Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
Molecular docking study, a method used in drug discovery, is used to estimate the interactions between small molecules and macromolecules. Docking can be divided into rigid and flexible docking where local and global docking is the subclass in the flexible approach. Two important criteria in docking are search algorithms and scoring functions. The former assesses the fitness of ligand poses within the protein’s binding site, while the latter explores different ligands “conformations until the point at which the least energy convergence is achieved.” Three user-friendly global docking programs (AutoDock Vina, MOE, and DOCK6) are utilized to study ligand behaviors against Enterovirus A71 3C protease (3Cpro), which causes hand-foot-mouth disease in children. The results suggested that the DOCK6 gives the fastest output, and all of the ligands correctly bind to the active site of 3Cpro. Rupintrivir is a good candidate for serving as a positive control in all three tools for binding site identification because it shows broad resistance to viruses. In comparison to AutoDock Vina and MOE, DOCK6 exhibits superior conformational space search efficiency and speed across the three docking technologies used in our investigation. AutoDock Vina, however, is typically regarded as being more appropriate for novices.
School of Biotechnology, International University, Hochiminh City, Vietnam
Vietnam National University, Ho Chi Minh City, Vietnam
Tu H.T. Nguyen
School of Biotechnology, International University, Hochiminh City, Vietnam
Vietnam National University, Ho Chi Minh City, Vietnam
Phuc-Chau Do*
School of Biotechnology, International University, Hochiminh City, Vietnam
Vietnam National University, Ho Chi Minh City, Vietnam
*Address all correspondence to: dnpchau@hcmiu.edu.vn
1. Introduction
In the fields of molecular biology and pharmacology, studying how ligands interact with proteins is crucial because it plays a key role in comprehending basic biological processes and developing therapeutic treatments. Ligands can be tiny molecules, medications, or signaling molecules. Its function is to interact with proteins to influence a variety of biological processes (e.g., enzymatic activity, signal transmission, and gene expression). Scientists can gain crucial understanding of the fundamental processes of disease etiology and possible paths toward treatment by clarifying the molecular processes that underlie these interactions. Knowing how ligands interact with proteins makes it easier to predict drug toxicity, efficacy, and side effects, as well as to identify adverse reactions. This knowledge aids in optimizing drug candidates for better pharmacokinetic and bioavailability during drug discovery and development. Precision medicine techniques are made possible by the rational creation of innovative treatments that are specifically targeted at molecular targets or certain disease pathways, which is made possible by a thorough understanding of ligand-protein interactions. The investigation of ligand-protein interactions is fundamental to the advancement of pharmaceutical research, propelling breakthroughs in drug discovery and development, and to our comprehension of biology and disease.
Biological systems are incredibly flexible and adaptive; they can respond to a variety of stimuli and maintain equilibrium. The intricate nature of signaling pathways and transcriptional networks is demonstrated by the dynamic regulation of gene expression. Their intricacy is highlighted by the way that many elements interact, such as through protein-protein interactions and metabolic pathways. Understanding emergent behaviors is hampered by breaking down distinct components. Understanding the dynamic nature of biological systems requires the application of integrated viewpoints. Due to their transient and reversible character, as well as their dynamic conformational changes, protein-ligand interactions present difficulties in these kinds of systems. Interactions are further complicated by varying cellular architecture and subcellular localization patterns. Temperature, pH, and posttranslational changes are among the factors that make it difficult to measure interaction specificity precisely.
Drug development problems require multidisciplinary approaches that combine computational modeling and experimental methods, such as structural biology. In order to assist identify leads against macromolecules, virtual screening and molecular docking have become effective methods for high-throughput screening. Molecular docking is famous for identifying novel lead compounds rapidly against macromolecules since 1980s due to the significant growth in computing power and abundant availability of molecular structures [1]. These methods provide insights into molecular mechanisms and binding affinities by modeling interactions between proteins and ligands. To aid in the search for new drugs, docking studies forecast ligand selectivity and off-target effects. Understanding protein-ligand interactions in biological systems is improved by combining experimental techniques with docking simulations [1].
To perform docking, two approaches can be applied, namely, rigid and flexible docking. A report carried out by Xuan-Yu Meng and his colleagues defined a rigid docking where ligands and proteins maintain fixed conformations during docking process, neglecting the flexibility and dynamic movements of the molecules. Then, Fischer proposed a new approach, which is to measure the geometric fit between their shapes is directly comparable to their affinity, and is known as the lock-and-key theory [2]. Specifically, in the simplest rigid-body systems, to fit the protein’s binding site, ligands undergo the search in a six-dimensional space including rotational and translational changes via multiple interactions (e.g., van der Waals forces, coulombic interactions, and hydrogen bonds). As a result, docking poses are generated as the output, which will be ranked according to their scores, and the most favorable ones will be selected, which might imply the active site of that protein. However, the rigid body docking does not satisfy the experimental reality that molecules, especially proteins, are crystallized separately. The three-dimensional structure of protein primarily determined its function. This is defined by intricate macromolecules composed of linear chains of amino acids, is highly dynamic, and constantly changes conformations, which can significantly affect the overall shape complementary and interactions with ligands [3]. Moreover, assessment of accuracy in docking is preferable in bound complexed rather than unbound ones, indicating that treating proteins as entire rigid body is not certified [1]. The induced fit approach was first introduced by Koshland et al. in 1963. It suggested that proteins and ligands be flexible during docking to accommodate the dynamic nature of the system [4]. Partial or fully flexible methods, where local changes or entire protein conformations are allowed to vary, are included in flexible docking. While fully flexible docking incorporates side chain and backbone changes, partial flexibility concentrates on particular regions of the molecular structure. Through the investigation of various conformational changes, these techniques bolster the binding mode’s accuracy as well as predict binding affinity efficiently. Large conformational spaces can be efficiently explored with the help of technological advancements, such as graphics processing units (GPUs), which increase computing efficiency for extensive searches [1].
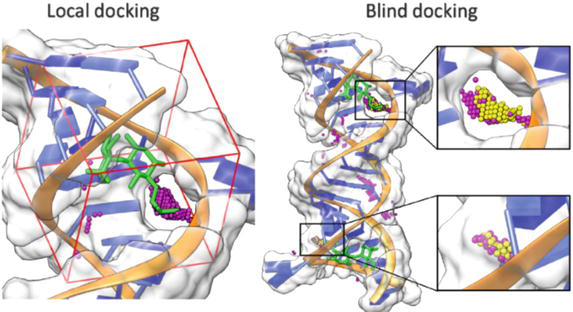
Global docking or blind docking refers to a method where a ligand (small molecules) will be docked to a whole surface of a target receptor (typically a protein) without knowing prior to the receptor’s binding site. To be more specific, the process searches for the entire conformational space of both ligand and receptor to determine the most favorable interaction and binding poses (Figure 1). While local docking focuses on flexibility at the specific site of docking, global docking allows all the rigid, flexibility, and orientation of both molecules, which might require more resource requirements and running time.
Figure 1.
Local docking and global docking of an aminoglycoside antibiotic, gentamicin (green), and the 16SrRNA a site of bacterial ribosome. Pink and yellow residues are the binding sites estimated by RLDOCK [5].
Good global docking results require careful consideration of numerous crucial parameters. The precision of the protein structure employed for docking is critical. Docking accuracy improves when both the protein and the ligand are properly prepared, including the removal of water molecules and the adjustment of protonation states. Furthermore, it is critical to select appropriate scoring systems that accurately measure binding affinity and to incorporate flexibility in both the protein and the ligand during simulation. Docking results can be validated against experimental data to ensure trustworthiness, while efficient sampling techniques and taking into account solvent effects enhance prediction accuracy. Recently, there have been more than 60 docking programs and servers appearing to be available for academic or commercial purposes [6, 7, 8]. The differences among these docking tools are a scoring function and a search algorithm in estimating the binding modes and affinities between proteins and ligands (Table 1). The capability of sufficiently sampling the degrees of freedom (rotation and translation) within the complex system to ensure the actual binding modes are counted has proven to be an effective characteristic of basic search algorithms. Meanwhile, scoring function’s role is to predict the binding free energies (ΔG bind) within a specific range, followed by the evaluation of the true binding modes and finally ranking poses based on predicted binding affinities.
Random Forest-based scoring function for Virtual Screening
Monte Carlo-Based Search, Lamarckian Genetic Algorithm, Iterated Local Search, Local Optimization
Free
Table 1.
Selected global docking tools.
2.1 Searching algorithm
Effective methods are essential for precise global docking, wherein the root mean square deviation (RMSD) is employed to measure ligand position in relation to experimental data. Due to computational limitations (considering just six degrees of freedom: rotation, translation, and conformational change), rigid body approximation was initially popular. However, it has constraints in describing particular interactions between ligands and binding sites. Certain programs maximize computing resources, improve interaction realism, and provide complete ligand variation while restricting the flexibility of proteins. Conformational alterations in ligands or receptors are referred to as flexibility in docking. The three types of flexible ligand docking methodologies are simulation, stochastic, and systematic searches.
The aim of the systematic search methods is to systematically cover the entire search area of possible ligand’s configurations. They are subsequently divided into three subcategories, namely, exhaustive, fragmentation, and conformational ensemble methods. The first method, exhaustive search, involves the rotation at 360° of all possible rotatable bonds in ligand at predefined intervals, but some challenges arise with combinatorial explosion, restricting its practicality in exclusively small and flexible ligands [6]. Meanwhile, fragmentation approach splits the ligands into modular parts, and one of its fragments is placed immovably in the receptor’s binding site, followed by adding other flexible parts to complete the ligand structure. Regarding conformational ensemble approach, despite minimizing the computational costs because pre-formed ligand conformations are used, generating such conformational ensembles requires additional tools and bioactive structures may sometimes be neglected.
In the context of stochastic algorithms, random alterations based on predefined probability functions are either accepted or rejected to seek ligand binding orientations and conformations. Hence, a global energy minimum has risen due to a variety of conformational space of ligands, as well as energy landscapes discovered. However, as a large search space is examined, this results in computational extensive. There are four basic categories: Genetic Algorithm (GA), Monte Carlo (MC), Ant Colony Optimization (ACO), and Tabu Search (TS). The first group-MC method was primarily applied in the minimization step in molecular dynamic (MD) simulations (e.g., GROMACS [9]). Its principle lies in applying the Boltzmann probability function to calculate the chance of accepting random fluctuations in thermodynamically accessible states, which decreases the chances of being trapped in local minima [10]. Secondly, GAs are derived from evolutionary programming algorithms (employ evolution-driven computational models) designed to determine the precise conformation to the global energy minimum. Meanwhile, TS method is an iterative optimization process and a variation of GA method [1]. It functions as a meta-heuristic method that can switch from one solution to another while keeping previously visited solutions in memory. However, TS separates itself from local search strategies by integrating a memory structure (Tabu list) to prevent the return of already guessed solutions and to encourage further exploration of new regions [11]. The principle of the last ACO type aims to apply pheromone trails for marking the low-energy ligand structures, which are altered in further repetitions to produce apparently low-energy conformations [12].
While systematic search focuses on thoroughly exploring the whole conformational space in a methodical manner and the objective of stochastic search is to select the optimal ligand conformations based on a predetermined probability function, simulation search involves solving Newton’s equations of motion. MD and pure energy minimization methods stand out as the two main types of this approach. The former considers forces and motions in the molecular behaviors over the time, but it is limited in locating rugged hypersurfaces and productively sampling different ligand structures within an appropriate simulation period. Meanwhile, the latter requires the most robust system possibly by reducing the potential energy of the system. It comprises diverse techniques such as direct searches, gradient methods, or conjugate-gradient methods (e.g., Fletcher-Reeves) and is often used complementarily with other docking algorithms [13, 14, 15].
Several docked structures with favorable surface complementarity are built on the topic of rigid body approximation and then reorganized in relation to free energy. The fast Fourier transform (FFT) correlation method explores systems docked structures via electrostatic interactions or a combination of electrostatic and solvation terms [6]. Other features, such as atomic interaction properties, relative shape, dissociation energies, polar Fourier interactions, and advanced software/hardware technologies to accelerate outputs, are incorporated in some FFT-based methods [16, 17]. Another algorithm utilized in rigid body docking, which aims to determine complementarily molecular shapes using such surface patches or 3D Zernike descriptors and thus aid better alignment of local geometric feature, is known as the geometric hashing (GH) method [18]. Likewise, shape matching (SM) algorithms evaluate geometric overlap in identifying optimal matching between ligands and receptor’s binding site [19]. DOCK tool – a pioneer in employing both SM and GH algorithms to recognize potential binding sites – sphere centers [20].
2.2 Scoring function
With the advance in computational hardware, the primary challenges have shifted from search-related algorithms to those involving scoring. Specifically, the challenges in ΔG bind’s prediction should be considered, such as complex physical interactions – entropy and enthalpy (e.g., interacting bonds, solvent molecules, entropy loss due to rotation and translation of ligands). This leads to the idea of being simplified and assumptive in scoring functions to decrease the complexity of the systems, as well as balancing the speed and accuracy are crucial to avoid errors. Ideally, an optimal search algorithm comes along with the best scoring function, which is contrary to current docking tools depending on the specific characteristics of receptor’s binding sites and studied ligands, so making the method become impractical [10]. In the realm of scoring functions, it is categorized into three basic groups, namely, physics-based, empirical, and knowledge-based functions to forecast how strong the interactions between proteins and ligands are (Table 2). Each class contributes to advancing our understanding of molecular docking and holds implications for various applications from lead optimization to virtual screening in drug discovery.
Aspects
Physical-based
Empirical
Knowledge-based
Principle
Enthalpy contribution is considered along with the sum of bonded and non-bonded interactions within the complex.
The addition of different energetic terms ranging from hydrogen bonds to hydrophobic effects is then multiplied with a coefficient derived from linear regression analysis.
The distance-dependent pairwise potentials are transformed from the atom pairs using the Boltzmann law after obtaining the occurrence frequencies of the protein-ligand complexes’ structural information in a database
Advantages
Evaluation the binding poses quickly, suitable for high-throughput docking. Alignment with modern force fields Obtaining from fundamental physics principles Computational cost-effective
Simple energy term treatment makes faster computation Preferable for virtual screening
Affordable for large scale database Robust to training set diversity Effective differing the binding modes. Extension to many-body interactions More variable in ligand flexibility
Challenges
Restriction in approximations Ignoring the long-range binding effects, solvent effects and entropic terms makes inaccurate for complex and real biological systems. Rigid body assumptions are challenges in ligand and protein variations
Not consider the involvement of solvents explicitly Multiple energy terms are double-counting issue. Limited description of flexibility Reliant on training set quality causes lack of generality
Reference state determination Issues relating to potential energy accuracy. Less sensitive to solvent effects Entropic energy makes difficulty in accurate calculation. Sensitive to ligand positions
Recent advances
Refinements in electrostatic interactions, ligand desolvation energies. Post-scoring docking applying the treatment of solvent molecules as continuum dielectric medium to accelerate force field models.
Introduction of continuous, non-linear terms and ligand poses’ variation in functions leads to the Hammerhead and Surflex functions. Machine-learning approaches in some empirical scoring functions
Iterative methods for improving accuracy in ITScore, including solvation effects and configurational entropy. Quantitative structure-activity relationships (QSAR)-Machine-learning (ML) approach based on distance-dependent atom pair occurrences.
Examples
Goldscore, AutoDock, Generalized Born/surface area model
Chemscore, Glidescore, ChemPLP
Drugscore, RF-score, ITscore.
Table 2.
Comparison among three basic scoring function [1, 10, 21, 22, 23].
In addition to the basic scoring functions mentioned above, many improved scoring approaches have been evaluated, but they are limited to a specific task [1]. Thus, scientists have raised up the idea of combining multiple scoring functions to tackle the errors from individual function, which is termed the consensus scoring method. In order words, best-docked pose of each compound will be reassessed using diverse scoring functions and the compounds that frequently ranked top scored across all functions are determined as potential candidates for further assays [10]. Take X-SCORE as an example of consensus scoring technique that combines various scoring functions or algorithms such as ChemScore, FlexX, DOCK-like, and GOLD-like [24]. Despite being concerned about error amplification, consensus scoring function still proves its capability in ranking effectiveness or false positives reduction. Another approach that is developed these recent years is to apply ML – the random forests, and the convolutional neutral network – in scoring function, principally introducing QSAR analysis to assess the interaction between proteins and ligands [1]. This method involves multiple properties of the complex such as atom pairs, geometric factors, and ligand-based characteristics to build models for evaluating the binding scores. The process involves two stages. Initially, ML automatically learns the known structure and binding information from a training dataset, which is similar to empirical scoring approach. Then, ML will be trained and builds its own mathematical formulas to rank and screen with better accuracy than other basic scoring approaches. Some relevant ML scoring functions are random forest-based score, and support vector machine score [25].
Above algorithms are generally applied for flexible ligand docking, which could be employed for flexible receptor docking, but obtaining the results at a reasonable timeframe is not yet addressed due to an increased in dimensionality and hence computationally intensive for a larger search space. One solution is to use intricated algorithms combined with simulations to effectively locate the expanded search space, but this still accounts for too much computational resources. Other well-known approaches, including rotamer libraries, soft-receptor modeling, and ensembles of protein conformations have been developed with the purpose of simplifying the system’s approximation, accompanied by a reduction in computational costs. The transition to using different global docking tools on a given system necessitates a thorough examination of each tool’s performance characteristics. This evaluation ought to consider a variety of factors such as the accuracy of anticipated binding poses and binding affinities, the productivity of computing resources employed, and the algorithm’s resistance to fluctuations in input parameters. Furthermore, assessing each tool’s virtues and drawbacks reveals important information about its underlying algorithms and approaches. Such extensive comparison studies allow researchers to make educated judgments about which docking tool is most suited to their individual research needs, enhancing the accuracy and usefulness of docking results to research objectives.
In this book chapter, we carried out a case study on the 3C protease (3Cpro) of Enterovirus (EV) – A71 that causes hand-foot-mouth disease (HFMD) in mostly children worldwide. A series of ligands, which have been assessed for their antiviral properties against different mutated EV-A71 3Cpro, were tested using three common docking tools AutoDock Vina [26, 27], MOE [28], and DOCK6 [20]. Subsequently, their docking poses were assessed to identify the active site based on their proximity to the binding site. Ultimately, a comparative analysis on different criteria (e.g., execution time, setting, process on multiple ligands) was also carried out for further conclusion.
HFMD, which is primarily caused by EV and Coxsackievirus (CV), belonging to Picornaviridae family, is a pediatric disease mostly targeting children under the age of five [29]. Reports found out that EV-A71 displays more severe consequences such as dysfunction in the central nervous system, cardiopulmonary failure, or vesicular eruptions on feet, hands, and oral mucosa than CV-A16 or other strains [30]. EV-A71 was initially discovered in 1957 in New Zealand and then isolated from an encephalitis female’s face in 1969 in California [30, 31]. Then, in the late twentieth century, many outbreaks of HFMD had been detected in all over the world, and most large expansions of this serious disease occurred in Asia-Pacific areas, which resulted in unattainable severity tracking. Unfortunately, there are limitations in available vaccines against these strains of EV-A71 and only symptomatic-supportive treatments are available [36]. Thus, these have raised an emergency alarm for thorough studies on different strains with existing inhibitors as well as leveraging for new potential candidates against EV-A71.
EV-A71, which attaches to host cells by different receptors, has a single-stranded RNA genome. This facilitates the uncoating and viral replication processes [32]. There are four categorized regions in EV-A71 genome denoting from P1–P3 in which P1 is responsible for VP1–VP4 while seven non-structural proteins belong to P2 and P3 groups. During the maturation of EV-A71, 3C protease (3Cpro) – one of the P3’s subclasses, which is a chymotrypsin-like fold structure, involves in cleaving the viral polyprotein at eight distinct sites [33]. Another role of 3Cpro is the cleavage of essential eukaryotic initiation factors, which results in preventing host cap-dependent translation [34]. A report carried out by Wang et al. revealed that the binding site of 3Cpro contains three key amino acid residues (His40, Glu71, and Cys147) which is responsible for cooperatively promoting substrate cleavage [35]. Additional to the catalytic important loop is the 3Cpro cleavage site involving such several main amino acid residues: Tyr122, Phe124, Glu126, Leu125, Leu127, Ser128, Thr142, His161, Ile162, Gly164, and Phe170 [36]. In our previous study, these two mentioned binding sites, which had been discovered earlier by Wang and Lu’ groups, are preferable for most inhibitors [35, 37]. Thus, these binding sites could be considered as the active site of 3Cpro proteins.
Furthermore, viral mutations are unavoidable, resulting in the formation of new viral variants with changed antigenic features, which may lead to immune evasion and reduced efficiency of current vaccines and treatments, as well as research chemicals. Furthermore, some mutations may give resistance to antiviral medications, reducing their effectiveness in treating viral infections. Thus, understanding how mutations affect the binding of ligands globally is crucial in order to cope with the viral evolution and variations in 3Cpro sequences.
In our study case, three models of 3Cpro proteins are selected, including the wild-type strain (PDB ID: 5C1U) and two mutant strains (PDB ID: 5GSW, 3QZQ). Besides, five prospective ligands are considered for docking. Rupintrivir, one of the synthetic drugs used to treat HFMD, has proven to pass the first phase of clinical test successfully. Meanwhile, two additional synthetic compounds – an isopropyl-substituted 4-iminooxazolidin-2-one moiety (FIOMC) and an α-keto amide derivative (8x), alongside with two naturally occurring compounds belonging to the flavonoid family (rutin and luteoloside) – are chosen due to their potent inhibitory effects on the 3Cpro proteins [36]. The active site of 3Cpro proteins was identified in our prior work and displayed in Figure 2 below. Hence, ligands residing at the peptide’s position are recognized as the promising antiviral substances against 3Cpro proteins.
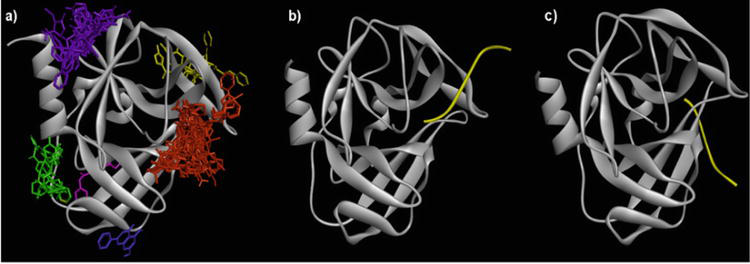
Figure 2.
Binding sites of 3Cpro protein (PDB ID: 5C1U) with a) six clusters generated when 5C1U docked by AutoDock Vina with 21 control candidates, b) experimental 3SJ9, and c) 3SJK between 3Cpro (gray color) with its targeted peptide (yellow color) [36].
3.1 AutoDock Vina
AutoDock Vina is widely used due to its user-friendly interface for molecular docking and virtual screening. In 2009, Dr. Oleg Trott and his team at molecular graphics lab at the Scripps Research Institute, USA, developed this tool from AutoDock 4.0 (AD4.0) [38]. Then, AutoDock Vina 1.2.0 was reported by Jerome et al. with additional support in the treatment of water molecule explicitly and multiple ligands docking (cite). Currently, a newest version of AutoDock Vina 1.2.5 is available at center for computational structural biology website (https://vina.scripps.edu/), belonging to Dr. Oleg’s lab. Alongside with other docking engines in AutoDock Suite package [38, 39, 40], AutoDock Vina recognizably outperforms with AD4.0 for being 100 times faster, as well as significantly reduce the running times via exploiting multiple CPUs or CPU cores. Moreover, its accuracy in binding mode predictions is also upgraded by employing efficient search algorithms and assessing fewer scoring functions. In addition, some parameters, that have proven to be well-performed in diverse scenarios, are automatically set. This allows the software to be more accessible and straightforward for either experts or nonexperts in the field.
AutoDock Vina’s configuration file has adjustable options that accommodate different system sizes and docking goals. Coordinates and box size are adjusted in global docking scenarios to sufficiently encompass the target region. Docking performance can be adjusted with flexibility using parameters, such as exhaustiveness value (default = 8) and energy range (default = 4 kcal/mol). While more exhaustiveness improves conformational exploration, it also increases computing times. Energy range modification impacts docking diversity and fidelity, as well as posture selection strictness. Rupintrivir’s broad antiviral spectrum is shown by the AutoDock Vina results, which show preferential ligand binding to active sites (Figure 3).
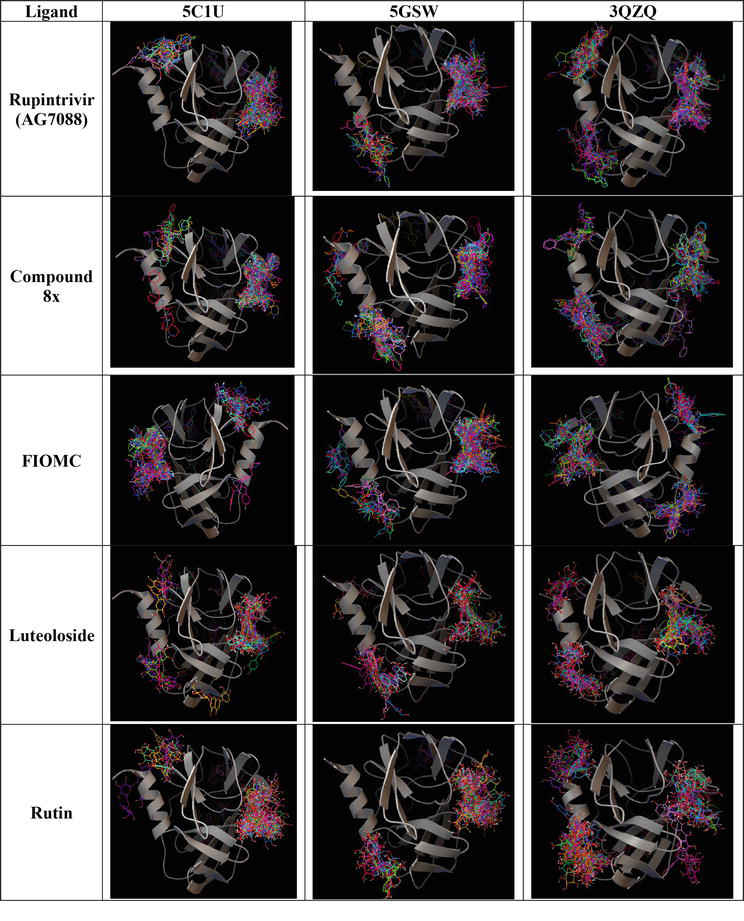
Figure 3.
Binding poses formed by AutoDock Vina between control ligands (line presentation) on three 3Cpro models (ribbon presentation).
As mentioned above, scoring functions and search algorithms are the major features in assessing the performance of a docking software. In the latest AutoDock Vina’s version, it includes both AD4.2 and Vina scoring functions. This is because Vina lacks some parameters that are present in a physics-based model – AD4.0 such as directional hydrogen bond potentials, electrostatic, and solvation [26]. Additionally, deriving from both knowledge-based and empirical scoring functions, the ligand conformational space and its interactions with receptors are assessed thoroughly. This effectively heightens the system’s accuracy. Pertaining to search algorithms, even though various stochastic global optimization approaches have been exploited in the evolution of Vina. Thus, a similar method developed by Abagyan et al. is known as Iterated Local Search global optimizer [27]. Thanks to the ability in using multicore CPUs in Vina, this intensive iteration can be done efficiently in an acceptable timeframe.
However, this is contradicted to what we researched earlier using the exhaustiveness value of 256 that FIOMC displays the best antiviral characteristic across the broad range of virus strains. Besides, the number of binding sites in the wild-type strain is more condensed than in the others. With regard to two mutant proteins, 5GSW is mutated from an asparagine to serine at position 69, while the glutamic acid is mutated to aspartic acid at position 71 in 3QZQ. It is noticeable that the ligands tend to be more dispersed than in the case of 5GSW, which agrees to our previous study [36]. Regarding the average time for one 3Cpro model with five ligands, it is estimated about 2 hours for a triplicate run with a default exhaustiveness value. This is because AutoDock Vina allows for maximum of 20 modes per single docking performance, so each pair of protein ligand is docked three times with different random seed numbers in order to produce more binding poses for a better evaluation, as well as reproducibility.
3.2 DOCK6
DOCK was initially built by Sheridan et al., implementing geometric matching and distance-based comparisons to find the most favorable binding poses in 1986 [41]. Since then, it has been remarkably evolving with additional essential features, such as Hawkins-Cramer-Truhlar Generalized Born/Surface Area (GB/SA) solvation scoring and Poisson–Boltzmann/Surface Area (PB/SA) solvation scoring through OpenEye’s Zap Library. This leads to the release of the newest version of DOCK6 developed by Therese Lang et al. and his collaborators in 2009 [20, 42, 43, 44, 45]. This new version has more functions, such as receptor flexibility consideration and conjugate-gradient minimization via accessing the NAB library. In addition, more scoring functions are also introduced in DOCK6 as described in the web manual. Regarding sampling methods, DOCK6 implements several advanced algorithms to study specific interactions between ligands and proteins (e.g., anchor-and-grow, de novo, GA, and Hungarian algorithm) [46].
For the purpose of selecting spheres and generating DMS (Database Management System) files to store structure management, DOCK6 needs UCSF Chimera. Docking box construction is dependent on selected spheres, as seen by the UCSF Chimera sphere table. When global docking conditions are met, the protein is covered entirely by the “0” option. For steric overlaps and fast score evaluation, grid creation uses bump and scoring grids, which are modifiable based on the task at hand. Every scoring algorithm operates independently, with results stored in separate files. Ligand energy minimization – which can be adjusted using DOCK6 scripts – is essential to lessen atomic collisions. For best results, flexible ligand docking requires modifying parameters, such as number_scored_conformers and rank_ligands [46]. Long docking periods and resource demands result from the search algorithm’s limited ability to manually customize boxes while effectively identifying 3Cpro binding locations.
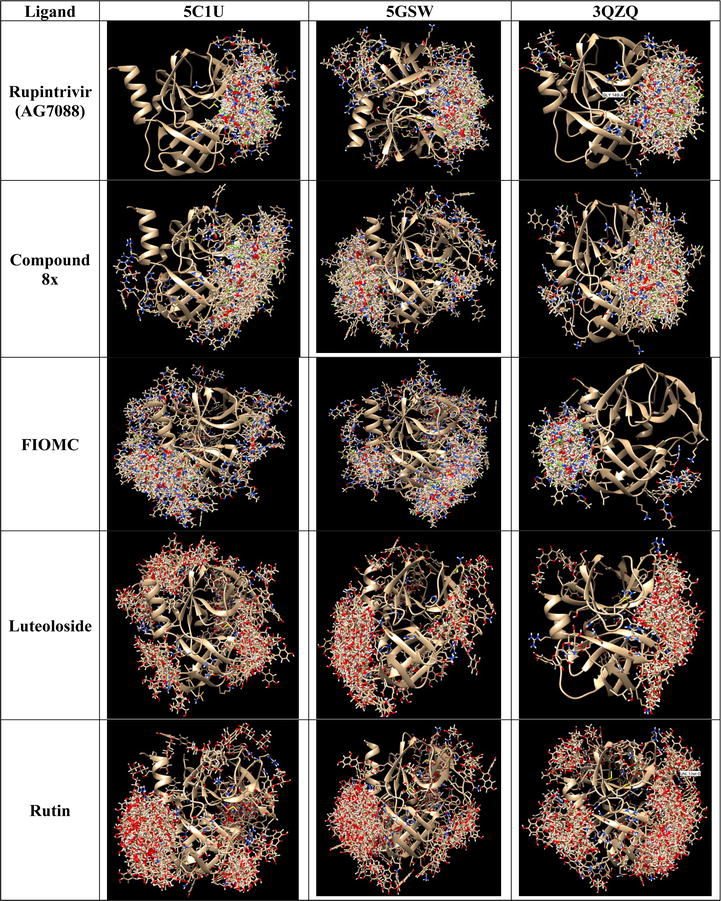
The docking tasks in DOCK6 are faster compared to Vina, even when utilizing default settings. The process takes approximately less than 30 minutes for a single system, comprising one protein model and five ligands. Although DOCK6 is implemented with multiple scoring functions, a default grid score is used in our study case. Furthermore, DOCK6 utilizes an anchor number to determine the generated modes of ligands, making a single run adequate for assessment. Looking at the docking results in Figure 4, it is observable that all of ligands interact at the active site of 3Cpro protein within three studied models. As a result, DOCK6 is more efficient in defining the active site of the targeted protein via evaluating the spheres covered the favorable binding sites in comparison to AutoDock Vina. Furthermore, we notice that rupintrivir binds more accurately at the active site than other ligands, which is similar to the above AutoDock Vina’ results. Overall, there is not much different among these models when using DOCK6 for docking.
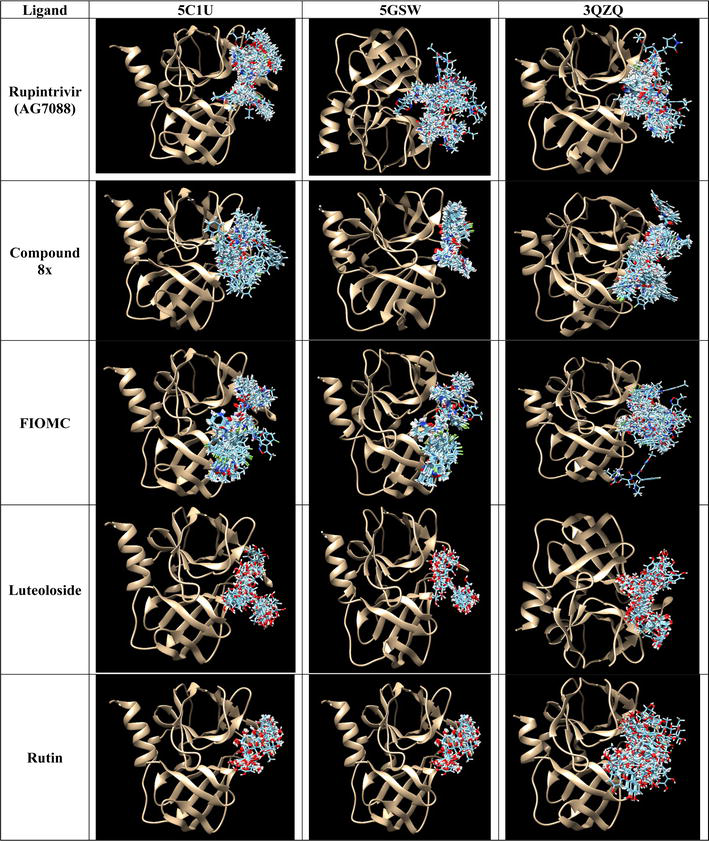
Figure 4.
Binding poses formed by DOCK6 between control ligands (line presentation) on three 3Cpro models (ribbon presentation).
3.3 MOE
Paul Labute, the President and CEO of Chemical Computing Group, which was established in 1994, is located in Montreal, Quebec, Canada (cite web). The team’s goal is to generate innovative tools that not only confront the existing norms but also transform scientific approaches. Thus, the launch of MOE demonstrates the company’s dedication to the generation of cutting-edge solutions that remarkably assist in the development of computational chemistry research area. Its operation can carry out in multiple systems (https://www.chemcomp.com/Products.htm).
In regard to the docking time, it is noticeable that MOE requires the longest time (≥ 1.5 hours) for a pair of protein ligand to produce 100 binding poses. When assessing the percentage of poses at the active site, Rupintrivir ranks the best potent compound in interacting with 3Cpro proteins at that site, while FIOMC and rutin bind at the opposite site (Figure 5).
Figure 5.
Binding poses formed by MOE between control ligands (line presentation) on three 3Cpro models (ribbon presentation).
Moreover, ligands tend to scatter in all cases due to default search algorithm setting. Apart from the wild-type strain, two other mutant strains show opposite results compared to two previous tools. In particular, ligands distributed more neatly in 3QZQ case, compared to 5GSW, where three out of five ligands do not bind at the active site. This is contrast to our earlier study that mutation in 5GSW did not show many impacts on the binding of ligand rather than that of 3QZQ [36].
The rational in choosing the parameter depends on the purpose of docking. Among them, the pharmacophore only supports the commercial version. Meanwhile, the other scoring functions can be changed depending on the interested interactions (e.g., Atom-Sphere Exclusion, Affinity ΔG scoring, Alpha Hydrogen Bond scoring, and GBVI/WSA), alongside with several crucial factors, including atom-atom interactions, geometric fit, H-bonding, and energy contribution. In our study case, default settings are employed for all systems that use triangle matcher in placement; London ΔG in rescoring, and no refinement is set.
In conclusion, this study examined several scoring functions and search algorithms in docking software, as well as the field of docking approaches, identifying local and global docking. This discussion centered on global docking, more especially on complete and partial flexible docking. In order to identify protein binding sites, three popular docking tools – AutoDock Vina, MOE, and DOCK6 – were evaluated using a case study containing the EV-A71 3C provirus that causes HFMD. Interestingly, DOCK6 outperformed the others in displaying concentrated ligand poses in the 5C1U protein’s active regions, while AutoDock Vina provided information about how mutations affect ligand interactions.
In order to evaluate the quality of docking results and support the logical design of new therapies, post-docking analysis requires accurate binding energy prediction. However, this is not possible at this level of global docking because the main goal is to evaluate receptor binding locations. These binding energy values must be ascertained by additional analyses, such as MD simulation or calculations utilizing the molecular mechanics (MM)/PBSA or MM/GBSA methods. Meanwhile, by utilizing the AMBER score and a basic MM-GB/SA method, DOCK6 assesses binding energy.
Regarding the study case, rupintrivir (AG7088) consistently showed excellent inhibitory activity across both wild-type and mutant 3Cpro animals, establishing it as a reliable positive control despite the variation in the data. All ligands docked at the active site of 3Cpro proteins in DOCK6 with the fastest time in less than 5 minutes for each pair of protein ligand when 100 poses are generated. Meanwhile, MOE does not appear to support a comprehensive evaluation of a putative binding site, depending on the quantity of produced ligands posed at various binding sites. Furthermore, the likelihood that ligands will bind to 3Cpro proteins is affected by mutations in either 5GSW or 3QZQ. As a result, the docking findings produced by MOE show something different from what we saw in AutoDock Vina’s previous studies (Table 3).
System requirement
Advantages
Disadvantages
AutoDock Vina
Support for 64-bit Linux, Mac OS X 10.15 (Catalina) or later versions, SGI IRIX, and Microsoft Windows
Hardware: ≥ 4GB RAM
Disk space: ≥ 100 MB
Compatible with CPUs only
Simple command–line interface: easy use
MGLtools is perquisite for preparing ligand and receptor docking inputs
Less settings: grid box, exhaustiveness value
Perform docking for multiple protein -ligand systems at once with a Perl script
An open–source software with widely used globally promotes scientific cooperation and continual progress to address any difficulties
Flexible ligand docking options
Academic free
Ligands and proteins in must PDBQT format, which is not ready to use
Higher exhaustiveness value requires extensive computational resources.
Complicated systems require longer timescale.
Limited side chains of receptors flexibility.
Solvation models by hydrated docking, prefer for AutoDock4 rather than Vina
DOCK6
Support for Linux or Windows (Unix-like environment – Cygwin); Mac OS with GNU compilers and configuration files
Hardware: ≥ 4GB RAM
Disk space: ≥ 200 MB
CPUs for basic dockings and GPU compiler
Command-line interface: easy to use
Fast docking process for a default setting
Efficient search algorithms
Decrease sampling
Various scoring functions help reducing scoring failure
Solvation models and flexible receptors docking: tackled by AMBER score
Flexible ligand docking option
Available free
Limited in the manual customization of the box
Multiple parameter settings to evaluate
Require intensive computational resources for AMBER score docking
Need to use with other programs
MOE
Support in Windows, Linux, MacOS
Hardware: ≥ 4GB RAM
Disk space: ≥ 1GB
Potential energy computations: 8 CPUs requirement
Command-line and graphical interface unit: easy to use
Allow multi-tasking with command line
Can customize wall constraint for each protein
Unlimited number of poses per experiment
Support many file formats
Flexibility of receptors with several limited side chains
Flexibility of ligands with ≤ 10 rotatable bonds
Increases adaptability by several search strategies
Maintains original geometry to improve search efficiency and precision
Uncommon command language SVL
More time is needed for a complex system
More pharmacophore is required for ≥ 10 movable bonds
Solvation models handling for the commercial version solely
Require ring structures to be generated in advance
Commercial tool
Table 3.
An analysis of the three docking systems’ respective system requirements, benefits, and drawbacks.
Default settings are frequently used since developers propose them for the purpose of tool comparison. However, as many possibilities have been covered previously, it is crucial to choose settings that are appropriate for the research aims in order to produce the best results. To guarantee a more complete understanding of protein-ligand interactions in the quest for drug discovery and design, researchers may investigate other technologies in the future to improve and build upon current discoveries. Efficient molecular docking for big chemical libraries is made possible by developments in computational resources, such as cloud and distributed computing. Processing operations on GPUs are accelerated, particularly when large conformational landscapes are being explored. Advancements such as cryo-electron microscopy offer abundant three-dimensional information, augmenting understanding of protein structure and maybe boosting docking accuracy. Pose prediction accuracy is increased by methods, including interaction fingerprints, better MM/PBSA analysis, and pharmacophore modeling integration. The development of drug delivery devices is aided in comprehending complex dynamics and binding processes by combining docking with MD simulations. Enhancing docking technologies, such as optimizing metaheuristics for accurate pose prediction, is possible with AI, especially with deep learning and machine learning. By iteratively retraining using informative data, active learning minimizes the need for screening resources while optimizing prediction reliability. Support vector machines and random forests are examples of machine-learning algorithms that outperform traditional score methods in terms of accuracy, which helps in ligand ranking. Drug research and design are improved by deep learning methods, such as convolutional and recurrent neural networks, which extract spatial and sequential information from 3D structures. The combination of AI and ML has the potential to transform docking tools, speed up the hunt for novel therapies, and enable more efficient drug discovery and design than is possible with current methods.
1.Stanzione F, Giangreco I, Cole JC. Use of molecular docking computational tools in drug discovery. In: Progress in Medicinal Chemistry. Netherlands: Elsevier B.V; 2021. pp. 273-343
2.Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: A powerful approach for structure-based drug discovery. Current Computer-Aided Drug Design. 2011;7(2):146-157
3.Li D, Ji B. Protein conformational transitions coupling with ligand interactions: Simulations from molecules to medicine. Medicine in Novel Technology and Devices. 2019;3:100026
4.Koshland DE. Correlation of structure an function in enzyme actio theoretical and experimental tools are leading correlations between enzyme structure and function. Science. 1963;142:1533-1541
5.Zhou Y, Jiang Y, Chen SJ. RNA–ligand molecular docking: Advances and challenges. Wiley Interdisciplinary Reviews: Computational Molecular Science. 2022;12:e1571
6.Pagadala NS, Syed K, Tuszynski J. Software for molecular docking: A review. Biophysical Reviews. 2017;9:91-102
7.Kellenberger E, Rodrigo J, Muller P, Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins: Structure, Function and Genetics. 2004;57(2):225-242
8.Wang Z, Sun H, Yao X, Li D, Xu L, Li Y, et al. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Physical Chemistry Chemical Physics. 2016;18(18):12964-12975
9.Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845-854
10.Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: Current status and future challenges. Proteins: Structure, Function and Genetics. 2006;65:15-26
11.Dias R, Filgueira De Azevedo W. Molecular docking algorithms. Current Drug Targets. 2008;9:1040-1047
12.Korb O. Efficient ant colony optimization algorithms for structure- and ligand-based drug design. Chemistry Central Journal. 2009;3(S1):O10
13.Abagyan R, Totrov M, Kuznetsov D. ICM-A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation docking and global energy optimization. Journal of Computational Chemistry. 1994;15(5):994
14.Ewing TJ, Makino S, Geoffrey Skillman A, Kuntz ID. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. Journal of Computer-Aided Molecular Design. 2001;15:411-428
15.Trosset JY, Scheraga HA. PRODOCK: Software package for protein modeling and docking. Journal of Computational Chemistry. 1999;20(4):412427
16.Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014;30(12):1771-1773
17.Macindoe G, Mavridis L, Venkatraman V, Devignes MD, Ritchie DW. HexServer: An FFT-based protein docking server powered by graphics processors. Nucleic Acids Research. 2010;38(SUPPL. 2):W445-W449
18.Venkatraman V, Yang YD, Sael L, Kihara D. Protein-protein docking using region-based 3D Zernike descriptors. BMC Bioinformatics. 2009;10:407
19.Huang SY, Zou X. Advances and challenges in protein-ligand docking. International Journal of Molecular Sciences. 2010;11:3016-3034
20.Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT, et al. DOCK 6: Impact of new features and current docking performance. Journal of Computational Chemistry. 2015;36(15):1132-1156
21.Verdonk ML, Ludlow RF, Giangreco I, Rathi PC. Protein-ligand informatics force field (PLIff): Toward a fully knowledge driven “force field” for biomolecular interactions. Journal of Medicinal Chemistry. 2016;59(14):6891-6902
22.Jain AN. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. Journal of Medicinal Chemistry [Internet]. 2003;46(4):499-511. DOI: 10.1021/jm020406h
23.Huang SY, Grinter SZ, Zou X. Scoring functions and their evaluation methods for protein-ligand docking: Recent advances and future directions. Physical Chemistry Chemical Physics. 2010;12(40):12899-12908
24.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nature Reviews Drug Discovery. 2004;3:935-949
25.Li J, Fu A, Zhang L. An overview of scoring functions used for protein–ligand interactions in molecular docking. Interdisciplinary Sciences – Computational Life Sciences. 2019;11:320-328
26.Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and python bindings. Journal of Chemical Information and Modeling. 2021;61(8):3891-3898
27.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry. 2009;31:455-461
28.Molecular Operating Environment (MOE). [Internet]. 2022.02 Chemical Computing Group ULC 910-1010 Sherbrooke St. W., Montreal, QC, Canada. 2024. Available from: https://www.chemcomp.com/ [Accessed: February 27, 2024]
29.Koh WM, Badaruddin H, La H, Chen MIC, Cook AR. Severity and burden of hand, foot and mouth disease in Asia: A modelling study. BMJ Global Health. 2018;3(1):e000442
30.Ong KC, Wong KT. Understanding enterovirus 71 neuropathogenesis and its impact on other neurotropic enteroviruses. Brain Pathology. 2015;25(5):614-624
31.Nayak G, Bhuyan SK Bhuyan R, Sahu A, Kar D, Kuanar A. Global emergence of enterovirus 71: A systematic review. Beni-Suef University Journal of Basic and Applied Sciences. 2022;11:78, 11 pages
32.Hu K, Onintsoa Diarimalala R, Yao C, Li H, Wei Y. EV-A71 mechanism of entry: Receptors/Co-receptors, related pathways and inhibitors. Viruses. 2023;15:785
33.Cui S, Wang J, Fan T, Qin B, Guo L, Lei X, et al. Crystal structure of human enterovirus 71 3C protease. Journal of Molecular Biology. 2011;408(3):449-461
34.Sun D, Chen S, Cheng A, Wang M. Roles of the picornaviral 3c proteinase in the viral life cycle and host cells. Viruses. 2016;8(3):82
35.Wang J, Fan T, Yao X, Wu Z, Guo L, Lei X, et al. Crystal structures of enterovirus 71 3C protease complexed with rupintrivir reveal the roles of catalytically important residues. Journal of Virology. 2011;85(19):10021-10030
36.Le TTV, Do PC. Molecular docking study of various enterovirus—A71 3C protease proteins and their potential inhibitors. Frontiers in Microbiology. 2022;13:987801
37.Lu G, Qi J, Chen Z, Xu X, Gao F, Lin D, et al. Enterovirus 71 and coxsackievirus A16 3C proteases: Binding to rupintrivir and their substrates and anti-hand, foot, and mouth disease virus drug design. Journal of Virology. 2011;85(19):10319-10331
38.Morris GM, Ruth H, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009;30(16):2785-2791
39.Santos-Martins D, Solis-Vasquez L, Tillack AF, Sanner MF, Koch A, Forli S. Accelerating AutoDock 4 with GPUs and gradient-based local search. Journal of Chemical Theory and Computation. 2021;17(2):1060-1073
40.Ravindranath PA, Forli S, Goodsell DS, Olson AJ, Sanner MF. AutoDockFR: Advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Computational Biology. 2015;11(12):e1004586
41.DesJarlais RL, Sheridan RP, Dixon JS, Kuntz ID, Venkataraghavan R. Docking flexible ligands to macromolecular receptors by molecular shape. Journal of Medicinal Chemistry. 1986;29(11):2149-2153
42.Prentis LE, Singleton CD, Bickel JD, Allen WJ, Rizzo RC. A molecular evolution algorithm for ligand design in DOCK. Journal of Computational Chemistry. 2022;43(29):1942-1963
43.Brozell SR, Mukherjee S, Balius TE, Roe DR, Case DA, Rizzo RC. Evaluation of DOCK 6 as a pose generation and database enrichment tool. Journal of Computer-Aided Molecular Design. 2012;26:749-773
44.Allen WJ, Fochtman BC, Balius TE, Rizzo RC. Customizable de novo design strategies for DOCK: Application to HIVgp41 and other therapeutic targets. Journal of Computational Chemistry. 2017;38(30):2641-2663
45.Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, et al. DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA. 2009;15(6):1219-1230
46.William Joseph Allen (TACC), Trent Balius (FNLCR), John Bickel (SUNY-Stony Brook), Brock Boysan (SUNY-Stony Brook), Scott Brozell (Rutgers University), Chris Corbo (SUNY-Stony Brook), et al. DOCK 6.11 Users Manual [Internet]. 2023. Available from: https://dock.compbio.ucsf.edu/DOCK_6/dock6_manual.htm [Accessed: February 27, 2024]
Written By
Vy T.T. Le, Tu H.T. Nguyen and Phuc-Chau Do
Submitted: 29 February 2024Reviewed: 13 March 2024Published: 26 April 2024