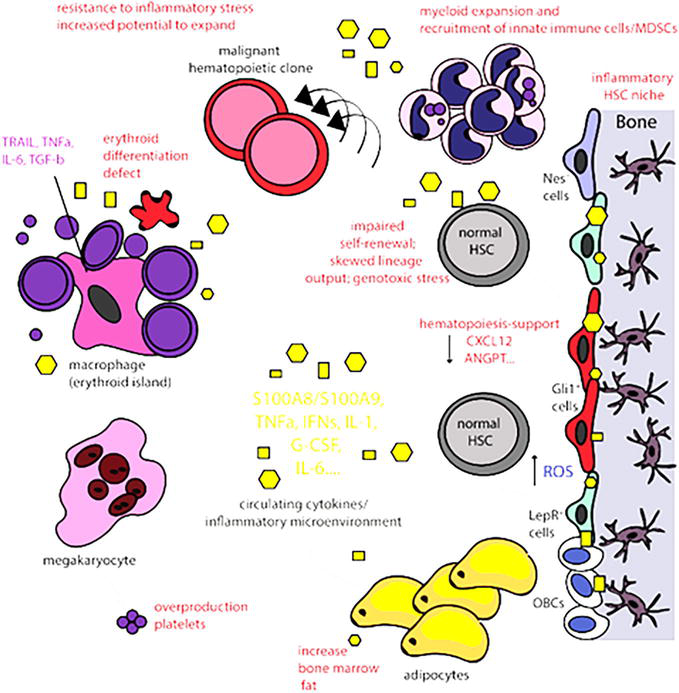
Figure 2.
The inflammatory bone marrow microenvironment. An inflammatory bone marrow microenvironment develops when MSCs acquire an inflammatory, secretory phenotype such as nestin+ (Nes+), Gli1+, leptin-receptor (LepR+) cells and release pro-inflammatory signals affecting both the HSC niche and the erythroblastic islands [
Bone marrow T cells can also modulate hematopoiesis in inflammatory conditions. At low neutrophil counts, interleukin 23 (IL-23) released by macrophages and DCs stimulates CD4 T cells, gamma delta T cells (ɣɗ-T cells), and NK cells, producing IL-17, which promotes granulopoiesis. At high neutrophil counts, inhibition of interleukin-23 (IL-23) by apoptotic neutrophils results in negative feedback [17]. Sometimes, the bone marrow is the target of effector T cells, such as in hematological malignancies, idiopathic thrombocytopenic purpura (ITP), and autoimmune diseases [17].
The brief neutropenia occurs early in sepsis and rapidly corrects by accelerated cell emigration and reactive neutrophilia follows mainly due to increased immature proliferative compartment of granulopoiesis. Immature neutrophils in the bone marrow also become resistant to inflammatory signals [24]. An early onset lymphopenia, characteristic of sepsis, is mainly due to massive lymphocyte apoptosis primarily of CD4+ T cells by activation of multiple cell death pathways [18]. Inflammatory cytokines also promote lymphocyte mobilization [24]. Homing and proliferation of CD4+ memory T cells occur in response to IL-7 at resolution of sepsis-induced lymphopenia.
4.3 Leukocyte dynamics in stroke and stress conditions
Stroke and stress often induce lymphopenia and immunosuppression while increasing myeloid cells.
Lymphopenia affects the entire B-cell lineage, including B-cell progenitors in the bone marrow, due to reduced proliferation and differentiation, and enhanced apoptosis. Lymphopenia in these cases is due to the hypothalamic-pituitary-adrenal (HPA) axis action via glucocorticoid receptors on hematopoietic cells. Both long-range and local microenvironmental signals link the neuroendocrine system to emergency hematopoiesis and impair the lymphopoiesis. The neutrophil-to-lymphocyte ratio in peripheral blood is a prognostic indicator that defines poor long-term outcomes in these patients [36].
Cell death and release of danger-associated factors, such as high mobility group box 1 (HMGB-1) and heat shock proteins (HSPs) in ischemic injuries, provoke an inflammatory response. In addition, systemic glucocorticoid levels caused by activation of the sympathetic nervous system (SNS) and the HPA axis increase the apoptosis of B-cell progenitors [37]. Furthermore, altered lineage decision of HSCs in ischemia is due to sympathetic signals to stem cell niches, favoring myelopoiesis.
4.4 Hematopoietic stem cell engraftment
In steady states, lymphocyte homeostasis is maintained by balancing production and lifespan rates. A full recovery of T cells is often slow after autologous HSC transplantation and may take several years [38].
T cells can promote HSC engraftment by eliminating residual host cells responsible for rejection and enhancing stromal function [17].
Although studying the dynamics of transplanted cells is often difficult, recent imaging techniques allow
4.5 Bone metastasis
The complex network of immune and bone cells of the bone marrow microenvironment increases the chance of metastases to bone than other sites [40]. The endosteal and vascular stem cell niches of the marrow microenvironment support the seeding and establishment of cancer cells [41].
The tumor antigen-primed T cells migrate to the bone marrow and initiate the pre-metastatic niche by the transfer of signals that alter bone homeostasis. The tumor cells then colonize the bone marrow and reside in the hematopoietic niches [19].
The interaction of tumor cells with bone is the main pathogenic mechanism of metastasis involving released signals that sustain a vicious cycle [42]. In addition, the release of immune-suppressive cytokines by tumor cells promotes the conversion of M1 macrophages and N1 neutrophils to tumor-associated M2 macrophages and N2 neutrophils, with tumor-promoting activity [40].
Another mechanism-promoting metastasis is immunosuppression, in which myeloid-derived suppressor cells (MDSCs) play a critical role. They are immature myeloid cells co-expressing CD11b and cluster of differentiation 33 (CD33) [43], originating in the bone marrow and migrating to secondary lymphoid organs, where they inhibit the CD8+ T antitumor response [41]. MDSCs also suppress T-cell function by impairing antigen recognition, releasing small soluble oxidizers, and depleting local essential amino acids. They suppress both CD4 and CD8 T cells while promoting activation and expansion of regulatory T cells (Tregs) [44]. The critical roles of MDSCs rationally explain their assessment as prognostic indicators in osteotropic tumors.
Meanwhile, cancer cell-released soluble factors stimulate MDSCs differentiation into activated osteoclasts. Both MDSCs and activated T cells produce pro-osteoclastogenic factors, including C-C chemokine receptor type 2 (CCR2) and RANK-L, respectively. Thus, tumor growth sustains the osteoclast activation by several mechanisms [41].
In summation, tumor-cell intrinsic traits and interaction with the specialized microenvironmental niches play critical roles in controlling tumor-cell colonization with initial seeding, dormancy, and outgrowth, as demonstrated in Figure 3 [45]. In the perivascular niche, tumor cells interact with CXCL12-expressing stromal cells and endothelial E-selectin promotes “mesenchymal-to-epithelial transition,” “stemness,” “survival,” and “growth.” Activated osteoclasts express Notch ligand, Jagged1 (JAG1), and VCAM-1, increasing the production of the osteoclast-stimulating factors, macrophage colony-stimulating factor (M-CSF), and receptor activator of nuclear factor κΒ ligand (RANK-L) which alter the bone turnover. In addition, bone resorption releases transforming growth factor-β (TGF-β) propagating a “vicious cycle” and promoting osteolytic bone metastasis.
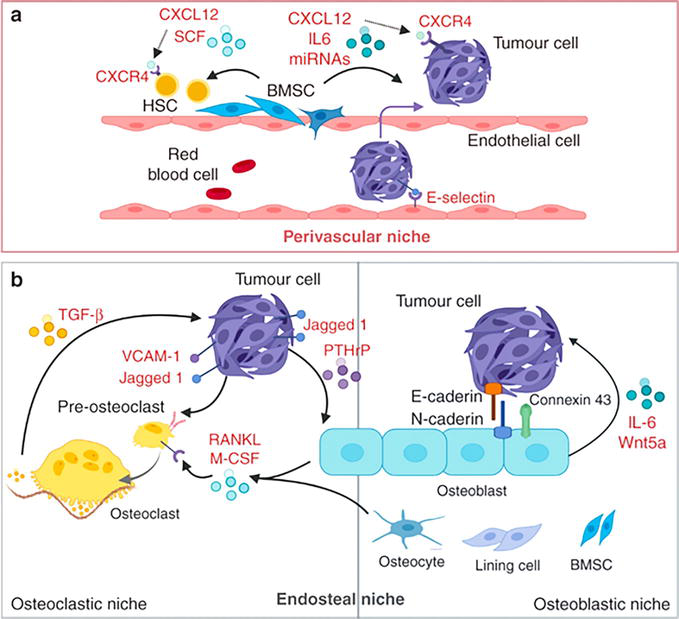
Figure 3.
Metastatic bone marrow niches. The perivascular and endosteal niches in bone metastasis [
On the other hand, osteoblasts secrete Wnt5a and interleukin (IL)-6 and form gap junctions, and E-cadherin/N-cadherin junctions, thus promoting bone metastasis [45].
Other cells contributing to the metastatic niche include megakaryocytes, adipocytes, and sympathetic nerve cells by altering the immune escape, dormancy, and proliferation of tumor cells . Understanding the intricate pathways of metastasis helps the evolution of new therapeutic targets [45].
5. Conclusion
In conclusion, the unique bone marrow’s organizational and cellular structure is critical to the immune system’s homeostasis and functions. Understanding the lymphocyte dynamics in the bone marrow and their regulations may clarify many of the pathologic changes in many physiologic and pathologic conditions and their prognostic impact. It can also provide potential venues for the prevention and treatment of some diseases.
References
- 1.
Immunopaedia. “Free Immunology Education.” 2022. Available from: www.immunopaedia.org.za/ - 2.
Moreau JM. Chapter 6. In: B Cell Dynamics within the Bone Marrow Microenvironment: Comparisons of Inflammation and Steady State [Thesis]. Canada. TSpace: Department of Immunology, University of Toronto; 2017. pp. 68-100. Available from: https://tspace.library.utoronto.ca/handle/1807/77479 - 3.
Christopher MJJ, Rao Liu F, Woloszynek JR, Link DC. Expression of the G-CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice. The Journal of Experimental Medicine. 2011; 208 (2):251-260. DOI: 10.1084/jem.20101700 - 4.
Kawano Y, Petkau G, Wolf I, Tornack J, Melchers F. IL-7 and immobilized, kit-ligand stimulate serum and stromal cell-free cultures of precursor B-cell lines and clones. European Journal of Immunology. 2017; 47 :206-212. DOI: 10.1002/eji.201646677. PMID: 17145957; PMCID: PMC2118173 - 5.
Pinho S, Tony MT, Yang E, Wei Q , Claus NC, Frenette PS, et al. Lineage-biased, hematopoietic stem cells are regulated by distinct niches. Developmental Cell. 2018; 44 :634-641, Cell Press. Elsevier Inc. DOI: 10.1016/j.devcel.2018.01.016 - 6.
Lucas D. Structural organization of the bone marrow and its role in hematopoiesis. Current Opinion in Hematology. 2021; 28 (1):36-42. DOI: 10.1097/MOH.0000000000000621 - 7.
Grosschedl R. Establishment and maintenance of B cell identity. In: Cold Spring Harbor Symposia on Qualitative Biology. Vol. LXXVIII, 78. Cold Spring Harbor Laboratory Press; 2013. pp. 23-30. DOI: 10.1101/sqb.2013.78.020057. Available from: https://www.researchgate.net/publication/261736570 - 8.
Kaminski N, Sulentic C. B lymphocytes. In: Encyclopedic Reference of Immunotoxicology. Berlin, Heidelberg: Springer; 2005. pp. 88-89. DOI: 10.1007/3-540-27806-0_158 - 9.
Salvo P, Vivaldi FM, Bonini A, Biagini D, Bellagambi FG, Miliani FM, et al. Biosensors for detecting lymphocytes and immunoglobulins. Biosensors. 2020; 10 :155. DOI: 10.3390/bios10110155https://pubmed.ncbi.nlm.nih.gov/33946495/ - 10.
Cherukommu S. Role of GSK-3 and T-Bet in Anti-Tumor Immunity [Thesis]. Canada: Université de Montréal; 1980]. Faculté de Médecine Mémoire présenté en vue de l’obtention du grade de Maitrise en biologie moléculaire. Papyrus: Institutional Repository. 2021. Available from: https://papyrus.bib.umontreal.ca/xmlui/handle/1866/25645 - 11.
Verstegen NJM, Pollastro S, Unger PA, Marsman C, Elias G, Jorritsma T, et al. Single-cell analysis reveals dynamics of human B cell differentiation and identifies novel B and antibody-secreting cell intermediates. Elife. 2 Mar 2023; 12 :e83578. DOI: 10.7554/eLife.83578. PMID: 36861964; PMCID: PMC10005767 - 12.
Mehr R, Shahaf G, Sah A, Cancro M. Asynchronous differentiation models explain bone marrow labeling kinetics and predict reflux between the pre-and immature B cell pools. International Immunology. 2003; 15 (3):301-312. The Japanese Society for Immunology. DOI: 10.1093/intima/dxg025 Available from:www.intimm.oupjournals.org - 13.
Zehentmeier S, Roth K, Cseresnyes Z, et al. Static and dynamic components synergize to form a stable survival niche for bone marrow plasma cells. European Journal of Immunology. 2014; 44 :2306-2317. DOI: 10.1002/eji.201344313 - 14.
Winter O, Moser K, Mohr E, et al. Megakaryocytes constitute a functional component of a plasma cell niche in the bone marrow. Blood. 2010; 116 (11):1867-1875, ISSN 0006-4971. DOI: 10.1182/blood-2009-12-259457 Available from:https://www.sciencedirect.com/science/article/pii/S000649712032989X - 15.
Belnoue E, Tougne C, Rochat AF, et al. Homing and adhesion patterns determine the cellular composition of the bone marrow plasma cell niche. Journal of Immunology (Baltimore, Md.: 1950). 2012; 188 (3):1283-1291. DOI: 10.4049/jimmunol.1103169 - 16.
Caraux A, Vincent L, Bouhya S, et al. Residual malignant and normal plasma cells shortly after high dose melphalan and stem cell transplantation. High-light of a putative therapeutic window in multiple myeloma? Oncotarget. 2012; 3 (11):1335-1347. DOI: 10.18632/oncotarget.650 - 17.
Di Rosa F. T-lymphocyte interaction with stromal, bone, and hematopoietic cells in the bone marrow. Immunology and Cell Biology. 2009; 87 :20-29. DOI: 10.1038/icb. Available from:http://www.nature.com/icb/journal/v87/n1/full/icb200884a.html - 18.
Skirecki T, Swacha P, Hoser G, et al. Bone marrow is the preferred site of memory CD4 + T cell proliferation during recovery from sepsis. JCI Insight. 2020; 5 (10):e134475. DOI: 10.1172/jci.insight.134475.insight.jci.org - 19.
Bonomo A, Monteiro AC, Balduíno A. Hematopoietic Stem Cells, Tumor Cells, and Lymphocytes — Party in the Bone Marrow. London, UK: InTechOpen; 2014. DOI: 10.5772/58843 - 20.
Bilwani FA, Knight KL. Adipocyte-derived soluble factor(s) inhibits early stages of B lymphopoiesis. London, UK: Journal of Immunology. InTechOpen; 2014; 189 (9):4379-4386. DOI: 10.4049/jimmunol.1201176 Epub 2012 Sep 21 - 21.
Kim SE, Kim HM, Doh J. Single-cell arrays of hematological cancer cells for assessment of lymphocyte cytotoxicity dynamics, serial killing, and extracellular molecules. Lab on a Chip. 2019; 19 :2009-2018. DOI: 10.1039/c91c00133f - 22.
Tsunokuma N, Yamane T, Matsumoto C, et al. Depletion of neural crest-derived cells leads to reduction in plasma noradrenaline and alters B Lymphopoiesis. Journal of Immunology. 2016; 198 (1):156-169. DOI: 10.4049/jimmunol.1502592 - 23.
Dudakov JA, Goldberg GL, Reiseger JJ, et al. Sex steroid ablation enhances hematopoietic recovery following cytotoxic antineoplastic therapy in aged mice. Journal of Immunology. 2009; 183 :7084-7094 - 24.
Ueda Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. Journal of Experimental Medicine. The Rockefeller University Press; 6 Jun 2005; 201 (11):1771-1780. DOI: 10.1084/jem.20041419. PMID: 15939792; PMCID: PMC1952536 - 25.
Pabst R. The bone marrow is not only a primary lymphoid organ: The critical role for T lymphocyte migration and housing of long-term memory plasma cells. European Journal of Immunology. 2018; 48 :1096-1100. DOI: 10.1002/eji.201747392 (2018) - 26.
Matthes-Martin S, Feuchtinger T, Shaw PJ, et al. European guidelines for diagnosis and treatment of adenovirus infection in leukemia and stem cell transplantation: Summary of ECIL-4. Transplant Infectious Disease. 2012; 14 :555-563 - 27.
Tikhonova AN, Dolgalev I, Hu H, et al. The bone marrow microenvironment at single-cell resolution. Nature. 2019; 569 (7755):222-228. DOI: 10.1038/s41586-019-1104-8 [Epub 2019, Apr 10]. Erratum in: Nature. 2019 Aug;572(7767): E6 - 28.
Schürch CM, Caraccio C, Nolte MA. Diversity, localization, and (patho)physiology of mature lymphocyte populations in the bone marrow. Blood. 2021; 137 (22):3015-3026. DOI: 10.1182/blood.2020007592 - 29.
Paramithiotis E, Cooper MD. Memory B lymphocytes migrate to bone marrow in humans. Proceedings of the National Academy of Sciences of the United States of America. 1997; 94 :208-212 - 30.
Gatter K, Brown D. Bone Marrow Diagnosis: An Illustrated Guide. 3rd ed. Chichester, UK: John Wiley & Sons Ltd; 2014. ISBN 10: 1118253655; ISBN 13: 9781118253656 - 31.
Fauci AS. Human bone marrow lymphocytes I. Distribution of lymphocyte subpopulations in the bone marrow of normal individuals. The Journal of Clinical Investigation. 1975; 56 :98-110 - 32.
Benet Z, Jing Z, David R. Fooksman DR. Plasma cell dynamics in the bone marrow niche. 2021, Cell Reports 34, 108733. DOI: 10.1016/j.celrep.2021.108733 - 33.
Campello S, Lacalle RA, Bettella M, et al. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. Journal of Experimental Medicine. Vol. 203, No. 13. The Rockefeller University Press; 25 Dec 2006. pp. 2879-2886. DOI: 10.1084/jem.20061877. Epub 2006 Dec - 34.
De Mol J, Kuiper J, Tsiantoulas D, Foks AC. The dynamics of B cell aging in health and disease. Frontiers in Immunology. 2021; 12 :733566. DOI: 10.3389/fimmu.2021.733566 - 35.
Leimkühler NB, Schneider RK. Inflammatory bone marrow microenvironment. Hematology. American Society of Hematology. Education Program. 2019; 2019 (1):294-302. DOI: 10.1182/hematology.2019000045 - 36.
Courties G, Frodermann V, Lisa Honold L, et al. Glucocorticoids regulate bone marrow B Lymphopoiesis after stroke. Circulation Research. 2019; 124 :1372-1385. DOI: 10.1161/CIRCRESAHA.118.314518 - 37.
Dai S, Mo Y, Wang Y, Xiang B, Liao Q , Zhou M, et al. Frontiers in Oncology. 2020; 10 :1492. DOI: 10.3389/fonc.2020.01492 - 38.
Baliu-Pique M, van Hoeven V, Drylewicz J, et al. Cell-density, independent increased lymphocyte production and loss rates post-autologous HSCT. eLife. 2021; 10 :e59775. DOI: 10.7554/eLife.59775 - 39.
Ahn S, Choe K, Lee S, Kim K, Song E, Seo H, et al. Intravital longitudinal wide-area imaging of dynamic bone marrow engraftment and multilineage differentiation through nuclear-cytoplasmic labeling. PLoS One. 2017; 12 (11):e0187660. DOI: 10.1371/journal.pone.0187660 - 40.
Xiang L, Gilkes DM. The contribution of the immune system in bone metastasis pathogenesis. International Journal of Molecular Sciences. 2019; 20 (4):999. DOI: 10.3390/ijms20040999 - 41.
D’Amico L, Roato I. The impact of immune system in regulating bone metastasis formation by Osteotropic tumors. Journal of Immunology Research. 2015; 2015 :143526. DOI: 10.1155/2015/143526 - 42.
Gadiyar V, Patel G, Chen J, Vigil D, Ji N, Campbell V, et al. Targeted degradation of MERTK and other TAM receptor paralogs by heterobifunctional targeted protein degraders. Frontiers in Immunology. 2023; 14 :1135373. DOI: 10.3389/fimmu.2023.1135373 - 43.
Georgoulis V, Papoudou-Bai A, Makis A, Kanavaros P, Hatzimichael E. Unraveling the immune microenvironment in classic Hodgkin lymphoma: Prognostic and therapeutic implications. Biology. 2023; 12 :862. DOI: 10.3390/biology12060862 - 44.
Satolli MA, Buffoni L, Spadi R, Roato I. Gastric cancer: The times, they area-changin. World Journal of Gastrointestinal Oncology. 2015; 7 (11):303-316. ISSN: 1948-5204 (online). DOI: 10.4251/wjgo.v7.i11.303. Available from:http://www.wjgnet.com/1948-5204/full/v7/i11/303.htm - 45.
Chen F, Han Y, Kang Y. Bone marrow niches in the regulation of bone metastasis. British Journal of Cancer. 2021; 124 :1912-1920. DOI: 10.1038/s41416-021-01329-6