 Open access peer-reviewed chapter
Open access peer-reviewed chapter
Abstract
Murine models are widely used in scientific research because they share many genetic similarities with humans, making them a valuable tool for studying various diseases. C57BL/6 is an experimental mouse model to study the demyelination and inflammation aetiology of multiple sclerosis (MS). Intracranial inoculation of neurotropic murine β-coronavirus strain of mouse hepatitis virus in C57BL/6 mice induces demyelination with or without axonal loss, providing many insights regarding the mechanism of MS as well as SARS-CoV-2-mediated pulmonary and neuropathology in humans. By selectively using knockout mice in the wild-type C57BL/6 background, researchers can gain insights into the immunomodulatory nexus and can identify pathways involved in immune regulation which further can be efficiently studied with CD4−/−, CD40−/−, and CD40L−/− mice. In addition, C57BL/6 mice can also be used to generate syngeneic mouse models to investigate the aetiology and mechanism of various cancers, including ovarian cancer. Similarly, along with C57BL/6 mice, different immunocompromised mice models, such as nude mice, SCID mice, and NOD/SCID mice, can be used to study the aetiology, host-tumour interaction, function of the microenvironment, and tumour heterogeneity in tumour metastasis.
Keywords
- experimental mouse model
- murine β-coronavirus
- mouse hepatitis virus
- multiple sclerosis (MS)
- SARS-CoV-2
- knockout mice
- syngeneic mouse model
- immunocompromised mice
- cancer
1. Introduction
Mice are frequently employed as experimental models for various ailments, including cancer [1], diabetes [2], cardiovascular disease [3], and neurological disorders [4]. Because of several reasons like short generation time, low cost and adequate availability, being able to manipulate their genetics, ethical considerations, etc., mice are a preferred model organism in experimental research. Most importantly, genomic investigations have underlined the strong genetic similarities between humans and mice [5]. The ongoing pandemic of COVID-19, caused by the SARS-CoV-2, has affected millions of individuals across the globe. Researchers have been examining various animal models that replicate the human disease to better understand this virus’s pathogenesis and develop effective remedies and vaccines, among which mice models are noteworthy. Different mouse genotypes have been utilized to investigate SARS-CoV-2 infection. These mouse models developed using certain coronaviruses, such as mouse hepatitis virus (MHV), have provided valuable insights into the SARS-CoV-2 infection mechanism and have been used to test potential therapeutics and vaccines. Multiple sclerosis (MS), the most prevalent neurological condition, affects the central nervous system (CNS) and frequently causes severe physical or cognitive incapacity and neurological difficulties in young adults [6]. Although the exact cause of MS is unknown, infiltrating leukocytes and macrophages into the CNS and preexisting environmental and genetic factors may contribute to the disease’s development [7, 8]. Several mouse models have been developed to investigate MS pathologies [9, 10]. Researchers can thoroughly understand the intricate interaction between the immune system and nervous system by using these animal models to comprehend the underlying mechanism of MS pathogenesis. In the 1980s, several research teams created methods to introduce exogenous coding DNA sequences into the mouse genome and impart Mendelian inheritable features [11]. In the second half of the 1980s, a novel technology was developed that made it possible to create mice models with gene-specific null mutations and/or models with gene-specific alterations at the endogenous chromosomal location of a particular gene [11, 12]. Numerous mechanistic studies utilizing these models have shown distinct cellular and molecular pathways and the independent and/or overlapping characteristics of these pathways in mammals. Studying complex interactions within biological systems, also known as systems biology, has become increasingly important in understanding the underlying causes of many human diseases. This is partly due to the emergence of new research fields such as genomics, proteomics, and metabolomics, collectively known as ‘omics’ [13]. One of the key contributors to this advancement has been the development of mice through gene targeting methods, enabling researchers to study the role of specific genes in disease development. Additionally, using gene-knockout mice in conjunction with transgenic mouse lines to produce ‘humanized’ mouse models as part of drug discovery methodologies has proven incredibly beneficial [14]. On the other hand, researchers can study the biology of cancers in complex, dynamic physiological systems by using various murine tumour models [1]. Since the introduction of gene targeting in mice, cancer researchers have been among the most frequent users of transgenic mouse models, significantly advancing the understanding of the origins and progression of cancer. Mice are used in more than 95% of
2. Versatility of mouse models in scientific research: an overview of common strains and their contributions
Different strains of mice are commonly used in scientific research, each with unique characteristics and advantages. C57BL/6 mice, BALB/c mice, SJL/J mice, nude mice, SCID mice, and NSG mice are some of the most commonly used experimental mice strains [16, 17]. C57BL/6 mice is an immunocompetent mouse model that can be utilised to identify the key players in several human diseases, including liver disease, kidney disease, neurodegenerative diseases such as multiple sclerosis (MS), and cancer. It is a versatile model that can be used for physiology, immunology, oncology, and genetics research. In addition, to study the function of specific genes and their role in diseases, a revolutionised method in the field of biology is creating gene-knockout mice [11, 12]. On the other hand, various immunocompromised mice models (such as Nude, SCID, and NSG mice) are used to investigate the fundamental mechanisms behind tumour formation and assess the effectiveness of anti-cancer therapies. Throughout this chapter, we will discuss the various mice models and their significant contributions to the respective field of research.
3. C57BL/6 mice: an experimental mouse model to study neuroinflammatory demyelination and axonal pathology of murine β-coronavirus infection, which mimics specific pathologies of human neurological disease MS
White matter injury, consisting of the loss of axons, myelin sheath, and oligodendrocytes, is prevalent in various neurological conditions. Multiple sclerosis (MS) is a neurodegenerative and chronic inflammatory central nervous system (CNS) disease characterised by inflammation, demyelination, and axonal pathology [18]. Despite substantial research efforts, the aetiology and the exact mechanisms underlying MS pathogenesis still need to be better understood. Experimental animal models of multiple sclerosis could assist in advancing the understanding of the disease progression and identifying potential treatments. C57BL/6 mice are commonly used experimental models for investigating various diseases, including infectious and autoimmune disorders, due to their well-defined genetics, immunology, and ease of breeding and maintenance. The infection of C57BL/6 mice with murine β-coronavirus is one model that shares many resemblances of the pathological characteristics of human MS, including inflammation, demyelination, and CNS axonal damage. Implementation of this model has revealed insightful information into the pathogenesis of MS and assisted researchers in discovering potential therapeutic. Many studies have been performed to know the detailed pathobiology of mouse hepatitis virus (MHV)-induced demyelination in C57BL/6 mice, and numerous possible mechanisms have been hypothesised to explain the demyelination observed. One such hypothesis is that MHV induces an immune response [19] in the CNS where oligodendrocyte and/or myelin sheath are the main targets of immune system-mediated destruction, as seen in MS. New research has revealed that axonal damage is also present and is expected to be a significant factor contributing to the long-term disability observed in MS (Figure 1).
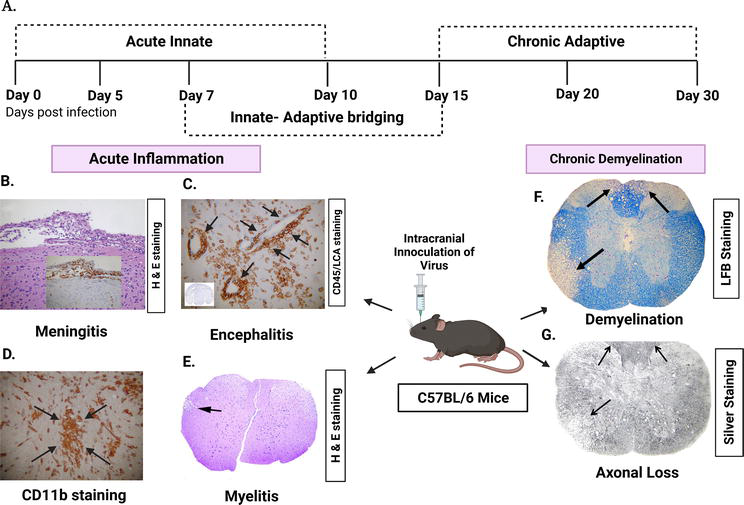
Figure 1.
Disease progression and pathological effects of intracranial inoculation with murine β-coronavirus (MHV-A59/RSA59) in C57BL/6 mice, a model to understand viral-induced demyelination concurrent with axonal loss. Coronal sections of MHV-A59 infected mice, taken on the 5th day after infection, stained using haematoxylin and eosin (H&E) or immunostained with antibodies for CD45 (a marker for inflammatory cells, also known as leukocyte common antigen) (LCA) and CD11b (a marker for microglia and macrophages). (A) Illustration of the timeframe of the infection, namely acute and chronic stages, along with the associated immune system. (B) H&E staining showing the presence of acute meningitis, and the inset displaying the existence of inflammatory cells that are positive for LCA. (C) Coronal sections of the brain stained for LCA indicating acute encephalitis, characterised by the formation of perivascular cuffing (arrows indicate perivascular cuffing). The inset indicates inflammatory infiltrates throughout the brain parenchyma. (D) In the MHV-A59-infected mice brain parenchyma, most inflammatory cells are positive for CD11b (arrows indicate microglial nodules). (E) Inflammatory lesion is observed in virus-infected mouse spinal cord at day 7p.i., stained with H & E (arrow indicates lesion areas in white matter). (F) LFB-stained sections showing loss of myelin in RSA59-infected mouse spinal cord at day 30p.i. (arrow indicates demyelinated area). (G) Bielchowsky silver impregnation showing loss of axons in RSA59-infected mouse spinal cord at day 30p.i. (arrows indicate the area of axonal loss). [adapted from [
Furthermore, different MHV strains may cause demyelination through unique mechanisms. One such study revealed that a neurovirulent JHM strain of MHV-induced demyelination could be eliminated in RAG-deficient mice that are unable to produce functional T and B cells [22]. In contrast, another study indicates that MHV-A59-induced demyelination is feasible in the absence of B and T cells [23]. Based on the aforementioned observation, it is probable that certain strains of MHV, when the immune system is not functioning effectively, can induce demyelination in the CNS by direct injury to oligodendrocytes (OLGs) that could be due to direct glial dystrophy. Thus, C57BL/6 mice infected with MHV can also serve as an experimental animal model to understand non-immune-mediated mechanisms of demyelination that specifically affect oligodendrocytes. MHV can induce inflammation within the CNS by directly infecting neural cells, and the associated responses of glial cells play a crucial role in demyelination and axonal dystrophy. Astrocytes and microglia release cytokines and chemokines [24] that aid the host immune system in combating with the MHV pathogenesis and eradicating the virus from CNS. During the acute phase of infection, specific cytokines and chemokines, such as IL-6, IL-2, IL-1α, and IL-1β, tumour necrosis factor-α (TNF-α), interferon α, β, and γ, and CXCL10 are produced [25], while demyelination is observed during the chronic phase of infection. Activation of glial cells in the CNS typically initiates an innate immune response, releasing proinflammatory cytokines and chemokines known as cytokine storm. This response is intended to combat intruding pathogens but can also damage CNS tissues. The influx of anti-inflammatory cytokines is vital for reducing inflammation, and the peripheral immune system plays a crucial role in this process. CD40-expressed microglia/macrophage (innate immune cell) interacts with CD40L-expressed CD4+T cell (adaptive immune cell) through CD40-CD40L. This intricate interaction helps eliminate viruses and restore homeostasis by secreting anti-inflammatory milieu. A recent study has shown that RSA59 (an isogenic recombinant strain of MHVA59)-infected CD40L knockout mice developed severe demyelination concomitant with axonal loss, indicating that CD40-CD40L interaction is a crucial host defence mechanism against virus-induced demyelination [26]. Likewise, CD4 knockout mice infected with RSA59 exhibited increased susceptibility to axonal degeneration in the CNS, as well as poliomyelitis and bulbar vacuolation observed, indicating that the CD4 gene is protective against immunopathological disorders [27]. However, interferon-induced tetratricopeptide repeats 2 (Ifit2), a protein strongly induced by interferons, plays a significant role in providing antiviral immunity and the function of Ifit2 has been studied thoroughly in mounting host defence by creating Ifit2 knockout mice [28]. Thus, researchers can gain knowledge about the specific function of a knocked-out gene in different physiological conditions by creating gene-specific knockout mice. Later in this chapter, we will discuss the knockout mice in greater depth. Now, it is evident that the MHV infection model in C57BL/6 mice provides a valuable tool for studying the fundamental process of demyelination and axonal loss regardless of the particular mechanism involved in the progression of the disease. A comparative study was conducted between natural and recombinant strains of MHV that cause demyelinating (DM) and non-demyelinating (NDM) diseases to comprehend the mechanisms of demyelination and axonal degeneration. The object was to identify the differences between the two strains [29, 30] and better understand how they contribute to the diseases where a specific strain of MHV-induced C57BL/6 mice was used as an experimental model. This model assisted researchers to pinpoint therapeutic targets. For example, research has shown that blocking the activity of proinflammatory cytokines such as IL-1β, IL-6, and TNF-α can reduce inflammation and demyelination in the CNS [31]. Moreover, treatments that promote remyelination, such as oligodendrocyte progenitor cells or drugs [32] that facilitate myelin repair, demonstrated encouraging results. In conclusion, the murine β-coronavirus-induced demyelinating disease model is a beneficial tool for investigating the pathogenesis of MS and identifying potential therapies. Nevertheless, caution should be exercised when extrapolating results from animal models to human disease, and additional research is required to explain the complexities of MS pathogenesis fully.
4. C57BL/6 mice: an experimental mouse model to study the disease progression and pathogenesis of EAE
Experimental autoimmune encephalomyelitis (EAE) is a common animal model for understanding the autoimmune pathobiology of MS [10]. Among the animal models, C57BL/6 mice are a popular experimental mouse model for investigating autoimmune diseases, such as experimental autoimmune encephalomyelitis (EAE), because they exhibit a uniform and stable disease course and clinical and pathological characteristics similar to those of human MS. Furthermore, these rodents have been exhaustively characterised, and their genetic and immunological backgrounds are well understood, making them an advantageous model for examining the pathogenesis of MS. This model has been extensively used to comprehend the disease progression and pathogenesis of MS, and it has yielded crucial insights into the immunological mechanisms underlying this disease. EAE could be induced by three different methods such as MBP-PLP fusion protein MP4, MOG peptide 35–55, and PLP peptide 178–191 in C57BL/6 mice, which display distinct pathologies in CNS. These three models differ in three aspects: tract specificity, motor neuron involvement, and demyelination kinetics. MP4, MOG 35–55, and PLP peptide 178–191 induced three different models of EAE showing differential spinal cord tract pathology [33]. Mainly, three tracts of the spinal cord (pyramidal tract responsible for motor function, dorsal tract for fine touch, and anterolateral tract responsible for pain and crude touch) get affected differentially after immunisation with three different methods. In the acute and chronic phase of EAE, all mice displayed damage of anterolateral tracts in all three models. On the other hand, MP4 and MOG 35–55 models showed a resemblance to each other in the dorsal tract in the acute phase, while the PLP peptide model did not. In MP4- and MOG-induced models, motor neuron alterations were prominent, but in the PLP model, no such alteration in motor neurons was observed. Differential demyelination patterns have been noticed in three distinct EAE models. Demyelination was evident in the acute stage of the MP4 model, while it was transient in the MOG 35–55-induced EAE model and absent in the PLP peptide model. So, the use of MP4, MOG peptide 35–55 and PLP peptide 178–191 induced EAE models in C57BL/6 background could provide a reasonable strategy for reproducing distinct features of CNS pathology to comprehend the complex mechanism of MS.
Four stages of disease progression are depicted in Figure 2 following subcutaneous infection with MOG 35–55 peptide, complete Freund’s adjuvant (CFA), and pertussis toxin in C57BL/6 mice [35].
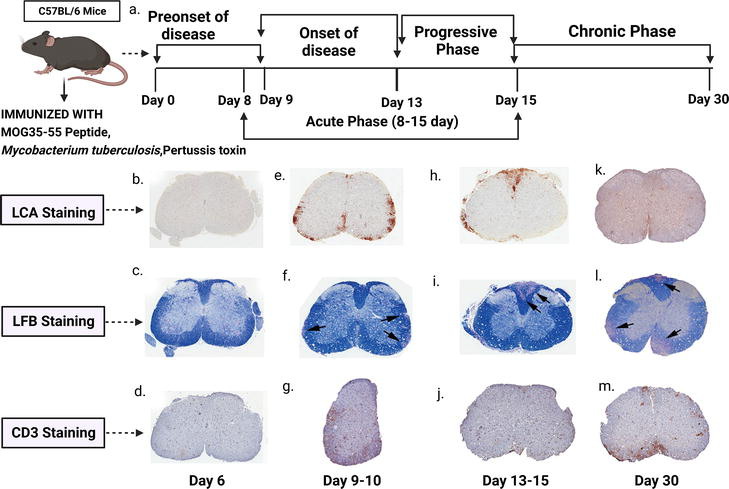
Figure 2.
Disease progression and pathological effects of C57BL/6 mice after immunisation with MOG 35–55. Representative sections (5 μm thick) of spinal cords from mice with EAE are taken at different time points after immunisation and stained with Luxol fast blue (LFB) for myelin, as well as histopathology for CD45 (LCA, to identify inflammatory cells) and CD3 (to identify T cells). (a) Demonstrating the timeline of disease progression of C57BL/6 mice after being immunised with MOG 35–55. A hemicord is depicted in (g). [(b)–(d)] Before EAE onset (day 6 after immunisation). [(e)–(g)] EAE onset (day 9–10 after immunisation). [(h)–(j)] Progressive phase (day 13–15 after immunisation), [(k)–(m)] chronic phase (day 30 after immunisation) [(b), (e), (h), (k)] LCA stain. [(c), (f), (i), (l) Luxol fast blue. [(d), (g), (j), (m)] CD3. Spinal cords of days 9–10 of EAE mice show an increase in LCA and CD3 positive cells in the white matter compared to spinal cords of day 6. The Luxol fast blue stain signifies the presence of early demyelinating plaques in the white matter. During the progressive phase, the size of demyelinating plaques increased (arrows denote locations of demyelinating plaques), and ongoing LCA immunoreactivity was observed, while CD3 immunoreactivity decreased. In the chronic phase, LCA immunoreactivity decreased, while CD3 immunoreactivity increased. The number and size of demyelinating plaques continued to increase during this phase. [adapted from [
Mice immunised with MOG display waddling gait, tail limpness, hind limb paralysis, and ascending paralysis [36]. The pathogenesis of EAE in C57BL/6 mice involves activating autoreactive CD4+ T cells, which recognise myelin antigens and become activated in the periphery [37]. The upregulation of integrins, such as the lymphocyte function-associated antigen (LFA-1) and the very late antigen-4 (VLA-4), allows these activated T cells to cross the blood-brain barrier (BBB). Autoreactive T cells reencountering antigens in the CNS become reactivated and produce inflammatory cytokines. These cytokines stimulate nearby immune and neural cells, attracting more inflammatory cells to the central nervous system (CNS). This process was seen in macrophages, believed to cause damage to the CNS either directly or indirectly [38]. Activated CD4+ T cells may differentiate into T helper 1 (Th1) and T helper 17 (Th17) cells [39], which are the primary effector cells responsible for developing EAE, with IFN-γ and IL-17A being the key cytokines produced by these cells, respectively. IFN-γ, IL-2, and TNF are inflammatory cytokines produced by CD4+ Th1 cells. In multiple sclerosis (MS) patients, the severity of the disease was linked to the levels of IFN-γ and IL-12 expression in both the cerebrospinal fluid (CSF) and CNS [40]. Th17 cells release proinflammatory cytokines, particularly IL-17 and IL-22 [41].
Additionally, these cells stimulate other types of cells to produce proinflammatory factors, such as granulocyte/macrophage colony-stimulating factor (GM-CSF), cytokines such as IL-6, and several chemokines like CXCL8, indicating that Th17 cells play an essential role in promoting inflammation in the CNS [42]. This model’s consistency and reliability, which permits the standardisation of experimental conditions and the comparison of results within studies, is one of its considerable advantages. In addition, the recurring and predictable disease course of EAE in C57BL/6 mice and the similarities to human MS in terms of clinical and pathological features make this model a useful tool for elucidating the underlying mechanisms of MS in order to develop novel therapies. This model has been used to test therapeutic targets for over 3–4 decades. Further studies utilising this model are required to clarify the complex immunological and pathological processes involved in MS and identify new therapeutic intervention targets. EAE can also be induced in SJL mice by immunisation with proteolipid protein (PLP), myelin basic protein (MBP), or peptides corresponding to the immunodominant epitopes of MBP, although here in this book chapter, we have only discussed about C57BL/6 mice as an experimental mouse model to understand the pathogenesis of EAE. While C57BL/6 mice are widely used to understand the inflammatory demyelinating disease as discussed in length, it can also serve as a neuroinflammatory model to understand the pathobiology of other coronaviruses like SARS-CoV-2, which is now a significant threat to human health. So, in the next section of this chapter, we will focus on how MHV-infected C57BL/6 mouse model could assist researchers in better understanding the pathobiology of SARS-CoV-2.
5. MHV-infected C57BL/6 mice as a model to understand SARS-CoV-2 pathobiology
While C57BL/6 mice proved to be a very efficient model for studying neurodegenerative disorders like MS, researchers have even extensively used mouse models to gain insights into various infectious diseases. The ongoing COVID-19 pandemic caused by SARS-CoV-2, a β-coronavirus, is keeping the human race in trauma. Significant drawbacks in SARS-CoV-2 research are the unavailability of BSL-3 facilities and limited patient data, which hinders research on SARS-CoV-2. In the peripheral systems, SARS-CoV-2 can primarily cause upper respiratory tract infection, followed by inflammation in the lungs leading to severe pneumonia and enteritis. Moreover, if it enters the CNS, it can cause meningitis and encephalitis. In some patients, it is reported to cause myelin loss too. It can also affect the peripheral nervous system (PNS), causing Guillain-Barré syndrome. There are reports of SARS-CoV-2 manifestation in the CNS as viral RNA could be found in the cerebrospinal fluid [43]. As human data are limited, in this context, mouse hepatitis virus (MHV) can be considered an excellent model to draw the parallel to understand the pathogenesis, as it is a β-coronavirus belonging to the same genus as SARS-CoV-2. Infection of mice with MHV shows varied severity depending on the strain of the virus used and the age of the mice. On the other hand, intracranial inoculation of MHV in C57BL/6 mice exhibits similar pathologies to SARS-CoV-2 with respect to CNS manifestations, such as meningitis, encephalitis, and myelin loss [20, 44]. Hence, MHV-infected C57BL/6 mice can be used as an experimental model to understand the pathobiology of moderate and severe COVID-19 cases, as both viruses exhibit similar pathogenesis, organ tropism, and immune response (Figure 3).
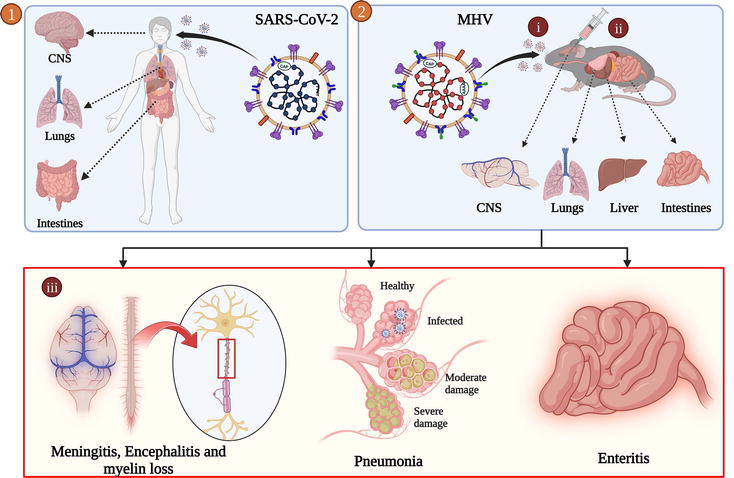
Figure 3.
Upon intranasal/intracranial inoculation (2-i and 2-ii), MHV infects the brain, lungs, and intestine, respectively, and similar organ tropism has also been found in SARS-CoV-2 infection [
A strain of MHV, MHV-A59-infected C57BL/6 mouse model can be considered an experimental model for mild-moderate cases of SARS-CoV-2. MHV-A59 infection starts with activating the brain resident astrocytes and microglia that help in the up-regulation of various cytokines and chemokines such as IL-6, IL-1β, IL-12, IL-15, TNF-α, and IFN-γ resulting in the infiltration of different peripheral innate immune cells such as neutrophils and monocytes/macrophages [45] as discussed earlier. Microglia/macrophages play an essential role in limiting viral replication and spread by recruiting several immune players of adaptive immunity, including CD4+ T cells, CD8+ T cells, and NK T cells into the CNS. Activation of the adaptive immune cells causes the secretion of even more cytokines. The release of these cytokines results in a cytokine storm, a phenomenon similar to SARS-CoV-2. Generally, infectious viral particles are cleared within 10 days p.i.; however, viral mRNA persists beyond day 10p.i. [20]. This persistent infection in mice gives rise to demyelination, which can be histopathologically confirmed by LFB staining [46]. This condition is similar in SARS-CoV-2 infection, where myelin loss is caused due to the activation of glial cells brought on by the proinflammatory state generated by the cytokine storm, primarily sustained by IL1, IL-6, and TNF-α [47]. To summarise, MHV-A59 causes a persistent infection with moderate neuroinflammation in C57BL/6 mice.
On the other hand, MHV-JHM.SD-infected C57BL/6 mice can be considered a model for severe COVID-19 cases due to vigorous viral replication, suboptimal CD4/CD8 T cell response, and the occurrence of cytokine storm [48]. These events result in severe pathologies of the disease and even death. MHV-JHM.SD infection in mice resulted in a longer innate immune response with upregulation of proinflammatory cytokines and chemokines with enhanced chemotaxis of innate immune cells such as macrophages, neutrophils, and NK cells into the CNS, leading to the destruction of the brain parenchyma. JHM.SD exhibits low T cell responses and IFN-γ levels [49]. Due to the high mortality rate of JHM.SD, an attenuated strain of MHV (2.2-V-1) is used to study the T cell response. So, to conclude, JHM.SD replicates severe pathogenesis as displayed by COVID-19 compared to MHV-A59, which mimics the mild-moderate pathogenesis of the SARS-CoV-2.
6. Gene-knockout mice and their role in studying neurodegenerative disease biology
Gene-knockout mice are genetically modified mice with a specific gene or genes deliberately removed or ‘knocked out’ to study the function of that gene. This is usually done by gene targeting, which involves introducing a modified version of the gene into embryonic stem cells, which are then used to create mice missing the targeted gene.
The use of gene-knockout mice has revolutionised the study of genetics and has provided valuable insights into the function of genes and their role in various diseases. They have been used to study multiple diseases, including cancer [50], Alzheimer’s disease [51], Parkinson’s disease [52], diabetes [53], and many others. For example, researchers have used knockout mice to study the role of the BRCA1 gene in breast cancer development [54, 55], the role of the amyloid precursor protein gene in Alzheimer’s disease, and the role of the insulin receptor gene in diabetes. Though these are the common knockout mice used by researchers, in this chapter, we will discuss the knockout of immunomodulatory molecules to understand their individual roles in the viral-induced demyelination model.
Intracranial inoculation of RSA59 (an isogenic recombinant strain of MHV-A59) results in sequential recruitment and activation of several immune players in the CNS. The outcome of this infection is determined by how RSA59 interacts with the innate immune system. When the virus replication is initially controlled, it triggers a robust immune response that includes proinflammatory and type I interferon responses and promotes an anti-inflammatory response. Maintaining a delicate balance between these opposing but mutually supportive immunity states is crucial for restoring tissue homeostasis. If the balance tips towards either extreme, it can be harmful and lead to immune-related health issues and tissue damage. The shift from innate to the adaptive immune response is controlled by a very important immune checkpoint regulator: CD40-CD40L dyad. Knocking out several immune system checkpoint genes such as CD4 [27], CD40R, CD40L [26], gave us an excellent idea about the consequences of disease biology (like neurodemyelinating diseases) in their absence.
Overall, knockout mice are a powerful tool for understanding the genetic basis of disease and developing new treatments and therapies. However, it is important to note that knockout mice do not perfectly replicate diseases in humans. Additional research is required to properly comprehend the implications of genetic changes in animal models for human health.
By studying the phenotype (observable traits) of mice that have had a specific gene knocked out, researchers can learn about the function of that gene and the consequences of its absence. This can help identify potential targets for drug development and develop new genetic disease treatments.
7. How are knockout mice made?
Gene knockout mice are created through gene targeting or gene disruption. This process involves embryonic stem cells (ES cells), pluripotent cells capable of developing into any cell in the body.
Researchers design a DNA construct containing a modified version of the gene of interest to create a gene-knockout mouse. This construct typically includes a selectable marker gene and a homology region for the target gene. The DNA construct is then introduced into embryonic stem cells, replacing the endogenous gene through homologous recombination.
The modified ES cells are then selected using the selectable marker gene, and those that have undergone successful homologous recombination are injected into mouse blastocysts. After that, these blastocysts are implanted into the uterus of a surrogate mother mouse.
If successful, the injected ES cells will give rise to chimeric mice with both regular and modified cells. The chimeric mice are then bred with wild-type mice to produce offspring carrying the altered gene in all their cells. These offspring are then screened to confirm that the targeted gene has been successfully disrupted [11, 56].
The resulting mice with the targeted gene disruption can then be studied to understand the gene’s function and role in disease or normal physiology. The detailed mechanism of creating a gene-knockout mouse using Neomycin cassette mutagenesis is described in Figure 4.
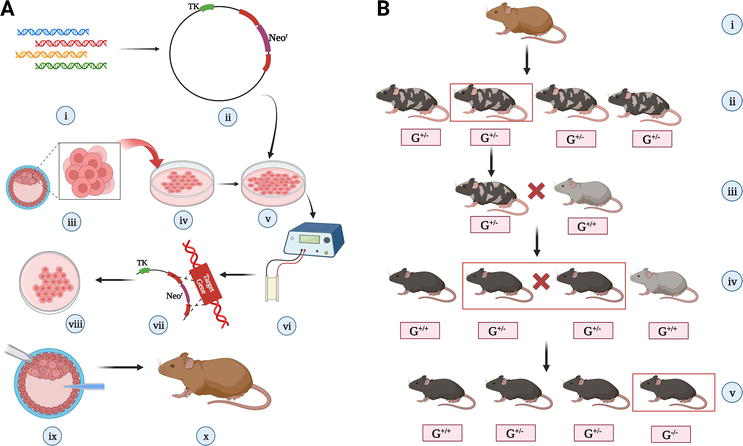
Figure 4.
Procedure of making gene-knockout mice using neomycin resistance cassette: (A) implantation of a particular gene-knockout blastocyst in a foster mother [(i) the gene which is to be knocked out is isolated from gene library; (ii) a cloning vector is designed in such a manner that it has a Neor gene and in its either side, there are terminal sequences of the target gene, and there even should be a thymidine kinase gene; (iii), (iv) embryonic stem cells are isolated from a donor mouse blastocyst and cultured
Another method is the creation of conditional mutant mice. This can be achieved by The Cre-loxP system [58], a genetic engineering technique commonly used in molecular biology research to manipulate and modify genes in living organisms, including bacteria, plants, and animals. It uses two components: Cre recombinase and loxP DNA sequences. By using Cre recombinase to selectively delete or modify specific genes in a tissue-specific or temporal-specific manner, researchers can investigate the role of these genes in different physiological processes. In a tissue-specific way, the absence of that particular gene can be studied conditionally.
8. Understanding the immunomodulatory nexus between CD4+ T cells and microglia/macrophages with the help of gene-knockout mice
During the neuroinflammatory process of virus-induced demyelination, communication between innate immune cells, such as microglia/macrophages (expressing CD40R), and adaptive immune cells, like activated CD4+ T cells (expressing CD40L), plays a significant role that serves crucial functions in fighting the body against infections and regulating tissue homeostasis. On one hand, CD4+ T cells can activate microglia and macrophages to enhance their phagocytic and antigen-presenting capabilities, allowing them to better clear pathogens and damaged cells. On the other hand, microglia and macrophages can also modulate the activity of CD4+ T cells by presenting antigens and secreting cytokines that either promote or suppress CD4+ T cell activation and differentiation. Depending on the cytokine environment and the type of virus, CD4+ T cells can differentiate into different subsets, including Th1, Th2, Th17, and Tfh cells.
Microglia are the cellular immunological occupants of the CNS, defending against invasive infections and damaged cells as the first line of defence. Conversely, macrophages are immune cells that can be recruited to the CNS from the blood in response to infection or injury.
Overall, the interplay between CD4+ T cells and microglia/macrophages is critical for maintaining immune homeostasis in the CNS and protecting against infections and injuries. Dysregulation of this interplay can lead to neuroinflammatory and neurodegenerative diseases in mice, highlighting the fact that the absence of any genes individually (like CD4, CD40, or CD40R) responsible for the proper functioning of this complex interaction is critical.
When the CD4 gene in C57BL/6 mice was functionally knocked down by deleting exon 5 (among the 11 exons) and then intracranially inoculated with the neurotropic virus RSA59 to study the demyelination and inflammatory aetiology of MS, the results were quite astonishing. In the absence of CD4, there was a significant decrease in the number of CD11b+ microglia/macrophages in the acute phase. At the same time, viral replication was observed with the presence of viral RNA transcripts even 30 days after infection. These infected CD4 knockout mice displayed symptoms such as poliomyelitis, bulbar vacuolation, and axonal degeneration and were more susceptible to chronic phase encephalitis and demyelination. However, during the chronic phase, CD11b+ phagocytic macrophages were present throughout the inflamed regions (including white and grey matter) of both the brain and spinal cord in the CD4 knockout mice suggesting a protective nature of the CD4 gene, that is, they help in reducing immunopathological disease and related morbidity and even aid in the clearance of the pathogen (Figure 5) [27].
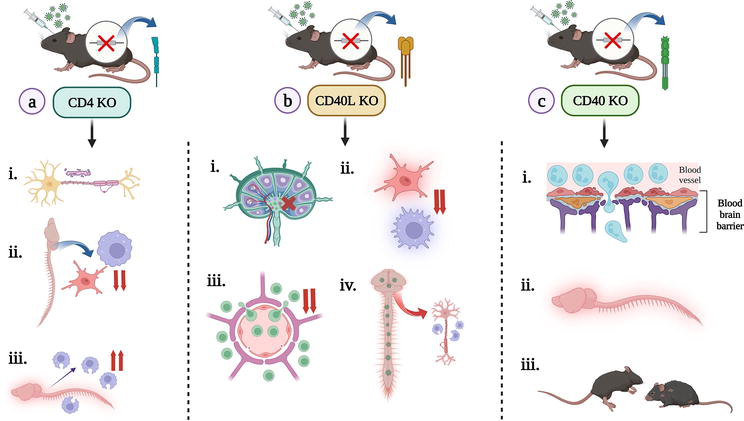
Figure 5.
Consequences of knocking out a particular gene and exploring its necessity in its absence, post-infection: (a) intracranial inoculation of RSA59 in a CD4 knockout (KO) mice [resulted in (i) axonal degeneration, bulbar vacuolation, poliomyelitis; (ii) significant decrease in CD11b+ microglia/macrophages during the acute phase; (iii) presence of phagocytic macrophages in the inflamed regions during chronic phase], (b) intracranial inoculation of RSA59 in a CD40L knockout (KO) mice [(i) impaired CD4+ T cell activation and priming in CLN; (ii) during the acute phase of the disease, microglia/macrophage activation got dampened; (iii) decreased infiltration of CD4+ T cells; (iv) myelin stripping by phagocytic macrophages during chronic phase], (c) intracranial inoculation of RSA59 in a CD40 knockout (KO) mice [(i) increased neutrophil infiltration in the acute phase; (ii) increased inflammation of CNS; (iii) increased clinical scores and morbidity] [created with
Similarly, functionally knocking out the CD40L gene (expressed primarily by activated T cells) of C57BL/6 mice by replacing exons 3 and 4 of the gene on the X chromosome with a PGK-Neo cassette, and intracranially inoculating with neurotropic virus RSA59 showed increased disease severity by dampening microglia/macrophage activation during the acute phase; reduced infiltration of T cells in the CNS and even impairing priming of T cells in the cervical lymph node. In the chronic phase, due to extensive replication of the virus in the CNS, severe demyelination and axonopathy were observed due to the presence of phagocytic microglia/macrophages (whose population got skewed due to the absence of the CD40L gene) (Figure 5) [26].
The CD40R is expressed by various haematopoietic cell types such as B cells, dendritic cells (DCs), monocytes, platelets, and macrophages and by non-haematopoietic cells such as myofibroblasts, fibroblasts, epithelial, and endothelial cells [59]. Because of its widespread prevalence, its absence will have detrimental effects on an individual. When this gene in C57BL/6 mice was functionally knocked out by replacing exon 3 (among the 9 exons) with neo-cassette and then infected with RSA59 intracranially, increased acute phase encephalitis was seen in the infected subjects as compared to the mock-infected ones, and the mice even showed high mortality rate. This is because, during the acute phase following the RSA59 infection, there was an increase in infiltrating neutrophils that led to heightened bystander neuroinflammation (unpublished data, JDS lab, IISER Kolkata, West Bengal, India) (Figure 5).
These studies suggest that the immunomodulatory nexus between CD4+ T cells and microglia/macrophages is highly interconnected and regulated by a complex network of genes and signalling pathways. And, if researchers are interested in understanding the role of a particular gene in the immune response to a specific pathogen (like viruses), they could create a knockout mouse in which that gene is disabled. They could then expose both the knockout mouse and a normal mouse to the pathogen and observe how their immune systems respond differently. Further research using gene-knockout mice will be necessary for elucidating the specific mechanisms governing this interaction and developing new therapies to modulate immune responses in various disease states. Mice have even played a critical role in advancing our understanding of cancer and developing new cancer treatments, and they will likely continue to be an essential tool in cancer research for many years to come. Hence, in the next section, we will discuss how different mice models can be efficiently used to study cancer in detail as they share many biological similarities with humans, making them excellent models for studying cancer development, progression, and treatment.
9. Mice as an experimental model in cancer research
Mice are frequently utilised as experimental models in cancer research due to their genetic and physiological similarities with humans. Various rodent models that mimic human cancers have been established to gain insight into the key mechanisms behind tumour formation and assess the effectiveness of anti-cancer therapies. The essential phases of tumour development and progression start with normal cells, and their development into aggressive tumours is recapitulated in these models. These models have helped identify the elements contributing to tumour metastasis, recurrence, and therapeutic resistance [60].
Tumour development is monitored in genetically engineered mice (GEM) models in which mutations, deletions, or overexpression of genes with links to human cancers have been introduced. This model allows researchers to examine tumour development and chemotherapy responses in a functional immune system context. However, human cancer xenografts in immunocompromised mice are another extensively used model because mouse tumour cells might not entirely replicate the tumorigenic mechanism in humans. These models use human tumour samples, cell lines, or cancer stem cells to develop tumours in various tissues. However, the roles of the tumour microenvironment, stroma, and conventional immune surveillance in tumour xenograft studies have not been investigated. This has led to several novel strains of humanised mouse models. In general, immunocompetent and immunocompromised mice models are the two main types of murine models used in cancer research [61].
9.1 Immunocompetent mice models
Immunocompetent mice have an immune system capable of mounting a normal immunological response to infections and foreign substances. This implies they have a fully working immune system, which allows researchers to explore the relationship between the immune system and various diseases. The use of immunocompetent mice is especially significant in cancer research because it provides a more realistic investigation of the complicated interactions between the immune system and tumours. Multiple immunocompetent murine models can be generated, which are discussed below.
9.1.1 Spontaneous tumour models
Spontaneous tumour models involve the genetic modification of mice to carry on mutations that imitate natural disease conditions in humans. The most therapeutically relevant tumour microenvironment for studying immunotherapeutic mechanisms can be found in these tumour models. Given the wide range of genes and cell targets that can be exploited, it has been discussed how generating mice with a targeted genetic mutation that causes spontaneous pancreatic cancer can be a helpful research tool. It was further reported that mice harbouring the mutant oncogene (G12D) spontaneously generate non-metastatic pancreatic cancers. Nevertheless, metastatic pancreatic tumours are produced when the p53 tumour suppressor gene is simultaneously mutated [62]. Another study summarised how this mouse model could examine the impact of oestrogens on mammary development and carcinogenesis. Further, they discussed the role of oestrogen receptors in controlling the proliferation, apoptosis, and differentiation of mammary epithelial cells. They have also looked into the bidirectional coordination between the epithelium and stroma in maintaining cancer cell stemness [63].
9.1.2 Genetically modified mouse models
The capacity to engineer mice to test the tumour progression is astounding, given the availability of the whole mouse genome sequence, genome manipulation technology, well-defined inbred strains, and thorough knowledge of the polymorphisms within strains. The function of a gene can now be altered, lost, mutated, under-expressed, or overexpressed in suitable cell types
To offer a systemic or tissue-specific expression of oncogenes, such as MYC and KRAS in breast cancer [64] or deletion of tumour suppressor genes, such as TP53 and PTEN in prostate cancer [65], GEM models are typically developed using transgenic technologies. The two major types of GEM models are germline and non-germline GEM models [66]. Germline GEM models contain mutations that result in the spontaneous development of malignant tumours. For instance, it has been found that a variety of solid and haematological tumours manifest in mice with a TP53 gene mutation [67]. Germline GEM models have enabled the investigation of mechanisms underlying tumour formation and progression. Still, it does not permit control over the time and location of tumour development [68]. On the other hand, non-germline GEM models offer spatiotemporal control of the development of tumours. Several mechanisms, such as the tamoxifen-inducible Cre-loxP system, can induce somatic mutations at a specific time and in a particular tissue. Hence any gene flanked by loxP sites can be deleted once the Cre-recombinase is activated by tamoxifen [69]. Furthermore, CRISPR/Cas9 technology has lately been extensively exploited for editing oncogenes, resulting in the development of models of lung cancer, hepatocellular carcinoma, and breast cancer [70, 71, 72].
9.1.3 Syngeneic mouse models
Traditionally, human tumour xenograft models were established by injecting cancer cell lines or transferring tissues from patients into immunodeficient mice. However, this model needed to be modified to comprehend the function of tumours and host immune factors in malignant transformation and metastasis. Thus, a syngeneic transplanted model has been established by introducing homologous cell lines into immunocompetent mice, which generates tumours rapidly while avoiding host rejection in an immunocompetent milieu [73]. C57BL/6 and BALB/c mice are suitable hosts for transplanting spontaneous, carcinogenic, or transgenic tumour cell lines [74, 75]. Cells implanted subcutaneously or intravenously multiply in the mice within a few weeks. Hence, the development of these kinds of models is rapid [76].
Researchers have used C57BL/6 mice to generate immunocompetent syngeneic allograft mouse models of paediatric diffuse midline glioma (DMG), a highly malignant and incurable brain tumour [77]. They created three genetically unique transplant models of the histone 3 wild-type (H3WT) and the K27M-mutant DMG (H3.3K27M and H3.1K27M). The histopathologic phenotype of these transplanted models matched that of their human counterparts. In the year 2000, potential syngeneic mouse models were developed for events related to ovarian cancer [78]. In a recent study, mouse epithelial ovarian cancer cell lines, ID8 and ID8-VEGF (overexpressing VEGF), were intraperitoneally injected in C57BL/6 female mice to generate a syngeneic ovarian cancer mouse model. It was observed that ID8-VEGF cells had a greater capacity to induce aggressive tumour growth as compared to ID8 cells, having basal levels of VEGF expression. On the other hand,
The effect of immunomodulating treatment is generally developed gradually and is measured by an increased survival rate [80]. Thus, the short time period provided by the rapid kinetics of tumour growth in syngeneic models is often insufficient to determine its efficacy. Moreover, the efficacy of immunotherapeutic medicines at earlier stages of tumour growth cannot be evaluated in syngeneic models either [81]. Now, we will discuss the various immunodeficient mice models used in cancer research.
9.2 Immunocompromised mice models
To determine whether a patient’s tumour will respond to a specific therapeutic agent, the anti-cancer response to a human tumour, rather than a mouse tumour, must be examined. This is where a human tumour xenograft on nude mice, SCID mice, or NOD/SCID humanised mice can be helpful. Particularly when
Over time, various immunocompromised mice models have been developed (Figure 6). These include nude mice that lack T lymphocytes due to abnormal thymus formation. However, nude mice remain restricted in their application to many diseases due to their relatively low levels of immunodeficiency. Mice with a mutated Prkdc gene constitute the severe combined immunodeficiency (SCID) models.
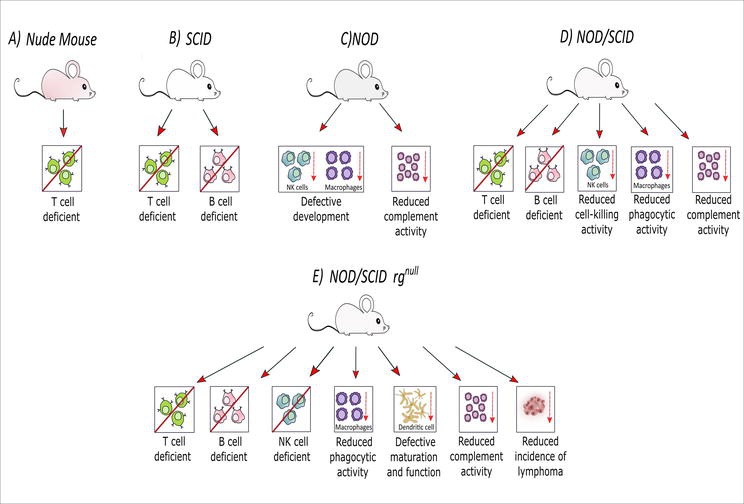
Figure 6.
Different types of immunocompromised mice models: (A) T cell-deficient nude mice; (B) T cell and B cell-deficient SCID mice; (C) NOD mice with defective development of NK cells and macrophages along with reduced complement activity; (D) NOD/SCID mice lacking T and B cells along with defective development of NK cells and macrophages and reduced complement activity; (E) T cell, B cell, and NK cell-deficient NOD/SCID rgnull mice with reduced phagocytic activities of macrophages, defective maturation and function of dendritic cells, reduced complement activity, and incidence of lymphoma [created with
Natural killer (NK) cells are present in SCID mice despite the absence of T and B lymphocytes. Further, introducing the SCID mutation into NK cell-deficient non-obese diabetic (NOD) mice, NOD/SCID mice developed. However, these animals have a short lifespan, impaired NK cell function, and a high rate of spontaneous thymic lymphoma. As a result, they are not used frequently as humanised animal models. To improve this situation, the IL-2 receptor gamma chain was knocked out in the NOD/SCID mice to generate the NOD/SCID rgnull mice [16]. These mice had a higher implantation rate of human cells without spontaneous thymomas and are presently the global standard immunocompromised mouse model.
9.2.1 Nude mice models
The earliest immunodeficient mouse model is nude mice, which Flanagan first described in 1966 [83]. Due to a Foxn1 gene malfunction, a mutation on chromosome 11 hinders the proper development of the thymus, which results in the reduced formation of mature T lymphocytes. IgM is the most abundant immunoglobulin in these mice, with very little or no IgA [84]. As a result, they do not show signs of rejecting allogeneic tissue. The most often used strains, Swiss-nu, BALB/c-nu, NIH-nu, and NC-nu, are extensively employed in researching immunological disorders and malignancies. They cannot, however, fully accept the engraftment of human immune cells since they still have B cells and NK cells, making them unsuitable for use as an ideal humanised mouse model [16].
9.2.2 SCID and NOD/SCID mice models
Inbred CB-17 mice with recessive mutations in a single gene on chromosome 16 were discovered in 1983. This mutation altered the recombination enzyme activity of the sequence encoding the mouse lymphocyte antigen receptor gene, making it challenging to produce IgG, T, and B lymphocyte receptors [85]. Because of this mutation, T and B cell receptors have more difficulty repairing and recombining. The capacity of these cells to differentiate and mature is similarly hindered. There are thus fewer mature T and B lymphocytes and fewer immunoglobulins in the blood and lymphoid tissues of SCID mice. However, NK cells and macrophages functioned normally in SCID animals [86]. Additionally, ‘leakage’ was seen, meaning that T and B cells returned in some mice as they aged [87].
Non-obese diabetic (NOD) mice were developed for the first time by scientists in 1980 through inbreeding and selective breeding [88]. Similar pathology alterations and symptoms to those seen in humans with diabetes were seen in these animals. Low NK cell and macrophage activity and no complement activity in the blood are symptoms of a weakened innate immune system in NOD mice. It was hypothesised that crossing NOD mice with animals carrying the SCID mutation would result in a strain of mice with compromised adaptive and innate immune systems known as NOD/SCID mice [89]. Therefore, NOD/SCID mice developed a deficiency in T and B lymphocytes and other immune cells, including NK cells. As a result, their already-weak immune systems became even more compromised. Recently, NOD/SCID mice have not been frequently utilised as humanised mice models due to the larger amount of haematopoietic stem cells required for engraftment. Instead, the more advanced and effective NOD/SCID rgnull mice are being used now.
9.2.3 NOD/SCID rgnull mice
The interleukin-2 (IL-2) receptor gamma chain is one of the significant components of the receptors for cytokines such as IL-2, IL-4, IL-7. These cytokines are essential for the development and maturation of T, B, and NK cells. Thus, NOD/SCID rgnull mice lacking the gamma chain of IL-2 receptors are significantly compromised in their innate and adaptive immune responses [90]. These mice have reduced phagocytic activities of macrophages along with defective maturation and function of dendritic cells. NOD/SCID rgnull mice are classified as either NOG or NSG mice based on the type of mutation in the IL-2 gamma chain. The IL-2 gamma chain targeted mutation is completely null in NSG mice leading to no expression of the IL-2 gamma chain. The IL-2 gamma chain mutation in NOG mice results in an expressed protein that binds cytokines but cannot transduce the signal. Transplantation success was more significant in NOG and NSG mice than in SCID or NOD/SCID mice, making these mouse strains the best models for human cell and tissue transplantation [91].
9.3 Generation of xenograft and allograft transplantation models
Transplanted tumours are most commonly used to test the therapeutic efficacy of a wide range of anti-cancer agents. Different laboratories currently use two major tumour transplantation models: allograft transplants and xenograft transplants (Figure 7).
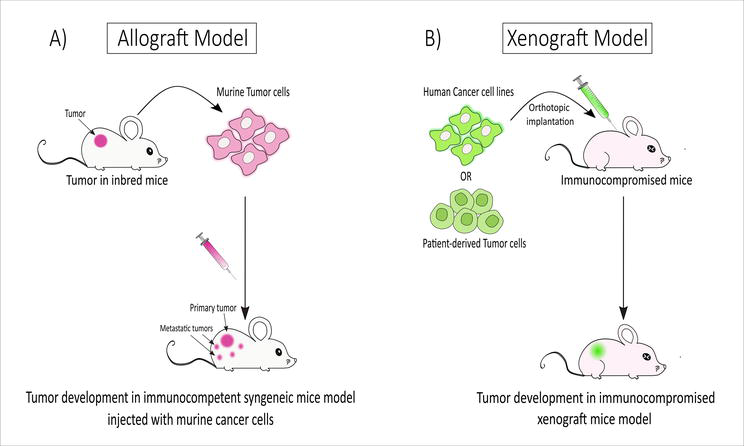
Figure 7.
Generation of allograft and xenograft transplantation models: (A) allograft transplant: murine tumour cells isolated from inbred donor mice are injected into immunocompetent mice resulting in tumour development. (B) Xenograft transplant: human cancer cells or patient-derived tumour cells are implanted into immunocompromised mice, resulting in tumour development [created with
Cancer cells from one mouse are transplanted into another mouse with a desired genetic feature in allograft (or syngeneic) transplantation models. Mice with functional immune systems accept the transplant when the cancer cells and the recipient share a common origin. This allows us to study the effects of therapeutic interventions in a setting very similar to that in which the tumour develops in an immunocompetent host. However, there is a possibility that the complexities of human malignancies will not be adequately represented by transplanted mouse tissue. Problems with GEM models, such as convoluted breeding plans and ineffective chemotherapy drugs, can be sidestepped with this transplanting approach [60].
To prevent the rejection of human cells, athymic nude mice or SCID mice are used to transplant human tumour cells subcutaneously or into the organ type (orthotopic) from where the tumour originated. This model involves implanting human cancerous cells or solid tumours into a mouse. The mice’s immune systems have been damaged to prevent cellular rejection by the host’s immune system. Orthotopic transplants place the tumour in its natural location on the host, while subcutaneous transplants place it just under the recipient’s skin. The human origin of the xenograft means that it shares some of the characteristics and complexities of human cancer. Once the tumour has reached a sufficient size, its response to therapy regimens can be performed according to the quantity of injected cells. As a result, these two transplantation models can be employed for a wide range of cancer-related experiments [60].
10. Conclusion
Human diseases have long been studied using mouse models. While these models have limitations and do not always accurately replicate actual disease, they have considerably aided in understanding disease mechanisms and developing new treatments. Mice are commonly used in medical research as study subjects due to their adaptability in replicating various human diseases and conditions in the lab. Mice are preferred models due to several factors, including their relative ease of handling, low cost, wide availability, and potential for genetic manipulation. Most importantly, genomic investigations have underlined the strong genetic similarities between humans and mice. Understanding complex human diseases might be challenging; hence, knockout mice can be used to understand the role of genes in complex diseases. Apart from knockout mice, immunocompromised mice can be efficiently used as models for cancer, immunology study as well as can be used for infectious diseases. Different types of immunocompromised mice exist, such as nude mice, NOD mice, SCID mice, and NOD/SCID mice. By virtue of their compromised immunity, these mice can be ideally used for xenograft mice models. Due to their smaller size and well-known genetic system, mice can be an appropriate model to study several complex diseases and disorders ranging from neurodegeneration to cancer, as discussed in this chapter.
The laboratory mouse: a heroic figure in the field of science
On the grounds of Novosibirsk’s Institute of Cytology and Genetics in southwestern Siberia, there lies a monument that serves as a tribute to the mice who were utilised in genetic research to gain insights into biological and physiological processes that could lead to the development of new medications and treatments for various illnesses [92].
Acknowledgments
The authors would like to express their sincere gratitude to the Indian Institute of Science Education and Research, Kolkata, for their valuable support and assistance. Neurotropic m-CoV research in an experimental animal model has provided a platform to write this book chapter. This work has been a collaborative effort of the Indian Institute of Science Education and Research Kolkata (IISER-K), India; the Indian Institute of Sciences (IISc), India; the University of Pennsylvania, USA; Thomas Jefferson University, USA; and the University of Colorado Denver, USA, for the past two decades. The bench research to study M-CoV biology was generously supported by the Department of Biotechnology (DBT) of India, the Council for Scientific and Industrial Research (CSIR) of India, the National Multiple Sclerosis Society, USA, the M.E. Groff Surgical Medical Research and Education Charitable Trust, and the Lindback Foundation Career Enhancement Award, USA. Indo-U.S. Science and Technology Forum (IUSSTF) also supported this work. The authors would also like to show their sincere gratitude to the System Medicine Cluster grant supported by the Department of Biotechnology (DBT) for cancer research. Finally, the success of this work has largely depended on the contribution and devotion of many researchers whose names are listed in the references.
Conflict of interest
The authors stated that they had no potential conflicts of interest with respect to the research, writing, and/or publication of this book chapter.
References
- 1.
Zhang W, Moore L, Ji P. Mouse models for cancer research. Chinese Journal of Cancer. 2011; 30 (3):149-152 - 2.
King AJ. The use of animal models in diabetes research. British Journal of Pharmacology. 2012; 166 (3):877-894 - 3.
Jia T, Wang C, Han Z, Wang X, Ding M, Wang Q. Experimental rodent models of cardiovascular diseases. Frontiers in Cardiovascular Medicine. 2020; 7 :588075 - 4.
Hafezparast M, Ahmad-Annuar A, Wood NW, Tabrizi SJ, Fisher EM. Mouse models for neurological disease. The Lancet Neurology. 2002; 1 (4):215-224 - 5.
Emes RD, Goodstadt L, Winter EE, Ponting CP. Comparison of the genomes of human and mouse lays the foundation of genome zoology. Human Molecular Genetics. 2003; 12 (7):701-709 - 6.
Compston A, Coles A. Multiple sclerosis. Lancet (London, England). 2008; 372 (9648):1502-1517 - 7.
Gourraud PA, Harbo HF, Hauser SL, Baranzini SE. The genetics of multiple sclerosis: An up-to-date review. Immunological Reviews. 2012; 248 (1):87-103 - 8.
Holmøy T, Hestvik AL. Multiple sclerosis: Immunopathogenesis and controversies in defining the cause. Current Opinion in Infectious Diseases. 2008; 21 (3):271-278 - 9.
Oleszak EL, Chang JR, Friedman H, Katsetos CD, Platsoucas CD. Theiler's virus infection: A model for multiple sclerosis. Clinical Microbiology Reviews. 2004; 17 (1):174-207 - 10.
Robinson AP, Harp CT, Noronha A, Miller SD. The experimental autoimmune encephalomyelitis (EAE) model of MS: Utility for understanding disease pathophysiology and treatment. Handbook of Clinical Neurology. 2014; 122 :173-189 - 11.
Doyle A, McGarry MP, Lee NA, Lee JJ. The construction of transgenic and gene knockout/knockin mouse models of human disease. Transgenic Research. 2012; 21 (2):327-349 - 12.
Menke DB. Engineering subtle targeted mutations into the mouse genome. Genesis (New York, NY: 2000). 2013; 51 (9):605-618 - 13.
Chen BS, Wu CC. Systems biology as an integrated platform for bioinformatics, systems synthetic biology, and systems metabolic engineering. Cell. 2013; 2 (4):635-688 - 14.
Nair RR, Corrochano S, Gasco S, Tibbit C, Thompson D, Maduro C, et al. Uses for humanised mouse models in precision medicine for neurodegenerative disease. Mammalian Genome : Official Journal of the International Mammalian Genome Society. 2019; 30 (7-8):173-191 - 15.
Onaciu A, Munteanu R, Munteanu VC, Gulei D, Raduly L, Feder RI, et al. Spontaneous and induced animal models for cancer research. Diagnostics (Basel). 31 Aug 2020; 10 (9):660 - 16.
Chen J, Liao S, Xiao Z, Pan Q , Wang X, Shen K, et al. The development and improvement of immunodeficient mice and humanized immune system mouse models. Frontiers in Immunology. 2022; 13 :1007579 - 17.
Croxford AL, Kurschus FC, Waisman A. Mouse models for multiple sclerosis: Historical facts and future implications. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2011; 1812 (2):177-183 - 18.
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. The New England Journal of Medicine. 2000; 343 (13):938-952 - 19.
Wang F-I, Stohlman SA, Fleming J. Demyelination induced by murine hepatitis virus JHM strian (MHV-4) is immunologically mediated. Journal of Neuroimmunology. 1990; 30 :31-41 - 20.
Das SJ. A mechanism of virus-induced demyelination. Interdisciplinary Perspectives on Infectious Diseases. 2010; 2010 :109239 - 21.
Shindler KS, Kenyon LC, Dutt M, Hingley ST, Das Sarma J. Experimental optic neuritis induced by a demyelinating strain of mouse hepatitis virus. Journal of Virology. 2008; 82 (17):8882-8886 - 22.
Wu GF, Dandekar AA, Pewe L, Perlman S. CD4 and CD8 T cells have redundant but not identical roles in virus-induced demyelination. Journal of Immunology (Baltimore, Md : 1950). 2000; 165 (4):2278-2286 - 23.
Matthews AE, Lavi E, Weiss SR, Paterson Y. Neither B cells nor T cells are required for CNS demyelination in mice persistently infected with MHV-A59. Journal of Neurovirology. 2002; 8 (3):257-264 - 24.
Li Y, Fu L, Gonzales DM, Lavi E. Coronavirus neurovirulence correlates with the ability of the virus to induce proinflammatory cytokine signals from astrocytes and microglia. Journal of Virology. 2004; 78 (7):3398-3406 - 25.
Buchmeier MJ, Lane TE. Viral-induced neurodegenerative disease. Current Opinion in Microbiology. 1999; 2 (4):398-402 - 26.
Saadi F, Chakravarty D, Kumar S, Kamble M, Saha B, Shindler KS, et al. CD40L protects against mouse hepatitis virus-induced neuroinflammatory demyelination. PLOS Pathogens. 2021; 17 (12):e1010059 - 27.
Chakravarty D, Saadi F, Kundu S, et al. CD4 deficiency causes poliomyelitis and axonal Blebbing in murine coronavirus-induced neuroinflammation. Journal of Virology. 2020; 94 (14):e00548-20 - 28.
Das Sarma J, Burrows A, Rayman P, Hwang M-H, Kundu S, Sharma N, et al. Ifit2 deficiency restricts microglial activation and leukocyte migration following murine coronavirus (m-CoV) CNS infection. PLOS Pathogens. 2020; 16 (11):e1009034 - 29.
Kenyon LC, Biswas K, Shindler KS, Nabar M, Stout M, Hingley ST, et al. Gliopathy of demyelinating and non-demyelinating strains of mouse hepatitis virus. Frontiers in Cellular Neuroscience. 2015; 9 :488 - 30.
Biswas K, Das SJ. Effect of microtubule disruption on neuronal spread and replication of demyelinating and nondemyelinating strains of mouse hepatitis virus in vitro. Journal of Virology. 2014; 88 (5):3043-3047 - 31.
Hart BAT, Brok HPM, Remarque E, Benson J, Treacy G, Amor S, et al. Suppression of ongoing disease in a nonhuman primate model of multiple sclerosis by a human-anti-human IL-12p40 Antibody1. The Journal of Immunology. 2005; 175 (7):4761-4768 - 32.
Sariol A, Mackin S, Allred MG, Ma C, Zhou Y, Zhang Q , et al. Microglia depletion exacerbates demyelination and impairs remyelination in a neurotropic coronavirus infection. Proceedings of the National Academy of Sciences of the United States of America. 2020; 117 (39):24464-24474 - 33.
Kuerten S, Gruppe TL, Laurentius LM, Kirch C, Tary-Lehmann M, Lehmann PV, et al. Differential patterns of spinal cord pathology induced by MP4, MOG peptide 35-55, and PLP peptide 178-191 in C57BL/6 mice. APMIS: Acta Pathologica, Microbiologica, et Immunologica Scandinavica. 2011; 119 (6):336-346 - 34.
Kishore A, Kanaujia A, Nag S, Rostami AM, Kenyon LC, Shindler KS, et al. Different mechanisms of inflammation induced in virus and autoimmune-mediated models of multiple sclerosis in C57BL6 mice. BioMed Research International. 2013; 2013 :589048 - 35.
Giralt M, Molinero A, Hidalgo J. Active induction of experimental autoimmune encephalomyelitis (EAE) with MOG (35-55) in the mouse. Methods in Molecular Biology (Clifton, NJ). 2018; 1791 :227-232 - 36.
Sarma JD, Ciric B, Marek R, Sadhukhan S, Caruso ML, Shafagh J, et al. Functional interleukin-17 receptor A is expressed in central nervous system glia and upregulated in experimental autoimmune encephalomyelitis. Journal of Neuroinflammation. 2009; 6 (1):14 - 37.
Keller CW, Sina C, Kotur MB, Ramelli G, Mundt S, Quast I, et al. ATG-dependent phagocytosis in dendritic cells drives myelin-specific CD4 (+) T cell pathogenicity during CNS inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2017; 114 (52):E11228-E11e37 - 38.
Takeshita Y, Ransohoff RM. Inflammatory cell trafficking across the blood-brain barrier: Chemokine regulation and in vitro models. Immunological Reviews. 2012; 248 (1):228-239 - 39.
Lovett-Racke AE, Yang Y, Racke MK. Th1 versus Th17: Are T cell cytokines relevant in multiple sclerosis? Biochimica et Biophysica Acta. 2011; 1812 (2):246-251 - 40.
Gutcher I, Becher B. APC-derived cytokines and T cell polarization in autoimmune inflammation. The Journal of Clinical Investigation. 2007; 117 (5):1119-1127 - 41.
Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annual Review of Immunology. 2009; 27 :485-517 - 42.
Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nature Medicine. 2002; 8 (5):500-508 - 43.
Viszlayová D, Sojka M, Dobrodenková S, Szabó S, Bilec O, Turzová M, et al. SARS-CoV-2 RNA in the cerebrospinal fluid of a patient with long COVID. Therapeutic Advances in Infectious Disease. 2021; 8 :20499361211048572 - 44.
Zanin L, Saraceno G, Panciani PP, Renisi G, Signorini L, Migliorati K, et al. SARS-CoV-2 can induce brain and spine demyelinating lesions. Acta Neurochirurgica. 2020; 162 (7):1491-1494 - 45.
Yu D, Zhu H, Liu Y, Cao J, Zhang X. Regulation of proinflammatory cytokine expression in primary mouse astrocytes by coronavirus infection. Journal of Virology. 2009; 83 (23):12204-12214 - 46.
Das Sarma J, Kenyon LC, Hingley ST, Shindler KS. Mechanisms of primary axonal damage in a viral model of multiple sclerosis. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2009; 29 (33):10272-10280 - 47.
Shabani Z. Demyelination as a result of an immune response in patients with COVID-19. Acta Neurologica Belgica. 2021; 121 (4):859-866 - 48.
Bender SJ, Weiss SR. Pathogenesis of murine coronavirus in the central nervous system. Journal of Neuroimmune Pharmacology : the Official Journal of the Society on NeuroImmune Pharmacology. 2010; 5 (3):336-354 - 49.
Rempel JD, Murray SJ, Meisner J, Buchmeier MJ. Differential regulation of innate and adaptive immune responses in viral encephalitis. Virology. 2004; 318 (1):381-392 - 50.
Jiang Y, Yu Y. Transgenic and gene knockout mice in gastric cancer research. Oncotarget. 2017; 8 (2):3696-3710 - 51.
Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rülicke T, et al. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 2000; 20 (21):7951-7963 - 52.
Lee Y, Dawson VL, Dawson TM. Animal models of Parkinson's disease: Vertebrate genetics. Cold Spring Harbor Perspectives in Medicine. 2012; 2 (10):a009324 - 53.
Bunner AE, Chandrasekera PC, Barnard ND. Knockout mouse models of insulin signaling: Relevance past and future. World Journal of Diabetes. 2014; 5 (2):146-159 - 54.
Diaz-Cruz ES, Cabrera MC, Nakles R, Rutstein BH, Furth PA. BRCA1 deficient mouse models to study pathogenesis and therapy of triple negative breast cancer. Breast Disease. 2010; 32 (1-2):85-97 - 55.
Shapiro AM, Miller-Pinsler L, Wells PG. Breast cancer 1 (BRCA1)-deficient embryos develop normally but are more susceptible to ethanol-initiated DNA damage and embryopathies. Redox Biology. 2016; 7 :30-38 - 56.
Hall B, Limaye A, Kulkarni AB. Overview: Generation of gene knockout mice. Current Protocols in Cell Biology. Sep 2009;Chapter 19:Unit 19.12 19.12.1-17 - 57.
Ney A, Canciani G, Hsuan JJ, Pereira SP. Modelling pancreatic neuroendocrine cancer: From bench side to clinic. Cancers (Basel). 28 Oct 2020; 12 (11):3170 - 58.
Kim H, Kim M, Im SK, Fang S. Mouse Cre-LoxP system: General principles to determine tissue-specific roles of target genes. Laboratory Animal Research. 2018; 34 (4):147-159 - 59.
Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunological Reviews. 2009; 229 (1):152-172 - 60.
Datta A, Mondal D. Chapter 5 - development of mouse models for cancer research. In: Verma AS, Singh A, editors. Animal Biotechnology. San Diego: Academic Press; 2014. pp. 73-94 - 61.
Walrath JC, Hawes JJ, Van Dyke T, Reilly KM. Genetically engineered mouse models in cancer research. Advances in Cancer Research. 2010; 106 :113-164 - 62.
Ding Y, Cravero JD, Adrian K, Grippo P. Modeling pancreatic cancer in vivo: From xenograft and carcinogen-induced systems to genetically engineered mice. Pancreas. 2010; 39 (3):283-292 - 63.
Pelekanou V, Leclercq G. Recent insights into the effect of natural and environmental estrogens on mammary development and carcinogenesis. The International Journal of Developmental Biology. 2011; 55 (7-8-9):869-878 - 64.
Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. Coexpression of MMTV/v-ha-ras and MMTV/c-myc genes in transgenic mice: Synergistic action of oncogenes in vivo. Cell. 1987; 49 (4):465-475 - 65.
Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005; 436 (7051):725-730 - 66.
Heyer J, Kwong LN, Lowe SW, Chin L. Non-germline genetically engineered mouse models for translational cancer research. Nature Reviews Cancer. 2010; 10 (7):470-480 - 67.
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992; 356 (6366):215-221 - 68.
Kersten K, de Visser KE, van Miltenburg MH, Jonkers J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Molecular Medicine. 2017; 9 (2):137-153 - 69.
Shibata H, Toyama K, Shioya H, Ito M, Hirota M, Hasegawa S, et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science (New York, NY). 1997; 278 (5335):120-123 - 70.
Weber J, Öllinger R, Friedrich M, Ehmer U, Barenboim M, Steiger K, et al. CRISPR/Cas9 somatic multiplex-mutagenesis for high-throughput functional cancer genomics in mice. Proceedings of the National Academy of Sciences of the United States of America. 2015; 112 (45):13982-13987 - 71.
Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014; 159 (2):440-455 - 72.
Annunziato S, Kas SM, Nethe M, Yücel H, Del Bravo J, Pritchard C, et al. Modeling invasive lobular breast carcinoma by CRISPR/Cas9-mediated somatic genome editing of the mammary gland. Genes & Development. 2016; 30 (12):1470-1480 - 73.
Wong DY, Feinberg SE. Incidence of growth of syngeneic oral squamous cell carcinoma in C57B1 bg/bg beige mice. Zhonghua yi xue za zhi =Chinese medical journal; Free China ed. 1990; 45 (1):53-59 - 74.
Zhu H, Kauffman ME, Trush MA, Jia Z, Li YR. A simple bioluminescence imaging method for studying cancer cell growth and metastasis after subcutaneous injection of Lewis lung carcinoma cells in syngeneic C57BL/6 mice. Reactive Oxygen Species (Apex, NC). 2018; 5 (14):118-125 - 75.
Jungwirth U, van Weverwijk A, Melake MJ, Chambers AF, Gao Q , Fivaz M, et al. Generation and characterisation of two D2A1 mammary cancer sublines to model spontaneous and experimental metastasis in a syngeneic BALB/c host. Disease Models & Mechanisms. 2018; 11 (1) - 76.
Ngiow SF, Loi S, Thomas D, Smyth MJ. Mouse models of tumor immunotherapy. 2016; 130 :1-24 - 77.
du Chatinier A, Meel MH, Das AI, Metselaar DS, Waranecki P, Bugiani M, et al. Generation of immunocompetent syngeneic allograft mouse models for pediatric diffuse midline glioma. Neuro-Oncology Advances. 2022; 4 (1):vdac079 - 78.
Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. 2000; 21 (4):585-591 - 79.
Mulchandani V, Banerjee A, Vadlamannati AV, Kumar S, Das SJ. Connexin 43 trafficking and regulation of gap junctional intercellular communication alters ovarian cancer cell migration and tumorigenesis. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2023; 159 :114296 - 80.
Olson B, Li Y, Lin Y, Liu ET, Patnaik A. Mouse models for cancer immunotherapy research. Cancer Discovery. 2018; 8 (11):1358-1365 - 81.
Gulley JL, Drake CG. Immunotherapy for prostate cancer: Recent advances, lessons learned, and areas for further research. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2011; 17 (12):3884-3891 - 82.
Lei ZG, Ren XH, Wang SS, Liang XH, Tang YL. Immunocompromised and immunocompetent mouse models for head and neck squamous cell carcinoma. OncoTargets and Therapy. 2016; 9 :545-555 - 83.
Flanagan SP. ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genetical Research. 1966; 8 (3):295-309 - 84.
Tani N, Kuchiba K, Osada T, Watanabe Y, Umemoto T. Effect of T-cell deficiency on the formation of periapical lesions in mice: Histological comparison between periapical lesion formation in BALB/c and BALB/c nu/nu mice. Journal of Endodontics. 1995; 21 (4):195-199 - 85.
Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983; 301 (5900):527-530 - 86.
Zhang B, Duan Z, Zhao Y. Mouse models with human immunity and their application in biomedical research. Journal of Cellular and Molecular Medicine. 2009; 13 (6):1043-1058 - 87.
Bosma GC, Fried M, Custer RP, Carroll A, Gibson DM, Bosma MJ. Evidence of functional lymphocytes in some (leaky) scid mice. The Journal of Experimental Medicine. 1988; 167 (3):1016-1033 - 88.
Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Jikken dobutsu Experimental animals. 1980; 29 (1):1-13 - 89.
Hesselton RM, Greiner DL, Mordes JP, Rajan TV, Sullivan JL, Shultz LD. High levels of human peripheral blood mononuclear cell engraftment and enhanced susceptibility to human immunodeficiency virus type 1 infection in NOD/LtSz-scid/scid mice. The Journal of Infectious Diseases. 1995; 172 (4):974-982 - 90.
Takahashi T, Katano I, Ito R, Goto M, Abe H, Mizuno S, et al. Enhanced antibody responses in a novel NOG transgenic mouse with restored lymph node organogenesis. Frontiers in Immunology. 2017; 8 :2017 - 91.
Brooks DG, Kitchen SG, Kitchen CM, Scripture-Adams DD, Zack JA. Generation of HIV latency during thymopoiesis. Nature Medicine. 2001; 7 (4):459-464 - 92.
This Russian Mouse Honors The Humble Lab Mouse [Internet]. 2017. Available from: https://www.smithsonianmag.com/smart-news/russian-statue-honoring-laboratory-mice-gains-renewed-popularity-180964570/ [Accessed: 2023-04-23]