 Open access peer-reviewed chapter
Open access peer-reviewed chapter
Abstract
Recent studies have emphasized the multiple aspects of non-coding micro-RNAs in the regulation of pancreatic ß cells in type 2 diabetic patients. Thus, highlighting the significance of non-coding regions of the genome in regulating pancreatic endocrine cells. Functional dysregulation of pancreatic endocrine cells increases the incidence of metabolic disorders in otherwise healthy individuals. A precise understanding of the molecular biology of metabolic dysregulation is important from cellular and clinical perspectives. The current chapter will highlight the important recent findings from type 2 diabetic human patients and aims to enhance our current understanding of ß cell pathophysiology from a clinical perspective for the development of novel therapeutic approaches to control this global incidence.
Keywords
- diabetes mellitus
- ß cells
- glucose
- insulin
- micro-RNAs
1. Introduction
Diabetes mellitus in principle is a pathophysiological condition in which the ability of cells to metabolize glucose is compromised primarily because of insulin deficiency or compromised insulin signaling (Figure 1). Diabetes mellitus is considered a global issue as it is estimated to impact around 700 million individuals worldwide by the year 2040 [1]. Hyperglycemia is the subsequent aftermath of diabetes mellitus where an abnormally high concentration of glucose persists at cellular and plasma levels. The persistent high concentration of glucose results in the activation of the polyol pathway and formation of advanced glycation end products (AGEs) along with an increase in AGE receptors (RAGE) [2, 3]. Activation of the polyol pathway enhances the mitochondrial production of reactive oxygen species (ROS) and increases the cytosolic concentration of ROS [4].
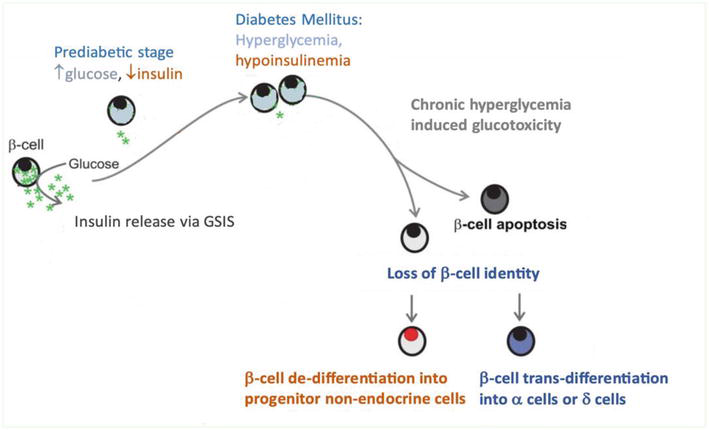
Figure 1.
Description of progressive ß cell decline during the course of diabetes mellitus and the subsequent loss of the cellular identity resulting in the trans- or de- differentiation of ß cells.
Type 2 diabetes mellitus (T2DM) is the most common metabolic disorder occurring globally [5, 6, 7]. Diets rich in carbohydrates and fats along with lack of exercise are highlighted as major risk factors for the increasing incidence of T2DM globally [8]. Obesity with visceral fat deposition and increased body mass index (BMI) have been shown to play a central role in the progression of type 2 diabetes complications [9].
2. Pathophysiology of type 2 diabetes mellitus (T2DM)
Regardless of the etiology, the T2DM progression is characterized either by a slow, progressive, and yet significant decline in the insulin secretion from ß cells or by a disruptive insulin signaling at the insulin-responsive tissues (muscles, fat, and liver) resulting in hyperinsulinemia which is clinically manifested as insulin resistance [10, 11, 12, 13]. The inability of insulin to properly bind and activate insulin receptors (IRs) on the cell membrane of muscle cells, adipocytes, and hepatocytes has been attributed as the main reason for hyperinsulinemia and insulin resistance [14, 15].
As activation of cytosolic carbohydrate metabolism is induced by insulin and results in glucose phosphorylation and subsequent formation of glucose-6-phosphate inside the cells [16, 17]. Insulin signaling enhances the enzyme activities of hexokinases in muscle and fat tissue and glucokinase (GCK) activity in the ß cells and hepatocytes [18, 19, 20, 21]. The activated cell surface insulin receptors stimulate cytosolic adaptor proteins called insulin receptor substrates (IRS1, IRS2) [22, 23]. Phosphorylation of IRS proteins activates the phosphoinositide2-kinase enzyme (PI3K) which further recruits ATP molecules to ultimately activate AKT protein (serine and threonine kinase) [24]. AKT activation helps the insulin to regulate multiple steps in glucose metabolism like a) increased cellular uptake of glucose via glucose transporter (GLUT4) in skeletal muscles [25], b) inhibition of glycogen synthase kinase 3 (GSK3) to stop glycogen metabolism [26, 27, 28], c) AKT induced activation of the mechanistic target of rapamycin (mTOR) resulting in the protein and lipid synthesis [29, 30, 31, 32, 33, 34], d) transcriptional control of gene expression through AKT induced inhibition of forkhead family box O (FOXO) transcription factor proteins [35, 36, 37]. Insulin also greatly helps to reduce plasma fatty acid levels by reducing adipocyte lipolysis rate and enhancing the ability of hepatocytes to produce very low-density lipoprotein (VLDL) [38, 39, 40, 41, 42, 43]. Insulin plays an important role in increasing skeleton muscle mass by enhancing the cellular intake of amino acids favoring protein synthesis and limiting the process of protein metabolism and urea formation [44, 45, 46, 47, 48, 49].
These above-mentioned metabolic consequences of insulin signaling on macronutrients like carbohydrates, proteins, and lipids ensure the maintenance of nutritional homeostasis at the cellular level under diverse physiological conditions. Diabetes mellitus is fundamentally a loss of this metabolic homeostasis at the cellular level. The single and most important endogenous factor for the loss of metabolic homeostasis is dysfunctional insulin signaling. Once the insulin receptors are activated through binding insulin molecules, these receptors follow a deactivation phase by the process of internalization and thus can no longer bind with insulin, a phenomenon known as insulin receptor endocytosis [50]. Insulin receptor endocytosis is the primary physiological mechanism through which the duration and intensity of insulin signaling are controlled [51]. Hyperinsulinemia leads to insulin resistance by promoting insulin receptor endocytosis rate and diminishing the number of insulin receptors at the plasma membrane which could potentially bind with plasma insulin molecules [52]. Apart from receptor internalization, insulin receptor kinase activity is also shown to be compromised in T2DM patients [53]. Lack of or reduced insulin signaling fails to activate glucose transporter proteins (GLUT) which limit cellular uptake of glucose resulting in high plasma glucose levels. Normally high plasma glucose levels stimulate the release of insulin from ß cells, a phenomenon known as glucose-stimulated insulin secretion (GSIS) [54, 55, 56]. Surprisingly a compromised GSIS responsiveness of ß cells in terms of insulin secretion from T2DM donors has been reported [57, 58, 59]. Interestingly it has been reported that T2DM patients also have high plasma levels of glucagon along with insulin resistance, hyperinsulinemia, and hyperglycemia [60]. Highlighting that the insulin is unable to downregulate glucagon secretion from islet α cells [61]. T2DM patients have also been reported to have decreased levels of gut incretin hormone known as glucagon-like peptide-1 (GLP-1) which normally induces the postprandial release of insulin from ß cells and downregulates glucagon secretion from α cells [62, 63]. These findings implicate the gut as an important regulator of insulin secretion during feeding and fasting and might contribute to the incidence of T2DM [64, 65]. Current treatment options for T2DM include newly developed GLP-1 receptor agonists to curtail hyperglycemic episodes contributed by either the lack of insulin and/or increased glucagon secretion [66, 67, 68].
2.1 Transcriptional modulation of ß cells in T2DM
Altered gene expression profiles of important proteins involved in the secretory process of insulin and insulin signaling are the major consequence of T2DM, as
Another important T2DM hallmark is the unusual loss of functional β cells, a process that starts during the prediabetic stage [73]. Almost half of the total population of functional β cells have been reported to be lost in T2DM patients [74, 75, 76, 77]. Two distinct mechanisms have been credited for this remarkable loss in functional β cells during T2DM namely: 1) apoptosis and 2) dedifferentiation. Interestingly the process of β cell apoptosis is initiated due to the hyperactivity of β cells which results in hyperinsulinemia. Prolonged exposure of β cells to high levels of insulin activates enzymes like caspases which initiate apoptosis, and nitric oxide synthase (iNOS) triggering excessive nitric oxide (NO) production, along with the formation of hydrogen peroxide (H2O2) [78, 79, 80]. Hyperglycemia along with high plasma levels of free fatty acids, known as glucolipotoxicity, also induces β cell apoptosis in T2DM [81, 82]. The deleterious effect of increased lipid accumulation in the β cells includes hyperactivation of lipid signaling pathways causing loss of functional β cells. Recently cellular dedifferentiation (cells switching back to the undifferentiated/progenitor stage from the differentiated/functional stage) has emerged as an important factor influencing the functional β cell mass in T2DM [83, 84, 85]. β cell dedifferentiation also propagates the appearance of other islet cell types indicating that β cells might trans-differentiate into α and δ cell types. The removal of epigenetic control on the transcription of glucagon and somatostatin genes in β cells during trans-differentiation limits the unique identity of β cells as insulin secretory cells. This phenomenon of trans-differentiation might help to clarify the increased plasma levels of glucagon observed in T2DM patients. Specifically, glucagon and somatostatin-positive cells have inactivated (cytoplasmic) FOXO1 and NKX6.1 (β cell-specific transcription factors) proteins in the islets of T2DM donors hinting towards the possibility of trans-differentiation of β cells [84]. Transcriptionally upregulated pluripotent genes and downregulated β cell-specific genes have been observed to assist the trans- and dedifferentiation of functional β cells. Down-regulation of specific genes like
2.2 Epigenetic regulation of β cell-specific gene expression profile
Aberration in β cell functionality is mainly attributed to deregulated gene expression control through epigenetic mechanisms [91, 92, 93, 94, 95, 96]. Specifically, chromatin modifications, DNA methylation, and post-translational modifications of histones are the classical epigenetic mechanisms through which gene expression profile of functional β cells are controlled. Alteration in the expression pattern of non-coding RNA sequences like microRNA (miRNA/miR) has also been shown to regulate the β cell function as well as their cellular identity [97, 98, 99, 100, 101, 102, 103]. Nonetheless, the recent findings that the majority of diabetes susceptibility loci are located in the non-coding regions of the human genome highlight the importance of epigenetic control in glucose homeostasis and β cell regulation [104, 105].
3. microRNA’s regulation of β cell function
Pancreatic β cells are highly adaptative cellular entities in nature [106, 107, 108, 109]. Adaptivity is specifically required to cater to different physiological states that demand different/opposing β cell responses. Like during feeding and fasting as well as during the high energy demands in exercise and pregnancy or during the intake of high carb high-fat diets. TF. Transcription upregulation of specific miRNAs during the embryonic developmental stage has been shown comprehensively to play an important part in the expression of a network of genes responsible for β cell development and function in mammals [110, 111]. However, the human data from diabetic adult patients suggests that the miRNAs might play a permissive role in the induction of β cell dysfunction [112]. On the other hand, certain miRNAs have been attributed to enhanced β cell mass/number in the pancreas, a phenomenon termed as β cell compensation [113, 114, 115]. These conflicting roles of miRNAs warrant a thorough understanding of the peculiar role of specific miRNAs under different physiological and pathological conditions. As miRNAs play a primary role in mRNA silencing and attenuate posttranscriptional regulation of gene expression in different physiological states of β cells. These fine-tuned variations are vital for glucose homeostasis. The loss of miRNA’s ability to fine-tune the gene expression led to β cell decompensation which results in abnormal insulin secretion and ultimately the development of diabetes. β cell decompensation leads to two main pathophysiological events: 1) impaired (reduced) β cell function in terms of glucose sensitivity and insulin secretion (Reduced GSIS), and 2) reduced β cell number/mass due to dedifferentiation or apoptosis. Impaired β cell function involves molecular defects in insulin biosynthesis, glucose uptake, and exocytosis of insulin granules. The downregulation of β cell-specific genes and upregulation of the non β-cells specific genes induce β cell dedifferentiation. T2DM patients have been shown to present these β cell-specific conditions [116, 117, 118]. A set of specific molecular pathways are involved in the secretion of insulin during GSIS and failure of either one of these molecular pathways can result in the GSIS decline and consequently the incidence of diabetes.
3.1 Involvement of different miRNAs in insulin biosynthesis and signaling: Recent ex-vivo
The first comprehensive report on the possible involvement of miRNAs in regulating mammalian β-cells ability to synthesize and secrete insulin came in 2011. Down regulation of a subset of miRNA genes induced a decline in insulin gene promoter activity and subsequently resulted in reduced insulin content in isolated murine islets and cultured β cells [119]. These specific miRNAs were miRNA-24, miRNA-26, miRNA-148, and miRNA-181. Soon after another study employed pancreatic islets isolated from normal and glucose-intolerant human donors [120]. An islet-specific miRNA network involved in insulin secretion in human islets was suggested and consists of miRNA-375, miRNA-127-3p, and miRNA-184. Later, in 2015, miRNA-375 (an islet-specific miRNA) was shown to play a decisive role in regulating the phenotype of human β cells, and upregulation of miRNA-375 during
3.2 microRNAs influencing insulin secretion from β cells
As discussed earlier, the magnitude of glucose uptake during GSIS regulates the rate of insulin release from β cell. Currently multiple miRNAs have been identified in humans to affect the glucose uptake ability of the β cells thus indirectly reducing the rate of insulin release. Considerable decline in the expression of specific proteins responsible for KATP and Ca2+ channel regulation and insulin granular exocytosis has been observed in the islets from T2DM donors [129]. T2DM donor islets also present glucose induced rise in different miRNA levels namely miRNA-130a, miRNA-130b, and miRNA-152 [130]. These specific miRNAs impair post transcriptional mRNA processing of glucokinase and impacts glucose metabolism. Proper glucose metabolism via glucokinase action is essential for ATP synthesis inside β cells. As insulin exocytosis requires the closure of ATP sensitive KATP channels. On the contrary certain miRNAs have been shown to support β cells to preserve their unique cellular identity. Monocarboxylate transporter (MCT-1) is a mitochondrial protein which is specifically inhibited in β cells to allow them to only utilize glucose as a precursor for metabolism. These specific miRNAs include miRNA-29a, miRNA-29b, and miRNA-124 which have been shown to selectively target human MCT-1 gene [131]. miRNA-129a have also been shown to be glucose sensitive. As islets isolated from human donors when incubated in high glucose medium (hyperglycemic conditions) resulted in upregulation of miRNA-29 in β cells [132]. miRNA-29 also enhance the expression of
4. microRNAs regulation by lipid accumulation in β cells
Another important and clinically relevant phenomenon of T2DM is the dysregulated lipid metabolism resulting in dyslipidemia (hypertriglyceridemia, reduced HDL and increased LDL particles) thus raising the incidence of obesity and cardiovascular disease (CVD) [134, 135, 136]. High plasma levels of triglycerides (hypertriglyceridemia) in T2DM patients result in substantial reduction of functional β cell numbers and significantly reduced insulin secretion [137]. Interestingly it has been demonstrated that microRNA sequences are altered during the β cell lipid metabolism by exposure to high lipids/fats [138, 139, 140, 141, 142, 143, 144, 145, 146, 147, 148, 149]. Specific proteins responsible for lipid metabolism and cholesterol homeostasis in β cells have been found to be transcriptionally controlled by several microRNAs. miRNA-33 has been shown to regulate cholesterol homeostasis by controlling sterol regulatory binding protein (SREBP) gene expression [150]. miRNA-34a has been reported to induce β cell lipotoxicity
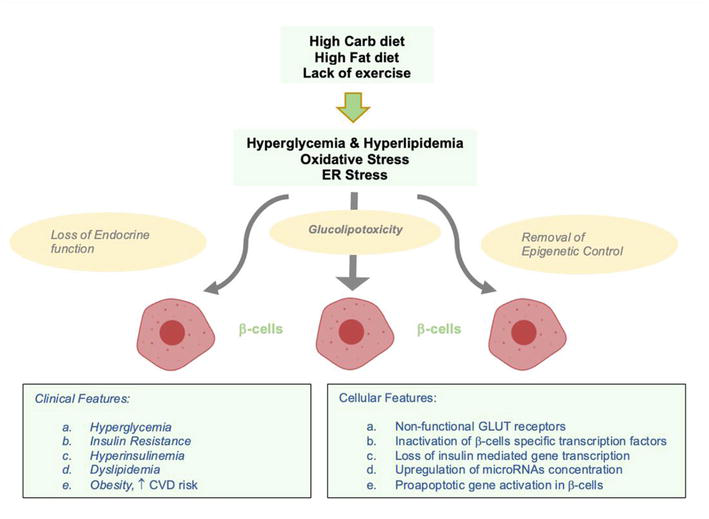
Figure 2.
Pathophysiology of type 2 diabetes mellitus.
5. Conclusion
Epigenetic regulation of β cells supported by the growing number of high-impact studies on human patients have established the fact that microRNAs play a key role in defining β cell identity. Pathological dysregulation of β cells is in part caused by aberrant microRNA expression helping in the progression of T2DM. As the processes of microRNA biogenesis are characterized hinting towards the possibility of microRNAs as attractive therapeutic targets. Diabetes and β cell-specific microRNAs also represent distinct biomarkers for the early detection of diabetes. Because of the biochemical stability of microRNAs in the extracellular fluids like blood and plasma/serum, microRNAs also represent an excellent biomarker for diagnostics. Comprehensive studies involving large cohorts of diabetic patients to assess the predictive values of serum levels of different microRNAs in conjunction with the specific diabetic stage are required to delineate the pathological and beneficial role of specific microRNAs in diabetes mellitus.
Abbreviation
| T2DM | Type 2 diabetes mellitus |
| Micro-RNA | Micro- Ribo-Nucleic-Acid |
| ROS | Reactive Oxygen Species |
| AGE | Advanced Glycation End product |
| RAGE | Receptor of Advanced Glycation End Product |
| BMI | Body Mass Index |
| IRS | Insulin Receptor Substrate |
| GLUT | Glucose Transporter |
| GSIS | Glucose Stimulated Insulin Release |
| GCK | Glucokinase |
| FOXO | Forkhead Family Box O |
| GLP-1 | Glucagon-like Peptide 1 |
| GIP | Gastric Inhibitory Polypeptide |
| PDX1 | Pancreatic Duodenal Homeobox 1 |
| NAD | Nicotinamide Adenine Dinucleotide |
| VLDL | Very Low Density Lipoprotein |
References
- 1.
Maiese K. New insights for oxidative stress and diabetes mellitus. Oxidative Medicine and Cellular Longevity. 2015; 2015 :875961 - 2.
Brownlee M. The pathobiology of diabetic complications: A unifying mechanism (banting lecture). Diabetes. 2005; 54 (6):1615-1625 - 3.
Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circulation Research. 2010; 107 (9):1058-1070 - 4.
Pi J, Zhang Q , Fu J, Woods CG, Hou Y, Corkey BE, et al. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicology and Applied Pharmacology. 2010; 244 (1):77-83 - 5.
Hanley SC, Austin E, Assouline-Thomas B, Kapeluto J, Blaichman J, Moosavi M, et al. β-Cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology. 2010; 151 (4):1462-1472 - 6.
Satin LS, Butler PC, Ha J, Sherman AS. Pulsatile insulin secretion, impaired glucose tolerance and type 2 diabetes. Molecular Aspects of Medicine. 2015; 42 :61-77 - 7.
Moin ASM, Dhawan S, Cory M, Butler PC, Rizza RA, Butler AE. Increased frequency of hormone negative and polyhormonal endocrine cells in lean individuals with type 2 diabetes. The Journal of Clinical Endocrinology and Metabolism. 2016; 101 (10):3628-3636 - 8.
Roden M, Shulman GI. The integrative biology of type 2 diabetes. Nature. 2019; 576 (7785):51-60 - 9.
Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB, et al. Pathophysiology of type 2 diabetes mellitus. International Journal of Molecular Sciences. 2020; 21 (17):1-34 - 10.
Zick Y. Insulin resistance: A phosphorylation-based uncoupling of insulin signaling. Trends in Cell Biology. 2001; 11 (11):437-441 - 11.
Chatterjee S, Khunti K, Davies MJ. Type 2 diabetes. Lancet. 2017; 389 (10085):2239-2251 - 12.
Lewis GF, Carpentier A, Vranic M, Giacca A. Resistance to insulin’s acute direct hepatic effect in suppressing steady-state glucose production in individuals with type 2 diabetes. Diabetes. 1999; 48 (3):570-576 - 13.
Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. The Journal of Clinical Investigation. 1999; 104 (6):787-794 - 14.
RA Haeusler TMDA. Biochemical and cellular properties of insulin receptor signaling. Nature Reviews. Molecular Cell Biology. 2018; 19 (1):31-44 - 15.
Boucher J, Kleinridders A, Ronald KC. Insulin receptor signaling in normal and insulin-resistant states. Cold Springer Harbor Perspective Biology. 2014; 6 (1):a009191 - 16.
Posner BI. Insulin signaling: The inside story. Canadian Journal of Diabetes. 2017; 41 (1):108 - 17.
Newsholme EA, Dimitriadis G. Integration of biochemical and physiologic effects of insulin on glucose metabolism. Experimental and Clinical Endocrinology & Diabetes. 2001; 109 (Suppl. 2):S122-S134 - 18.
Matschinsky FM, Wilson DF. The central role of glucokinase in glucose homeostasis: A perspective 50 years after demonstrating the presence of the enzyme in islets of Langerhans. Frontiers in Physiology. 2019; 10 :148 - 19.
Massa ML, Gagliardino JJ, Francini F. Liver glucokinase: An overview on the regulatory mechanisms of its activity. IUBMB Life. 2011; 6391 :1-6 - 20.
Iynedjian PB. Molecular physiology of mammalian glucokinase. Cellular and Molecular Life Sciences. 2009; 66 (1):27-42 - 21.
Matschinsky FM. Regulation of pancreatic-cell glucokinase: From basics to therapeutics. Diabetes. 2002; 51 (Suppl. 3):S394-S404 - 22.
Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signaling. Nature Reviews. Molecular Cell Biology. 2018; 19 (1):31-44 - 23.
White MF. IRS proteins and the common path to diabetes. American Journal of Physiology – Endocrinology Metabolism. 2002; 283 (3):46-43 - 24.
Kohn AD, Kovacina KS, Roth RA. Insulin stimulates the kinase activity of RAC-PK, a pleckstrin homology domain containing ser/thr kinase. The EMBO Journal. 1995; 14 (17):4288-4295 - 25.
Zorzano A, Palacín M, Gumà A. Mechanisms regulating GLUT4 glucose transporter expression and glucose transport in skeletal muscle. Acta Physiologica Scandinavica. 2005; 183 (1):43-58 - 26.
Wang L, Liu Q , Kitamoto T, Hou J, Qin J, Accili D. Identification of insulin-responsive transcription factors that regulate glucose production by hepatocytes. Diabetes. 2019; 68 (6):1156-1167 - 27.
den Boer MAM, Voshol PJ, Kuipers F, Romijn JA, Havekes LM. Hepatic glucose production is more sensitive to insulin-mediated inhibition than hepatic VLDL-triglyceride production. American Journal of Physiology. Endocrinology and Metabolism. 2006; 291 :1360-1364 - 28.
Hatting M, Tavares CDJ, Sharabi K, Rines AK, Puigserver P. Insulin regulation of gluconeogenesis. Annals of the New York Academy of Sciences. 2018; 1411 (1):21-35 - 29.
Charlton M, Nair KS. Protein metabolism in insulin-dependent diabetes mellitus. The Journal of Nutrition. 1998; 128 (2):323S-327S - 30.
Fukagawa NK, Minaker KL, Rowe JW, Goodman MN, Matthews DE, Bier DM, et al. Insulin-mediated reduction of whole body protein breakdown. Dose-response effects on leucine metabolism in postabsorptive men. The Journal of Clinical Investigation. 1985; 76 (6):2306-2311 - 31.
Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metabolism. 2009; 9 (4):311-326 - 32.
Tessari P, Nosadini R, Trevisan R, de Kreutzenberg S, Inchiostro S, Duner E, et al. Defective suppression by insulin of leucine-carbon appearance and oxidation in type 1, insulin-dependent diabetes mellitus. Evidence for insulin resistance involving glucose and amino acid metabolism. The Journal of Clinical Investigation. 1986; 77 (6):1797-1804 - 33.
Yoon MS. The emerging role of branched-chain amino acids in insulin resistance and metabolism. Nutrients. 2016; 8 (7):405 - 34.
Asghari G, Farhadnejad H, Teymoori F, Mirmiran P, Tohidi M, Azizi F. High dietary intake of branched-chain amino acids is associated with an increased risk of insulin resistance in adults. Journal of Diabetes. 2018; 10 (5):357-364 - 35.
Wang L, Liu Q , Kitamoto T, Hou J, Qin J, Accili D. Identification of insulin-responsive transcription factors that regulate glucose production by hepatocytes. Diabetes. 2019; 68 (6):1156-1167 - 36.
den Boer MAM, Voshol PJ, Kuipers F, Romijn JA, Havekes LM. Hepatic glucose production is more sensitive to insulin-mediated inhibition than hepatic VLDL-triglyceride production. American Journal of Physiology Endocrinology Metabolism. 2006; 291 :1360-1364 - 37.
Hatting M, Tavares CDJ, Sharabi K, Rines AK, Puigserver P. Insulin regulation of gluconeogenesis. In: Annals of the New York Academy of Sciences. Vol. 1411. Blackwell Publishing Inc.; 2018. pp. 21-35 - 38.
Kamagate A, Dong HH. FoxO1 integrates insulin signaling to VLDL production. Cell Cycle. 2008; 7 (20):3162-3170 - 39.
Kamagate A, Qu S, Perdomo G, Su D, Dae HK, Slusher S, et al. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. The Journal of Clinical Investigation. 2008; 118 (6):2347-2364 - 40.
Kim DH, Zhang T, Lee S, Calabuig-Navarro V, Yamauchi J, Piccirillo A, et al. FoxO6 integrates insulin signaling with MTP for regulating VLDL production in the liver. Endocrinology. 2014; 155 (4):1255-1267 - 41.
Scherer T, Ohare J, Diggs-Andrews K, Schweiger M, Cheng B, Lindtner C, et al. Brain insulin controls adipose tissue lipolysis and lipogenesis. Cell Metabolism. 2011; 13 (2):183-194 - 42.
Iwen KA, Scherer T, Heni M, Sayk F, Wellnitz T, Machleidt F, et al. Intranasal insulin suppresses systemic but not subcutaneous lipolysis in healthy humans. The Journal of Clinical Endocrinology and Metabolism. 2014; 99 (2):E246-E251 - 43.
Saltiel AR. Insulin signaling in the control of glucose and lipid homeostasis. Handbook Experimental Pharmacology. 2016; 233 :51-71 - 44.
Charlton M, Nair KS. Protein metabolism in insulin-dependent diabetes mellitus. The Journal of Nutrition. 1998; 128 (2):323S-327S - 45.
Fukagawa NK, Minaker KL, Rowe JW, Goodman MN, Matthews DE, Bier DM, et al. Insulin-mediated reduction of whole body protein breakdown. Dose-response effects on leucine metabolism in postabsorptive men. The Journal of Clinical Investigation. 1985; 76 (6):2306-2311 - 46.
Newgard CB, Ann J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metabolism. 2009; 9 (4):311-326 - 47.
Tessari P, Nosadini R, Trevisan R, de Kreutzenberg S, Inchiostro S, Duner E, et al. Defective suppression by insulin of leucine-carbon appearance and oxidation in type 1, insulin-dependent diabetes mellitus. Evidence for insulin resistance involving glucose and amino acid metabolism. The Journal of Clinical Investigation. 1986; 77 (6):1797-1804 - 48.
Yoon MS. The emerging role of branched-chain amino acids in insulin resistance and metabolism. Nutrients. 2016; 8 (7):405 - 49.
Asghari G, Farhadnejad H, Teymoori F, Mirmiran P, Tohidi M, Azizi F. High dietary intake of branched-chain amino acids is associated with an increased risk of insulin resistance in adults. Journal of Diabetes. 2018; 10 (5):357-364 - 50.
Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiological Reviews. 2018; 98 (4):2133-2223 - 51.
Kaksonen M, Roux A. Mechanisms of clathrin-mediated endocytosis. Nature Reviews. Molecular Cell Biology. 2018; 19 (5):313-326 - 52.
Hall C, Yu H, Choi E. Insulin receptor endocytosis in the pathophysiology of insulin resistance. Experimental & Molecular Medicine. 2020; 52 (6):911-920 - 53.
Caro JF, Ittoop O, Pories WJ, Meelheim D, Flickinger EG, Thomas F, et al. Studies on the mechanism of insulin resistance in the liver from humans with noninsulin-dependent diabetes. Insulin action and binding in isolated hepatocytes, insulin receptor structure, and kinase activity. The Journal of Clinical Investigation. 1986; 78 (1):249-258 - 54.
Komatsu M, Takei M, Ishii H, Sato Y. Glucose-stimulated insulin secretion: A newer perspective. Journal of Diabetes Investigation. 2013; 4 (6):511-516 - 55.
Straub SG, Sharp GWG. Glucose-stimulated signaling pathways in biphasic insulin secretion. Diabetes/Metabolism Research and Reviews. 2002; 18 (6):451-463 - 56.
Wang J, Gu W, Chen C. Knocking down insulin receptor in pancreatic beta cell lines with lentiviral-small hairpin RNA reduces glucose-stimulated insulin secretion via decreasing the gene expression of insulin, GLUT2 and Pdx1. International Journal of Molecular Sciences. 2018; 19 (4) - 57.
Solimena M, Schulte AM, Marselli L, Ehehalt F, Richter D, Kleeberg M, et al. Systems biology of the IMIDIA biobank from organ donors and pancreatectomised patients defines a novel transcriptomic signature of islets from individuals with type 2 diabetes. Diabetologia. 2018; 61 (3):641-657 - 58.
Lyon J, Manning Fox JE, Spigelman AF, Kim R, Smith N, O’Gorman D, et al. Research-focused isolation of human islets from donors with and without diabetes at the Alberta Diabetes Institute Islet Core. Endocrinology. 2016; 157 (2):560-569 - 59.
Deng S, Vatamaniuk M, Huang X, Doliba N, Lian MM, Frank A, et al. Structural and functional abnormalities in the islets isolated from type 2 diabetic subjects. Diabetes. 2004; 53 (3):624-632 - 60.
D’alessio D. The role of dysregulated glucagon secretion in type 2 diabetes. Diabetes, Obesity and Metabolism. 2011; 13 (Suppl. 1):126-132 - 61.
Philippe J, Knepel W, Waeber G. Insulin regulation of the glucagon gene is mediated by an insulin-responsive DNA element. Proceedings of the National Academy Science USA. 1991; 88 (16):7224-7227 - 62.
Eissele R, Göke R, Willemer S. Glucagon-like peptide-1 cells in the gastrointestinal tract and pancreas of rat, pig and man. European Journal of Clinical Investigation. 1992; 22 :283-291 - 63.
Holst JJ. The physiology of glucagon-like peptide 1. Physiological Reviews. 2007; 87 (4):1409-1439 - 64.
Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes. 2012; 61 (2):364-371 - 65.
Lund A. On the role of the gut in diabetic hyperglucagonaemia. Danish Medical Journal. 2017; 64 (4):B5340 - 66.
Drucker DJ. Glucagon-like Peptide-1 and the islet β-cell: Augmentation of cell proliferation and inhibition of apoptosis. Endocrinology. 2003; 144 :5145-5148 - 67.
Drucker DJ, Nauck MA. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006; 368 (9548):1696-1705 - 68.
Gloyn AL, Drucker DJ. Precision medicine in the management of type 2 diabetes. The Lancet Diabetes and Endocrinology. 2018; 6 (11):891-900 - 69.
Ottosson-Laakso E, Krus U, Storm P, Prasad RB, Oskolkov N, Ahlqvist E, et al. Glucose-induced changes in gene expression in human pancreatic islets: Causes or consequences of chronic hyperglycemia. Diabetes. 2017; 66 (12):3013-3028 - 70.
Segerstolpe Å, Palasantza A, Eliasson P, Andersson EM, Andréasson AC, Sun X, et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metabolism. 2016; 24 (4):593-607 - 71.
Marselli L, Piron A, Suleiman M, Colli ML, Yi X, Khamis A, et al. Persistent or transient human β cell dysfunction induced by metabolic stress: Specific signatures and shared gene expression with type 2 diabetes. Cell Reports. 2020; 33 (9):108466 - 72.
Marselli L, Thorne J, Dahiya S, Sgroi DC, Sharma A, Bonner-Weir S, et al. Gene expression profiles of Beta-cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PLoS One. 2010; 5 (7):e11499 - 73.
Weir GC, Bonner-Weir S. Five of stages of evolving β-cell dysfunction during progression to diabetes. Diabetes. 2004; 53 (Suppl. 3):S16-S21 - 74.
Khin PP, Lee JH, Jun HS. A brief review of the mechanisms of β-cell dedifferentiation in type 2 diabetes. Nutrients. 2021; 13 (5):1593 - 75.
Bensellam M, Jonas JC, Laybutt DR. Mechanisms of β;-cell dedifferentiation in diabetes: Recent findings and future research directions. The Journal of Endocrinology. 2018; 236 (2):R109-R143 - 76.
Sun T, Han X. Death versus dedifferentiation: The molecular bases of beta cell mass reduction in type 2 diabetes. Seminars in Cell & Developmental Biology. 2020; 103 :76-82 - 77.
Marrif HI, Al-Sunousi SI. Pancreatic β cell mass death. Frontiers in Pharmacology. 2016; 7 :83 - 78.
Rachdaoui N, Polo-Parada L, Ismail-Beigi F. Prolonged exposure to insulin inactivates Akt and Erk 1/2 and increases pancreatic islet and INS1E β-cell apoptosis. Journal of Endocrinology Society. 2018; 3 (1):69-90 - 79.
Bucris E, Beck A, Boura-Halfon S, Isaac R, Vinik Y, Rosenzweig T, et al. Prolonged insulin treatment sensitizes apoptosis pathways in pancreatic β cells. The Journal of Endocrinology. 2016; 230 (3):291-307 - 80.
Sampson SR, Bucris E, Horovitz- Fried M, Parnas A, Kahana S, Abitbol G, et al. Insulin increases H2O2-induced pancreatic beta cell death. Apoptosis. 2010; 15 (10):1165-1176 - 81.
Imai Y, Cousins RS, Liu S, Phelps BM, Promes JA. Connecting pancreatic islet lipid metabolism with insulin secretion and the development of type 2 diabetes. Annals of the New York Academy of Sciences. 2020; 1461 (1):53-72 - 82.
Lytrivi M, Castell AL, Poitout V, Cnop M. Recent insights into mechanisms of β-cell lipo- and glucolipotoxicity in type 2 diabetes. Journal of Molecular Biology. 2020; 432 (5):1514-1534 - 83.
Bensellam M, Jonas JC, Laybutt DR. Mechanisms of β-cell dedifferentiation in diabetes: Recent findings and future research directions. The Journal of Endocrinology. 2018; 236 (2):R109-R143 - 84.
Hunter CS, Stein RW. Evidence for loss in identity, De-differentiation, and trans-differentiation of islet β-cells in type 2 diabetes. Frontiers in Genetics. 2017; 8 :35 - 85.
Cinti F, Bouchi R, Kim-Muller JY, Ohmura Y, Sandoval PR, Masini M, et al. Evidence of β-cell dedifferentiation in human type 2 diabetes. The Journal of Clinical Endocrinology and Metabolism. 2016; 101 (3):1044-1054 - 86.
Brereton MF, Iberl M, Shimomura K, Zhang Q , Adriaenssens AE, Proks P, et al. Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nature Communications. 2014; 5 :4639 - 87.
Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic β cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metabolism. 2014; 19 (5):872-882 - 88.
Neelankal John A, Ram R, Jiang FX. RNA-Seq analysis of islets to characterise the dedifferentiation in type 2 diabetes model mice db/db. Endocrine Pathology. 2018; 29 (3):207-221 - 89.
Talchai C, Xuan S, Lin H, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012; 150 (6):1223-1234 - 90.
Spijker HS, Ravelli RBG, Mommaas-Kienhuis AM, van Apeldoorn AA, Engelse MA, Zaldumbide A, et al. Conversion of mature human β-cells into glucagon-producing α-cells. Diabetes. 2013; 62 (7):2471-2480 - 91.
Schuit F. Epigenetic programming of glucose-regulated insulin release. The Journal of Clinical Investigation. 2015; 125 (7):2565-2568 - 92.
Dhawan S, Tschen SI, Zeng C, Guo T, Hebrok M, Matveyenko A, et al. DNA methylation directs functional maturation of pancreatic β cells. The Journal of Clinical Investigation. 2015; 125 (7):2851-2860 - 93.
Spaeth JM, Walker EM, Stein R. Impact of Pdx1-associated chromatin modifiers on islet β-cells. Diabetes, Obesity & Metabolism. 2016; 18 (Suppl. 1):123-127 - 94.
Dayeh T, Ling C. Does epigenetic dysregulation of pancreatic islets contribute to impaired insulin secretion and type 2 diabetes? Biochemistry and Cell Biology. 2015; 93 (5):511-521 - 95.
Campbell SA, Hoffman BG. Chromatin regulators in pancreas development and diabetes. Trends in Endocrinology and Metabolism. 2016; 27 (3):142-152 - 96.
Astro V, Adamo A. Epigenetic control of endocrine pancreas differentiation in vitro: Current knowledge and future perspectives. Frontiers in Cell and Development Biology. 2018; 6 :141 - 97.
Latreille M, Hausser J, Stützer I, Zhang Q , Hastoy B, Gargani S, et al. MicroRNA-7a regulates pancreatic β cell function. The Journal of Clinical Investigation. 2014; 124 (6):2722-2735 - 98.
Dumortier O, Fabris G, Pisani DF, Casamento V, Gautier N, Hinault C, et al. microRNA-375 regulates glucose metabolism-related signaling for insulin secretion. The Journal of Endocrinology. 2020; 244 (1):189-200 - 99.
Wan S, Zhang J, Chen X, Lang J, Li L, Chen F, et al. MicroRNA-17-92 regulates beta-cell restoration after streptozotocin treatment. Frontiers in Endocrinology. 2020; 11 :9 - 100.
Rodríguez-Comas J, Moreno-Asso A, Moreno-Vedia J, Martín M, Castaño C, Marzà-Florensa A, et al. Stress-induced microrna-708 impairs b-cell function and growth. Diabetes. 2017; 66 (12):3029-3040 - 101.
Fu X, Dong B, Tian Y, Lefebvre P, Meng Z, Wang X, et al. MicroRNA-26a regulates insulin sensitivity and metabolism of glucose and lipids. The Journal of Clinical Investigation. 2015; 125 (6):2497-2509 - 102.
Baroukh N, Ravier MA, Loder MK, Hill E, Bounacer A, Scharfmann R, et al. MicroRNA-124a regulates foxa2 expression and intracellular signaling in pancreatic β-cell lines. The Journal of Biological Chemistry. 2007; 282 (27):19575-19588 - 103.
Xu Y, Huang Y, Guo Y, Xiong Y, Zhu S, Xu L, et al. MicroRNA-690 regulates induced pluripotent stem cells (iPSCs) differentiation into insulin-producing cells by targeting Sox9. Stem Cell Research & Therapy. 2019; 10 (1):59 - 104.
Mahanjan A et al. Fine-mapping type 2 diabetes loci to single variant resolution using high-density imputation and islet specific epigenome maps. Nature Genetics. 2018; 50 :1505-1513 - 105.
Vujkovic M et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcome among 1.4 million participants in a multi-ancestry meta-analysis. Nature Genetics. 2020; 52 :680-691 - 106.
De Jesus DF, Kulkarni RN. “Omics” and “epi-omics” underlying the β-cell adaptation to insulin resistance. Molecular Metabolism. 2019; 27 (Suppl):S42-S48 - 107.
Wortham M, Sander M. Mechanisms of β-cell functional adaptation to changes in workload. Diabetes, Obesity & Metabolism. 2016; 18 (Suppl 1):78-86 - 108.
Wortham M, Sander M. Transcriptional mechanisms of pancreatic β-cell maturation and functional adaptation. Trends in Endocrinology and Metabolism. 2021; 32 (7):474-487 - 109.
Kim H, Kulkarni RN. Epigenetics in β-cell adaptation and type 2 diabetes. Current Opinion in Pharmacology. 2020; 55 :125-131 - 110.
Kaviani M, Azarpira N, Karimi MH, Al-Abdullah I. The role of microRNAs in islet β-cell development. Cellular Biology International. 2016; 40 (12):1248-1255 - 111.
Eliasson L, Esguerra JLS. Role of non-coding RNAs in pancreatic beta-cell development and physiology. Acta Physiologica. 2014; 211 (2):273-284 - 112.
Kim KH, Hartig SM. Contributions of microRNAs to peripheral insulin sensitivity. Endocrinology. 2022; 163 (2):bqab250 - 113.
Jacovetti C, Abderrahmani A, Parnaud G, Jonas JC, Peyot ML, Cornu M, et al. MicroRNAs contribute to compensatory β cell expansion during pregnancy and obesity. The Journal of Clinical Investigation. 2012; 122 (10):3541-3551 - 114.
Cerf ME. Beta cell dynamics: Beta cell replenishment, beta cell compensation and diabetes. Endocrine; 44 (2):303-311 - 115.
Geach T. Diabetes: Preventing β-cell apoptosis and T2DM with microRNAs – A role for MIR-200? Nature Reviews Endocrinology. 2015; 11 (8):444 - 116.
Cinti F, Bouchi R, Kim-Muller JY, Ohmura Y, Sandoval PR, Masini M, et al. Evidence of β-cell dedifferentiation in human type 2 diabetes. The Journal of Clinical Endocrinology and Metabolism. 2016; 101 (3):1044-1054 - 117.
Khin PP, Lee JH, Jun HS. A brief review of the mechanisms of β-cell dedifferentiation in type 2 diabetes. Nutrients. 2021; 13 (5):1593 - 118.
Bensellam M, Jonas JC, Laybutt DR. Mechanisms of β-cell dedifferentiation in diabetes: Recent findings and future research directions. The Journal of Endocrinology. 2018; 236 (2):R109-R143 - 119.
Melkman-Zehavi T, Oren R, Kredo-Russo S, Shapira T, Mandelbaum AD, Rivkin N, et al. miRNAs control insulin content in pancreatic β-cells via downregulation of transcriptional repressors. EMBO Journal. 2011; 30 (5):835-845 - 120.
Bolmeson C, Esguerra JLS, Salehi A, Speidel D, Eliasson L, Cilio CM. Differences in islet-enriched miRNAs in healthy and glucose intolerant human subjects. Biochemical Biophysical Research Communication. 2011; 404 (1):16-22 - 121.
Nathan G, Kredo-Russo S, Geiger T, Lenz A, Kaspi H, Hornstein E, et al. MiR-375 promotes redifferentiation of adult human β cells expanded in vitro. PLoS One. 2015; 10 (4):e0122108-e0122108 - 122.
Xu G, Chen J, Jing G, Shalev A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nature Medicine. 2013; 19 (9):1141-1146 - 123.
Sebastiani G, Po A, Miele E, Ventriglia G, Ceccarelli E, Bugliani M, et al. MicroRNA-124a is hyperexpressed in type 2 diabetic human pancreatic islets and negatively regulates insulin secretion. Acta Diabetologica. 2015; 52 (3):523-530 - 124.
Jo SH, Chen J, Xu G, Grayson TB, Thielen LA, Shalev A. miR-204 controls glucagon-like peptide 1 receptor expression and agonist function. Diabetes. 2018; 67 (2):256-264 - 125.
Brunton SA, Wysham CH. GLP-1 receptor agonists in the treatment of type 2 diabetes: Role and clinical experience to date. Postgraduate Medicine. 2020; 132 (suppl. 2):3-14 - 126.
Nachawi N, Rao PP, Makin V. The role of GLP-1 receptor agonists in managing type 2 diabetes. Cleveland Clinic Journal of Medicine. 2022; 89 (8):457-464 - 127.
Li N, Jiang D, He Q , He F, Li Y, Deng C, et al. microRNA-181c-5p promotes the formation of insulin-producing cells from human induced pluripotent stem cells by targeting smad7 and TGIF2. Cell Death & Disease. 2020; 11 (6):1-12 - 128.
Bhushan R, Rani A, Gupta D, Akhtar A, Dubey PK. MicroRNA-7 regulates insulin signaling pathway by targeting IRS1, IRS2, and RAF1 genes in gestational diabetes mellitus. MicroRNA. 2022; 11 (1):57-72 - 129.
Rorsman P, Ashcroft FM. Pancreatic β-cell electrical activity and insulin secretion: Of mice and men. Physiological Reviews. 2018; 98 (1):117-214 - 130.
Ofori JK, Salunkhe VA, Bagge A, Vishnu N, Nagao M, Mulder H, et al. Elevated miR-130a/ miR130b/ miR-152 expression reduces intracellular ATP levels in the pancreatic beta cell. Scientific Reports. 2017; 7 (1):1-15 - 131.
Pullen TJ, da Silva XG, Kelsey G, Rutter GA. miR-29a and miR-29b contribute to pancreatic β-cell-specific silencing of Monocarboxylate transporter 1 (Mct1). Molecular and Cellular Biology. 2011; 31 (15):3182-3194 - 132.
Bagge A, Clausen TR, Larsen S, Ladefoged M, Rosenstierne MW, Larsen L, et al. MicroRNA-29a is up-regulated in beta-cells by glucose and decreases glucose-stimulated insulin secretion. Biochemical and Biophysical Research Communications. 2012; 426 (2):266-272 - 133.
Bagge A, Dahmcke CM, Dalgaard LT. Syntaxin-1a is a direct target of miR-29a in insulin-producing β-cells. Hormone and Metabolic Research. 2013; 45 (6):463-466 - 134.
Taskinen MR. Diabetic dyslipidemia. Atherosclerosis. Supplements. 2002; 3 (1):47-51 - 135.
Mahato RV, Gyawali P, Raut PP, Regmi P, Khelanand PS, Dipendra RP, et al. Association between glycaemic control and serum lipid profile in type 2 diabetic patients: Glycated haemoglobin as a dual biomarker. Biomedical Research. 2011; 22 (3):375-380 - 136.
Taskinen MR. Diabetic dyslipidaemia: From basic research to clinical practice. Diabetologia. 2003; 46 (6):733-749 - 137.
Tong X, Liu S, Stein R, Imai Y. Lipid droplets' role in the regulation of β-cell function and β-cell demise in type 2 diabetes. Endocrinology. 2022; 163 (3):bqac007 - 138.
Tarlton JMR, Patterson S, Graham A. MicroRNA sequences modulated by beta cell lipid metabolism: Implications for type 2 diabetes mellitus. Biology (Basel). 2021; 10 (6):534 - 139.
Lovis P, Roggli E, Laybutt DR, Gattesco S, Yang J-Y, Widmann C, et al. Alterations in microRNA expression contribute to fatty acid-induced pancreatic β-cell dysfunction. Diabetes. 2008; 57 :2728-2738 - 140.
Han Y-B, Wang M-N, Li Q , Guo L, Yang Y-M, Li P-J, et al. MicroRNA-34a contributes to the protective effects of glucagon-like peptide-1 against lipotoxicity in INS-1 cells. Chinese Medical Journal. 2012; 125 :4202-4208 - 141.
Lin X, Guan H, Huang Z, Liu J, Li H, Wei G, et al. Downregulation of Bcl-2 expression by miR-34a mediates palmitate-induced Min6 cells apoptosis. Journal Diabetes Research. 2014; 2014 :258695 - 142.
Lu H, Hao L, Li S, Lin S, Lv L, Chen Y, et al. Elevated circulating stearic acid leads to a major lipotoxic effect on mouse pancreatic beta cells in hyperlipidaemia via a miR-34a-5p-mediated PERK/p53-dependent pathway. Diabetologia. 2016; 59 :1247-1257 - 143.
Kong X, Liu C-X, Wang G, Yang H, Yao X-M, Hua Q , et al. LncRNA LEGLTBC functions as a ceRNA to antagonize the effects of miR-34a on the downregulation of SIRT1 in glucolipotoxicity-induced INS-1 Beta cell oxidative stress and apoptosis. Oxidative Medicine and Cellular Longevity. 2019; 2019 :4010764 - 144.
Liu Y, Dong J, Ren B. MicroRNA-182-5p contributes to the protective effects of thrombospondin 1 against lipotoxicity in INS-1 cells. Experimental and Therapeutic Medicine. 2018; 16 :5272-5279 - 145.
Guo R, Yu Y, Zhang Y, Li Y, Chu X, Lu H, et al. Overexpression of miR-297b-5p protects against stearic acid-induced pancreatic β-cell apoptosis by targeting LATS2. American Journal of Physiology Metabolism. 2020; 318 :E430-E439 - 146.
Yu Y, Guo R, Zhang Y, Shi H, Sun H, Chu X, et al. miRNA-mRNA profile and regulatory network in stearic acid-treated β-cell dysfunction. The Journal of Endocrinology. 2020; 246 :13-27 - 147.
Li Y, Xu X, Liang Y, Liu S, Xiao H, Li F, et al. miR-375 enhances palmitate-induced lipoapoptosis in insulin-secreting NIT-1 cells by repressing myotrophin (V1) protein expression. International Journal of Clinical and Experimental Pathology. 2010; 3 :254-264 - 148.
Wang J, Lin Z, Yang Z, Liu X. lncRNA Eif4g2 improves palmitate-induced dysfunction of mouse β-cells via modulation of Nrf2 activation. Experimental Cell Research. 2020; 396 :112291 - 149.
Lovis P, Roggli E, Laybutt DR, Gattesco S, Yang JY, Widmann C, et al. Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes. 2008; 57 (10):2728-2736 - 150.
Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, et al. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010; 328 (5985):1566-1569 - 151.
Han YB, Wang MN, Li Q , Guo L, Yang YM, Li PJ, et al. MicroRNA-34a contributes to the protective effects of glucagon-like peptide-1 against lipotoxicity in INS-1 cells. Chinese Medical Journal. 2012; 125 (23):4202-4208 - 152.
Lu H, Hao L, Li S, Lin S, Lv L, Chen Y, et al. Elevated circulating stearic acid leads to a major lipotoxic effect on mouse pancreatic beta cells in hyperlipidaemia via a miR-34a-5p-mediated PERK/p53-dependent pathway. Diabetologia. 2016; 59 (6):1247-1257 - 153.
Kong X, Liu C, Wang G, Yang H, Yao X, Hua Q , et al. LncRNA LEGLTBC functions as a ceRNA to antagonize the effects of miR-34a on the downregulation of SIRT1 in glucolipotoxicity-induced INS-1 beta cell oxidative stress and apoptosis. Oxidative Medicine and Cellular Longevity. 2019; 2019 :4010764 - 154.
Lin X, Guan H, Huang Z, Liu J, Li H, Wei G, et al. Downregulation of Bcl-2 expression by miR-34a mediates palmitate-induced Min6 cells apoptosis. Journal Diabetes Research. 2014; 2014 :258695 - 155.
Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J, et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biology. 2006; 4 (2):e31 - 156.
Solomon JM, Pasupuleti R, Lei X, McDonagh T, Curtis R, DiStefano PS, et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Molecular and Cellular Biology. 2006; 26 (1):28-38 - 157.
Lee J-H, Song M-Y, Song E-K, Kim E-K, et al. Overexpression of SIRT1 protects pancreatic β-cells against cytokine toxicity by suppressing the nuclear factor-κB signaling pathway. Diabetes. 2009; 58 (2):344-351 - 158.
Wu L, Zhou L, Lu Y, Zhang J, Jian F, Liu Y, et al. Activation of SIRT1 protects pancreatic β-cells against palmitate-induced dysfunction. Biochimica et Biophysica Acta. 2012; 1822 (11):1815-1825 - 159.
Lovis P, Roggli E, Laybutt DR, Gattesco S, Yang JY, Widmann C, et al. Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes. 2008; 57 (10):2728-2736 - 160.
Liu Y, Dong J, Ren BO. MicroRNA-182-5p contributes to the protective effects of thrombospondin 1 against lipotoxicity in INS-1 cells. Experimental and Therapeutic Medicine. 2018; 16 (6):5272-5279 - 161.
Guo R, Yu Y, Zhang Y, Li Y, Chu X, Lu H, et al. Overexpression of miR-297b-5p protects against stearic acid-induced pancreatic β-cell apoptosis by targeting LATS2. American Journal of Physiology. Endocrinology and Metabolism. 2020; 318 (3):E430-E439 - 162.
Li Y, Xu X, Liang Y, Liu S, Xiao H, Li F, et al. miR-375 enhances palmitate-induced lipoapoptosis in insulin-secreting NIT-1 cells by repressing myotrophin (V1) protein expression. International Journal of Clinical and Experimental Pathology. 2010; 3 (3):254 - 163.
Paterson MR, Kriegel AJ. MiR-146a/b: A family with shared seeds and different roots. Physiological Genomics. 2017; 49 (4):243-252 - 164.
Fred RG, Bang-Berthelsen CH, Mandrup-Poulsen T, Grunnet LG, Welsh N. High glucose suppresses human islet insulin biosynthesis by inducing miR-133a leading to decreased polypyrimidine tract binding protein-expression. PLoS One. 2010; 5 (5):e10843 - 165.
Jiang W, Liu J, Dai Y, Zhou N, Ji C, Li X. MiR-146b attenuates high-fat diet-induced non-alcoholic steatohepatitis in mice. Journal of Gastroenterology and Hepatology. 2015; 30 (5):933-943 - 166.
Compagnoni C, Capelli R, Zelli V, Corrente A, Vecchiotti D, Flati I, et al. MiR-182-5p is upregulated in hepatic tissues from a diet-induced NAFLD/NASH/HCC C57BL/6J mouse model and modulates Cyld and Foxo1 expression. International Journal of Molecular Sciences. 2023; 24 :9239 - 167.
Kumar A, Ren Y, Sundaram K, Mu J, Sriwastva MK, Dryden GW, et al. miR-375 prevents high-fat diet-induced insulin resistance and obesity by targeting the aryl hydrocarbon receptor and bacterial tryptophanase (tnaA) gene. Theranostics. 2021; 11 (9):4061-4077