
Abstract
Neurological disorders represent a complex spectrum of conditions, ranging from neurodegenerative diseases to acute injuries, each posing unique challenges to treatment. This chapter delves into the emerging role of the NRF2 transcription factor as a molecular beacon with therapeutic implications in the realm of neurological disorders. NRF2, a master regulator of cellular defense mechanisms, orchestrates antioxidant responses and mitigates oxidative stress—processes intricately linked to neuroprotection. The chapter explores the intricate interplay between NRF2 and neurological pathologies, emphasizing its influence on the progression of conditions such as Alzheimer’s, Parkinson’s, and ischemic stroke. By dissecting the molecular pathways through which NRF2 modulates inflammation, oxidative damage, and apoptosis in the nervous system, we gain insights into potential therapeutic strategies. Exciting research on NRF2 activators, both natural and synthetic, offers promising avenues for drug development. Furthermore, the chapter navigates through preclinical and clinical studies, highlighting the potential of NRF2-targeted interventions in preserving neuronal function and promoting recovery. As we unravel the molecular intricacies of NRF2 in neurological contexts, this chapter serves as a guide to understanding its therapeutic potential, paving the way for innovative strategies aimed at mitigating the burden of neurological disorders.
Keywords
- NRF2 transcription factor
- neurological disorders
- oxidative stress
- therapeutic implications
- molecular pathways
1. Introduction
The Nuclear factor erythroid 2-related factor 2 (NRF2) is a known transcription factor encoded in humans by the NFE2L2 gene that plays an important role in cellular protection against oxidative stress (OS) by activating genes that protect cells. Oxidative stress (OS) is a condition of an imbalance of production of reactive oxygen species (ROS) and nitrogen species (RNS) ratio and the ability of cells to protect them with antioxidants. Excessive production of reactive oxygen species (ROS) leads to changes in important cellular macromolecules, including proteins, DNA, and lipids. Many studies have demonstrated an association between oxidative stress (OS) and various neurodegenerative diseases [1, 2], including Alzheimer’s disease (AD) [3], Huntington’s disease (HD) [4], Parkinson’s disease (PD) [5], multiple sclerosis (MS) [6], and amyotrophic lateral sclerosis (ALS) [7]. ROS/RNS are generated from different sources within cellular compartments, either from normal physiological processes or because of exposure to damage or disease in the environment [8]. Superoxide (O2−) is the most severe form of reactive oxygen species (ROS) and is produced by the single electron reduction of O2 in mitochondria. Superoxide can also be produced by a group of NADPH oxidases (NOX) using oxygen and NADPH as substrates, where superoxide is rapidly removed [9]. Hydroxyl radicals (•OH) are important by-products of mitochondrial oxidative phosphorylation. These chemicals exhibit instability, high reactivity, and produce more reactive aldehydes through the process of membrane lipid peroxidation (LP), ultimately leading to cell death. Additionally, superoxide dismutase 1 (SOD1) rapidly produces reactive oxygen species (ROS) hydrogen peroxide (H2O2) in the cytoplasm, while extracellular H2O2 is produced by extracellular superoxide dismutase 3 (SOD3). Consequently, H2O2 may be a product of beta-oxidation of fatty acids by cytochrome P450 enzymes. When H2O2 is exposed to Fe2+ or Cu+ (Fenton reaction), it can transform into hydroxyl radicals, although they are stable and less active [10]. The main reactive nitrogen species (RNS) is ONOO−, which rapidly decomposes into HO•, nitrogen dioxide radicals (NO2•), and nitro cations (NO2+) [11]. This drug is neurotoxic. Targeted therapy for neurodegenerative diseases involves addressing the various mechanisms involved in the formation of the operating system (OS) and imbalances in the endogenous cellular protection of neuronal cells [12, 13].
NRF2 plays an important role in the development and activation of blood cells, a group of enzymes responsible for metabolizing drugs [14]. Proteins whose expression increases as a result of NRF2 signaling include heme oxygenase-1 (HO-1), SOD1, catalase, and enzymes involved in glutathione (GSH) metabolism, such as glutathione S-transferase (GST), glutathione cysteine. Ligase modifier subunit and glutathione cysteine ligase catalytic subunit (GCLC) [15, 16].
The ability of NRF2 to regulate central metabolism and mitochondrial function suggests that NRF2 activation is a good and effective strategy for the treatment of neurodegenerative diseases [17]. This review explores the importance of oxidative stress in neurodegenerative diseases and the benefits of targeting the NRF2 pathway as an antioxidant and cytoprotective response. It also proposes new therapies designed to treat oxidative damage and neuroinflammation by activating the NRF2 signaling pathway.
2. Recent findings in specific disorders
2.1 Alzheimer’s disease
Forgetfulness and cognitive impairment are hallmarks of Alzheimer’s, the most common type of dementia. AD is an age related neurodegenerative disease. Without changes in treatment, the number of AD patients in the United States is expected to increase from currently more than 5 million to approximately 13.8 million by 2050 [18]. AD is characterized by a variety of diseases, including brain atrophy due to neuronal and synaptic loss, senile plaques composed primarily of fibrillar amyloid (Aβ) peptide, and neuronal plaques composed of hyperphosphorylated tau, a cytoskeletal protein fiber tangle (NFT). β- and γ-secretase cleave amyloid precursor protein (APP) to form Aβ. Small oligomers of self-assembled peptides are thought to contribute to brain damage in AD [19]. Aging is an important risk factor for AD, and although most cases of AD are unrelated, a small proportion have a familial basis. Early onset familial disease is associated with mutations in APP, presenilin 1 (PS1) and presenilin 2 [20]. In addition to plaques and NFTs, AD brains also feature reactive gliosis, mitochondrial dysfunction, and oxidative damage to proteins and lipids [21, 36].
2.1.1 NRF2-related genes in AD cells
Early studies have shown that the NAD(P)H:quinone oxidoreductase 1 (NQO1) content in AD tissues is higher than in control tissue [22, 23, 24] A. The same is true for both was also observed for oxygenase 1 (HO-1), glutathione reductase, and glutathione peroxidase [25, 26, 27]. Additionally, AD brains have higher levels of p62, HO-1, and glutamate cysteine ligase modifier subunit (GCLM). In addition, Tanji and colleagues [28] found co-localization of Keap1 with NFTs, evidence of interaction between them, and the presence of both p62 and Keap1 in both the soluble and insoluble protein fractions in AD cells. According to a study examining NRF2 by immunohistochemistry, NRF2 was detected in the cytoplasm and nucleus of healthy hippocampal neurons, but only in the cytoplasm in AD [29]. This suggests that NRF2 activation is lower in AD cells than in previous studies. In fact, studies have shown that some of the same NRF2-dependent genes described previously are downregulated or unchanged in AD [30, 31, 32]. These conflicting results are not surprising and may have much to do with the stage of disease at the time the tissues were collected. Additionally, according to our unpublished findings, most NRF2-dependent genes and proteins and glutathione (GSH) levels in the brain and spinal cord are NRF2-dependent. It is not reduced in −/− mice (Figure 1) [33, 34].
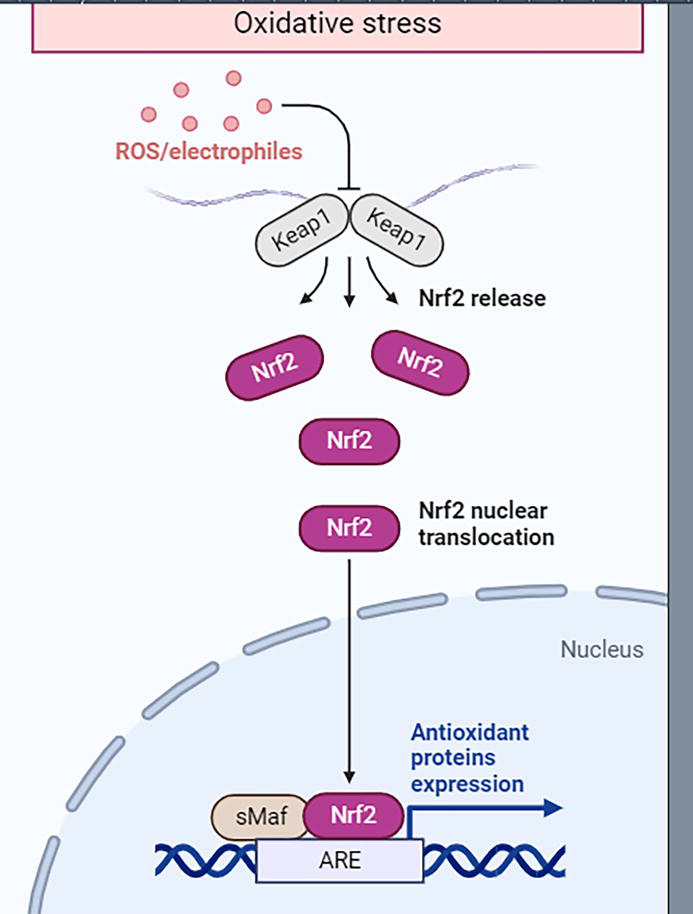
Figure 1.
The Keap1-NRF2 pathway is a signaling pathway involved in cellular responses to oxidative stress. The regulatory protein Keap1 regulates the activity of the transcription factor NRF2. Under conditions of oxidative stress, NRF2 is released from Keap1 and translocates to the nucleus, where it activates the expression of genes encoding antioxidant enzymes. This pathway plays an important role in cell defense against oxidative damage and redox maintenance of homeostasis.
2.2 Parkinson’s disease
Parkinson’s disease (PD) is the most common neurodegenerative disease. It is characterized by a variety of non-motor neuropsychiatric problems including motor symptoms such as tremor, bradykinesia, rigidity, and postural instability [35]. Loss of dopaminergic neurons in the substantia nigra pars compacta (SN), which reduces the number of dopamine nerve terminals projecting to the striatum, is one of the symptoms of Parkinson’s disease (PD). Another notable feature is the formation of intracellular inclusions or Lewy bodies (LB) [36, 37, 38] composed of ubiquitin-linked α-synuclein (SYN). Similar to AD, PD is generally rare, with familial onset accounting for approximately 15% of cases [39]. α-SYN, parkin, leucine-rich repeat kinase 2 (LRRK2), putative PTEN-induced kinase 1 (PINK1), and DJ1 are among many genes associated with familial Parkinson’s disease (PD) [40]. Just as Alzheimer’s has both sporadic and familial features, Parkinson’s disease tends to run in families. Therefore, the majority of familial PD and AD are early-onset, progressive diseases.
Studies have shown that NQO1 expression is increased in astrocytes, endothelial cells, and dopaminergic neurons in people with Parkinson’s disease [41]. HO-1 and peroxidase in the substantia nigra of Parkinson’s disease patients show a similar pattern [42]. Immunostaining demonstrated the presence of NRF2.
2.2.1 NRF2-related genes in PD cells
Nuclear localization of NRF2 was observed in dopaminergic neurons in the Parkinson’s disease brain [29]. Oxidative stress, mitochondrial dysfunction, protein carbonylation, and oxidative damage to substantia nigra DNA have been implicated in Parkinson’s disease [43, 44]. Glutathione concentrations are reduced in the brain of patients with Parkinson’s disease [45, 46]. In addition, Keap1 colocalizes with SYN and p62 in Lewy bodies in the Parkinson’s disease brain [28]. New research on induced pluripotent stem cells (iPSCs) from patients with Parkinson’s disease (PD) and control subjects provides further evidence of the role of NRF2 in Parkinson’s disease. Neurons derived from Parkinson’s disease-induced pluripotent stem cells (PD iPSCs) show decreased glutathione (GSH), increased oxidative stress, increased NRF2 activation, and increased NQO1 levels [47].
2.3 Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a serious motor disease in adults and is characterized by the deterioration of slow motor neurons [48]. Motor neurons have intracellular inclusions that are ubiquitin-positive, hyaline, and have a twisted appearance [49]. These include SOD1, which colocalizes with p62 [50]. ALS has also been shown to have TDP-43 (TAR DNA binding protein) and FUS-positive inclusions [51, 52]. Like AD and PD, most ALS patients have a variant form (sALS); Approximately 10–15% of cases are associated with a familial variant (fALS). The first genetic mutation found in familial amyotrophic lateral sclerosis (fALS) was in copper/zinc superoxide dismutase 1 (SOD1), which causes disease by increasing its activity [53]. More than 150 mutations have been identified in the SOD1 gene. Mice with an extreme hSOD1 mutant often have an ALS-like phenotype [54, 55]. The specific molecular mechanism responsible for the increase in dysfunction is currently unknown. However, toxicity to motor neurons requires expression of mutant SOD1 in both non-neuronal cells and motor neurons [56]. Over the last few years, researchers have discovered 24 additional genes associated with amyotrophic lateral sclerosis (sALS) and familial amyotrophic lateral sclerosis (fALS) [57]. One of these genes is p62, which directly affects Keap1/NRF2 and autophagy [58].
2.3.1 NRF2-related genes in ALS cells
The presence of NRF2 and Keap1 in the primary motor cortex and spinal cord of ALS patients was investigated [59]. NRF2 mRNA and protein levels were reduced in ALS patient tissues compared with control tissues, but Keap1 mRNA and protein levels were unchanged. NRF2 dependent genes were not analyzed. Furthermore, Keap1 has been shown to co-localize with intracellular inclusions in post-mortem ALS spinal motor neurons [28].
2.4 Huntington’s disease
Huntington’s disease (HD) is a neurological disease caused by the expansion of CAG repeats in the gene responsible for producing the Htt protein [60]. This leads to elongation of the N-terminal glutamine residue, and the intensity and duration of the disease are related to the length of the polyglutamine repeat. In addition, structural changes in mutant Htt lead to increased self-aggregation, which is associated with the appearance of intracellular inclusions [61]. Huntington’s disease (HD) is defined by pathological degeneration of the neostriatal regions (caudate and putamen) and cerebral cortex. This degeneration is thought to be the main cause of mobility, cognitive impairment, and psychiatric symptoms that worsen as the disease progresses. Oxidative stress due to mitochondrial dysfunction has been shown to be responsible in human patients [62]. According to data obtained from postmortem brain examination, dysfunction of mitochondrial complex II, III, and IV was detected in the striatum of deceased Huntington’s disease patients [63, 64].
2.4.1 NRF2-related genes in HD cells
Surprisingly, very little work has been done on HD tissues and NRF2. Only one study investigated the presence of NRF2 or NRF2-dependent genes in post-mortem HD cells [65]. This study demonstrated HO-1 immunohistochemical staining in HD cells.
2.5 Multiple sclerosis
Approximately 400,000 people in the United States and more than 2.3 million people worldwide are thought to be affected by multiple sclerosis (MS). Multiple sclerosis (MS) is a chronic disease characterized by inflammation of the nervous system. It begins with the activation of CD4+ T lymphocytes in the peripheral zone, which then cross the blood-brain barrier and initiate an autoimmune attack on myelin and oligodendrocytes in the central nervous system (CNS) [66]. Neurological disorders occur in a variety of conditions, including vision loss, coordination problems, fatigue, hearing loss, cognitive impairment, and bladder and bowel problems. Multiple sclerosis (MS) disease occurs as a result of the destruction of the immune system, and autoreactive CD4+ Th1 and Th17 cells (myelin sheath), which are the protective covering of the brain, emerge and myelin-producing cells (oligodendrocytes) emerge. This attack is also accomplished by infiltrating macrophages and resident microglia. As a result, nerve cells decrease and the number of stellate cells (a type of glial cell) increases in response to the damage. Activated microglia and stellate cells secrete inflammatory substances such as cytokines and chemokines, as well as reactive oxygen species and nitrogen. These drugs exacerbate oxidative stress and further exacerbate the deleterious tissue damage associated with neuroinflammation, leading to disease progression [67, 68]. Currently, the FDA has approved eight drugs to treat relapsing-remitting MS, the most common form of MS, affecting 87 percent of people with MS. The main purpose of this medication is to prevent the immune system from crossing the blood-brain barrier. However, due to the complexity and diversity of diseases, the evolution of treatment over time, and the general belief that oxidative stress plays a role in disease, further research is required to improve and/or supplement existing treatments [69].
2.5.1 NRF2-related genes in MS cells
Damage to brain tissue in multiple sclerosis causes extensive oxidative damage resulting from the production of reactive oxygen species and nitrogen species resulting from inflammation. Antioxidants and enzymes, as well as NRF2 protein levels, have been shown to serve as markers of cellular stress in active disease compared to normal white matter (NAWM) and normal tissue in the brain that are not neuronally controlled [70, 71, 72]. Therefore, initiation of NRF2-mediated antioxidant response may indicate the existence of cellular defense mechanisms that prevent damage from oxidative stress caused by long-term inflammation. Previous studies have shown that NQO1, an enzyme putatively regulated by NRF2, is increased in multiple sclerosis (MS). This stimulation was particularly evident in hypertrophic stellate cells and myelin-laden macrophages. Oligodendrocytes sometimes show NQO1 immunoreactivity, but neuronal NQO1 labeling is rare [70]. Additional studies have shown that the disease in multiple sclerosis (MS) exhibits strong resistance to markers of oxidative stress, including 4-HNE, 8-hydroxydeoxyguanosine, and nitrotyrosine. It has also been found that these active bacteria contain antioxidant enzymes such as HO-1, SOD1, SOD2, and catalase. Like NQO1 manifestations, this staining is seen in hypertrophic stellate cells and myelin-rich macrophages. Rare staining was observed in NAWM. Upregulation of NRF2 in MS lesions can be seen in the nuclei and cytoplasm of invasive macrophages and reactive stellate cells, with little expression in oligodendrocytes around active lesions [72].
3. Therapeutic scope of the activation or inhibition of the transcriptional factor NRF2 in neurological disorders
NRF2 activation mitigates multiple pathogenic processes involved in these neurodegenerative disorders through upregulation of antioxidant defenses, inhibition of inflammation, improvement of mitochondrial function, and maintenance of protein homeostasis.
3.1 Oxidative stress and Nrf2
NRF2 controls the production of many antioxidant enzymes and proteins that provide protection to cells. Studies show that enhancing NRF2-dependent antioxidant activities may enhance brain functions in illness models. The synaptic degeneration and neuronal death in Alzheimer’s disease are a result of various disrupted processes such as heightened oxidative stress, persistent inflammation, impaired mitochondrial function, buildup of amyloid-β (Aβ) 1–42 peptides from the amyloid precursor protein (APP), proteasome inhibition, mutations in APP, presenilin-1, and presenilin-2 genes, and accumulation of hyper-phosphorylated tau protein [73]. Oxidative stress is partly caused by oligomeric Aβ peptides and occurs before other metabolic changes, leading to persistent inflammation due to oxidative damage.
3.2 Neuroinflammation and Nrf2
It is a feature of neurodegenerative diseases and is caused primarily by activated microglia (inhabitants of the immune system). When activated, microglia release cytotoxic reactive oxygen species and nitrogen, which can affect neurons, especially in cases of excessive microglial activation and dysregulation. Additionally, the presence of dead or injured neurons leads to activation of microglia (known as reactive microgliosis) through recognition receptors such as Toll-like receptors. This causes a continuous cycle of neuronal cell death. The density of microglia in the adult brain varies by region and can range between 0.5 and 16.6%. They are usually found in a dormant state with branching-like patterns in the gray matter. Regions with the most cells include the hippocampus, olfactory telencephalon, basal ganglia, and substantia nigra [74, 75]. Therefore, it is not surprising that certain regions of the brain can be affected by microgliosis, as seen in many neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral multiple sclerosis (ALS) [76]. The main source of reactive chemicals produced by microglia is the activation of the NADPH oxidase (NOX) system, which produces more superoxide anions (•O2−). The NOX enzyme complex in the cell membrane normally enhances the elimination of pathogens when triggered by different stimuli such as pathogen-associated molecular patterns (PAMPs), inflammatory cytokines, and neurotoxins. It also causes changes in cell shape and movement and regulates gene activity. Sustained activation of NOX leads to oxidation of lipids and other macromolecules and causes apoptosis, ultimately leading to neuronal cell death and degeneration. Similarly, enzyme-inducible nitric oxide synthase (iNOS) is stimulated, and its activation produces nitric oxide (NO•), similar to •O2− which can produce neurotoxic effects when consistently present at high concentrations. An increase in the level of NO• can disrupt the balance of redox homeostasis, and NO• can also react with •O2−, leading to the formation of more harmful compounds called peroxynitrite (ONNO−) [50]. At rest, microglia constantly monitor its environment for bacteria, abnormal cells, or cellular debris. They also help reorganize and maintain synapses. When activated by risk, microglia become activated and subsequently release anti-inflammatory substances such as TNF, interleukin (IL)-1, IL-6, IL-12, and IL-23. This phase of microglial activation is often referred to as the classical proinflammatory phase and is referred to as M1. M2 is the stage after the M1 stage and is characterized by a long period and immunity or change. During this time, microglia are activated in response to inflammatory cytokines, including IL-4, IL-10, and IL-13. The body then produces the same cytokines while stimulating genes involved in wound healing, including arginase 1 (ARG1), mannose receptor (MMC), and chitinase-like 3 in domain 1 (FIZZ1)-1 (Ym1 in mice). In fact, the antioxidant properties of microglia maintain cellular homeostasis and keep microglia constantly alert. NF-kappaB (NF-kß) is a master regulator of the brain’s response to infection, the environment, and cellular damage. The redox state of microglia plays an important role in regulating the nuclear level of NF-kß as well as the flux of NF-kß kinases [74]. Microglia function well when the body’s antioxidant defense system maintains oxidative stress (OS) at normal levels. However, when OS or neuroinflammation (NS) persists, the NF-kß signaling pathway remains active. This causes more inflammation in microglia, making them more active and affecting the nervous system. Transcription of NRF2 is responsible for the activation of antioxidant genes and maintenance of redox homeostasis. It is a master regulator of microglia fate, protecting microglia from oxidative stress and preventing hyperactivation of the M1 phenotype. Activation of NRF2 specifically leads to increased expression of genes involved in various processes, including elimination of oxidative stress (SOD3, GPx, and Prx), production of reducing agents (GCLM/C or NADPH), and regeneration of cofactors. Proteins (GR, Trx-TrxR, Prx/thioredoxin), transport of redox molecules (xCT), metal chelation (MT1/2 or ferritin), and antioxidant production enzymes (HO-1, NQO1 and Trx). In addition, NRF2 activation also inhibits the expression of the Trx inhibitor TXNIP. Many studies have identified NRF2 as a useful target for the treatment of neurological diseases associated with neuronal protection secondary to neurodegenerative pathologies. In one study, mice lacking the NRF2 gene (NRF2−/−) were injected with lipopolysaccharide (LPS), a substance known to cause inflammation. These animals show sensitivity to lipopolysaccharide-induced neuroinflammation, especially in the hippocampal region. NRF2−/− mice have protein and mRNA levels of the microglial marker F4/80 in the hippocampal gyrus compared to WT animals. In addition, the inflammatory markers iNOS, IL-6, and TNF-α were also improved. Additionally, SFN-treated mice showed a two- to threefold increase in HO-1 levels, decreased microglia numbers, and decreased production of specific markers in response to LPS [77]. Mice treated with SFN after spinal cord injury showed an increase in NRF2 and GCL levels as well as a decrease in the cytokines IL-1ß and TNF-α. As a result, these mice had decreased volume and improved coordination [78]. In a mouse model of Parkinson’s disease (PD), MPTP injection into animals lacking the NRF2 gene (NRF2−/−) showed strong oxidative stress (OS). These mice had significantly more impaired dopaminergic activity in the basal ganglia than control mice with normal NRF2 genes (WT littermate controls). Although there was no significant difference in the number of Cd11b-positive/CD45 advanced cells between WT and NRF2−/− mice (indicating peripheral macrophage infiltration), NRF2−/− mice showed more astrocyte hyperplasia and microgliosis. This is determined by higher expression and protein expression of GFAP and F4/80 [79]. This study confirmed previous studies showing that NRF2−/− mice exposed to MPTP showed increased neuroinflammation caused by activation of astrocytes (GFAP) and microglia (Iba-1) [80]. In addition, levels of inflammatory markers associated with microglial activation (such as COX2, iNOS, IL-6, and TNF-α) were also increased in the M1 state. In contrast, markers associated with M2 status microglia, such as FIZZ-1, YM-1, arginase-1, and IL-4, were found to be decreased. Findings indicate that NRF2 plays an important role in regulating neuroinflammation after neurotoxic injury [78]. A mouse model of tauopathy was induced in the mouse hippocampus by stereotaxic injection of an adeno-disrupting vector expressing TAUP301L. In this model, NRF2−/− animals showed high levels of microgliosis and astrocytosis. Validation included measuring TNF and IL-6 mRNA levels in the hippocampus and immunohistochemical staining for Iba-1 and GFAP. In contrast to WT mice, NRF2−/− animals did not show an increase in hippocampal HO-1 and GCLC mRNA levels and did not show cellular staining for HO-1. These findings suggest that TAUP301L-induced disease activates the NRF2 signaling pathway as a regulatory response. In line with these observations and previous studies showing that neuronal production of the chemokine fractalkine (CX3CL1) affects TAU-mediated neuroinflammation, the authors of this study showed that CX3CL1 secreted by injured neurons was exclusively associated with an increase in microglia. Receptors are found in cells. This interaction activates the NRF2-ARE signaling pathway and reduces the incidence of TAUP301L-induced microglial proliferation. Subsequent studies were supported by the finding that mice lacking CX3CR1 had increased hippocampal microgliosis in response to TAUP301L compared with normal controls. Additionally, NRF2-dependent HO-1 immunohistochemical staining was absent in these animals. So, when neurons are injured, they produce a substance called CX3CL1, which then interacts with microglia through the CX3CR1 receptor. This interaction activates the NRF2-ARE pathway, which reduces the neuroinflammatory response to injury [81].
In a study with co-culture experiments using microglial conditioned media and mouse primary neurons, degenerating neurons exposed to neurotoxic oligomeric amyloid ß (oAß) were found to induce microglia to release milk fat globule-EGF factor 8. It was found to emit signal (MFG-E8). Release of MFG-E8 increases the phagocytic activity of microglia. Additionally, microglia treated with MFG-E8 was found to activate the NRF2 signaling pathway and HO-1. However, when examining neuronal-microglia co-cultures, retention of MFG-E8 was counteracted by application of the HO-1 inhibitor tin (IV)-mesoporphyrin IX dichloride (SnMP). This suggests that increased HO-1 in microglia is important for protecting neurons from toxicity [82]. These studies suggest that message exchange between neurons and microglia and stimulation of the NRF2 signaling pathway may be a new and effective way to protect neurons, especially in the context of neurodegenerative diseases.
3.3 Mitochondrial function and Nrf2
NRF2 controls cellular defense by adjusting mitochondrial function. NRF2 activation offsets the generation of reactive oxygen species by mitochondria and protects against toxins generated by mitochondria. NRF2 function is reduced in mitochondrial illnesses such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Friedreich’s ataxia (FRDA). Research conducted with isolated mitochondria and cultured cells has shown that a lack of NRF2 results in reduced mitochondrial fatty acid oxidation, respiration, and ATP generation. Chemical substances that activate NRF2 help maintain mitochondrial health by either stimulating mitophagy or providing protection against oxidative stress-induced damage to the mitochondrial permeability transition pore [83].
4. Association between NFE2L2 and KEAP1 haplotypes and neurodegenerative disorders
Genomic studies were performed to determine whether there were genetic differences in the genes encoding NRF2 and its repressor protein Keap1 (called NFE2L2 and KEAP1, respectively). As discussed in this review, this is based on significant evidence that the neurodegenerative effects of OS/NS in diseases such as PD, AD, and ALS can be inhibited or prevented by increasing NRF2 activity. The aim is to investigate whether genetics plays a role in the development or susceptibility to these diseases. Based on analysis of published SNPs and other genetic variants, the mutation frequency of human NFE2L2 was estimated to be 1 in every 72 base pairs. This suggests that genes may have many variations, called polymorphisms [84]. Von Otter et al. Initial data analysis in patients with Parkinson’s disease (PD) identified a protective NFE2L2 haplotype, GAGCAAAA. This haplotype contains a variant in the NRF2 promoter that is active in Swedish and Polish samples and is associated with high activity [85]. In a Swedish study, the presence of GAAAA haplotype alleles was associated with a higher estimated age of onset (AAO) of Parkinson’s disease (PD) by approximately 5 years per allele (p = 0.001). In the Polish study, individuals with the protective entire haplotype GAGCAAAA had a lower risk of developing PD; There was a difference of 0.4 and 0.2 for heterozygous and homozygous carriers, respectively (p = 2 × 10−6). Additionally, studies in Sweden found that GAGGG and GAAAG haplotypes were associated with an increased risk of Parkinson’s disease, with odds of 2.4 and 3.7 for each allele, respectively. Importantly, the promoter haplotype AGA tended to increase the risk of PD, whereas the entire promoter haplotype GAGAAGGG showed an overall risk (difference between each haplotype allele was 2.8). However, studies have shown that the entire haplotype allele GAGCAAAAA causes a delay in the age of onset of Parkinson’s disease for all haplotype alleles. Neither the Swedish nor the Polish samples found a significant effect on Parkinson’s disease risk for specific SNPs alone. Genetic polymorphisms of KEAP1 were also examined. However, there was no association with age at onset (AAO) or likelihood of Parkinson’s disease [85]. A recent meta-analysis investigated the genetic association of NFE2L2 variants encoding NRF2 with Parkinson’s disease (PD). The review included the literature mentioned above, as well as four new independent European patient-controlled studies from Malta, Germany and Italy, and a second Swedish study. The results of this analysis showed an association between the GAGCAAAAA haplotype, including the high-activity promoter haplotype AGC, and reduced risk of PD (odds ratio (OR) of none per allele 0.8, p = 0.012). Additionally, this haplotype is associated with delayed onset of PD (+1.1 years per allele, p = 0.048) [84]. The results of this study confirm the first haplotype study linking genetic variation in the NFE2L2 gene to the development of Parkinson’s disease. This latest study also reported four single nucleic acid polymorphisms (SNPs) found in haplotypes associated with age of onset (AAO) in Parkinson’s disease (PD). Three of the SNPs (rs7557529 G > A, rs35652124 A > G, rs2886161 A > G) were found to accelerate the onset of PD and each allele was associated with a reduction in AAO at 1.0, 1.1, and 1.2 years, respectively. The P values of these relationships were 0.042, 0.045, and 0.021, respectively. On the other hand, the fourth SNP (rs1806649 G > A) was shown to delay the onset of PD, with each allele being associated with an increase in AAO at 1.2 years. It is worth noting that the single nucleotide polymorphism (SNP) rs35652124 is functional and is located in the promoter region of the NFE2L2 gene [86]. Unlike the Parkinson’s disease (PD) studies mentioned above, analysis of the NFE2L2 promoter in a control group of Taiwanese patients did not reveal any genetic changes. Three single nucleotide polymorphisms (SNPs) in the NFE2L2 promoter were examined: rs35652124, rs6706649, and rs6721961. These SNPs can be detected alone or in combination with a genetic haplotype associated with susceptibility to Parkinson’s disease (PD). However, there was no association between these SNPs or haplotype variants and susceptibility to PD. Researchers have suggested that differences between studies in Taiwan and Europe may be due to differences in ethnic and environmental variables in different regions [87]. Previous studies using AD Swedish case-control data found no evidence supporting single SNPs or haplotypes in NFE2L2 or KEAP1 as genes that increase AD risk. However, genetic analysis in the NFE2L2 gene shows a significant association between the GAAAA haplotype and the onset of Alzheimer’s disease 2 years earlier than average (p = 0.013). This means that some common mutations in the NFE2L2 gene may affect Alzheimer’s disease and therefore the onset of the disease. In studies, the presence of NFE2L2 or KEAP1 gene mutations was not associated with Mini-Mental State Examination (MMSE) results or total tau and amyloid ß 1–42 (Aß42) levels in cerebrospinal fluid (CSF) [88]. A Swedish case study of NFE2L2 and KEAP1 in ALS disease showed that a specific NFE2L2 haplotype (GGGAC) was associated with a reduced risk of ALS (or one allele = 0.62, p = 0.015). Additionally, haplotypes in KEAP1 (CGC) were found to be associated with delayed onset of ALS (+3.4 years per allele, p = 0.015). Furthermore, the distribution of ALS revealed the existence of a specific group characterized by the NFE2L2-related haplotype GAGCAGA. This subgroup reveals three function-promoting SNPs associated with increased NRF2 protein expression. Each additional allele of this haplotype was associated with a 4.0-year delay in the onset of ALS (p = 0.008). Each additional allele of this haplotype was associated with a 4.0-year delay in the onset of ALS (p = 0.008) [89]. In a separate case-control study in Italian ALS patients, three NFE2L2 promoter polymorphisms (i.e., SNPs −653 A/G, −651 G/A, and −617 C/A) were tested. However, there was no significant difference between ALS cases and controls. The frequency of the −653 A/G promoter polymorphism was slightly higher in patients than in controls but did not reach significance. Additionally, there was no association between the NFE2L2 promoter polymorphisms tested and the oxidative stress biomarkers tested [84]. Similar studies have not been conducted in patients with Huntington’s disease (HD) or multiple sclerosis (MS).
5. Conclusion
The NRF2-ARE pathway is an important target for therapeutic intervention in neurodegenerative diseases, as demonstrated by the facts presented. Current data provide the highest level of evidence for Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS). However, future trials in Alzheimer’s disease (AD) and Huntington’s disease (HD) are needed to better understand the importance of NRF2 in these diseases. Cell and in silico high-throughput screens have identified new drugs that activate NRF2 [90]. Effective strategies may include the identification of non-covalently linked compounds with interactions between DLG or ETGE motifs and Keap1 [91, 92]. Now the US Food and Drug Administration (FDA) has approved Biogen Idec’s DMF (BG-12) for the treatment of multiple sclerosis (MS). As described previously, DIF has been evaluated in previous HD models. However, it is important to note that in previous models of the above diseases, only CDDO drugs were analyzed. Reata Pharmaceuticals developed the first CDDO molecule, bardoxolone methyl (also known as CDDO methyl ester), and initiated a Phase III study for the treatment of kidney disease. Unfortunately, this study was discontinued in 2012 due to a higher incidence of cardiovascular disease in the bardoxolone methyl group compared to the placebo group [93]. A new CDDO derivative called RTA 408 has been developed and is currently being tested in several non-neurodegenerative diseases [94, 95, 96, 97]. A clinical trial investigating the use of RTA 408 to treat Friedreich’s ataxia, a neurodegenerative disease that causes cerebellar ataxia, has recently begun. This condition is characterized by a decrease in the protein frataxin, resulting in a defect in mitochondrial respiration (ClinicalTrials.gov identifier NCT02255435). The inclusion of these future CDDO drugs in the major neurodegenerative diseases mentioned in this review may be an important first step, supported by the findings of previous CDDO history.
A new aspect of treatment strategies for this system is that some cells are more effective in protecting neurons from degeneration. Compared to the protection obtained by overexpression of NRF2 in stellate cells in models of ALS and PD, the lack of protection in vivo by overexpression of NRF2 in neurons or muscle cells suggests that not only the pharmacological target but also the cell specificity is still affected. Targetability may be important when considering NRF2-dependent therapeutic approaches.
References
- 1.
Reynolds A, Laurie C, Mosley RL, Gendelman HE. Oxidative stress and the pathogenesis of neurodegenerative disorders. International Review of Neurobiology. 2007; 82 :297-325. DOI: 10.1016/S0074-7742(07)82016-2 - 2.
Seminotti B, Grings M, Tucci P, Leipnitz G, Saso L. Nuclear factor erythroid-2-related factor 2 signaling in the neuropathophysiology of inherited metabolic disorders. Frontiers in Cellular Neuroscience. 2021; 15 :785057. DOI: 10.3389/fncel.2021.785057 - 3.
Tejo FV, Quintanilla RA. Contribution of the NRF2 pathway on oxidative damage and mitochondrial failure in Parkinson and Alzheimer’s disease. Antioxidants. 2021; 10 :1069. DOI: 10.3390/antiox10071069 - 4.
Moretti D, Tambone S, Cerretani M, Fezzardi P, Missineo A, Sherman LT, et al. NRF2 activation by reversible KEAP1 binding induces the antioxidant response in primary neurons and astrocytes of a Huntington’s disease mouse model. Free Radical Biology & Medicine. 2021; 162 :243-254. DOI: 10.1016/j.freeradbiomed.2020.10.022 - 5.
Petrillo S, Schirinzi T, Di Lazzaro G, D’Amico J, Colona VL, Bertini E, et al. Systemic activation of NRF2 pathway in Parkinson’s disease. Movement Disorders. 2020; 35 :180-184. DOI: 10.1002/mds.27890 - 6.
Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, et al. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain. 2012; 135 :886-899. DOI: 10.1093/brain/aws012 - 7.
Xiao Y, Karam C, Yi J, Zhang L, Li X, Yoon D, et al. ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharmacological Research. 2018; 138 :25-36. DOI: 10.1016/j.phrs.2018.09.019 - 8.
Finkel T. Signal transduction by mitochondrial oxidants. The Journal of Biological Chemistry. 2012; 287 :4434-4440. DOI: 10.1074/jbc.R111.283280 - 9.
Nauseef WM. Detection of superoxide anion and hydrogen peroxide production by cellular NADPH oxidases. Biochimica et Biophysica Acta - General Subjects. 2014; 1840 :757-767. DOI: 10.1016/j.bbagen.2013.10.029 - 10.
Rojkind M, Domínguez-Rosales JA, Nieto N, Greenwel P. Role of hydrogen peroxide and oxidative stress in healing responses. Cellular and Molecular Life Sciences. 2002; 59 :1872-1891. DOI: 10.1007/PL00012511 - 11.
Niedzielska E, Smaga I, Gawlik M, Moniczewski A, Stankowicz P, Pera J, et al. Oxidative stress in neurodegenerative diseases. Molecular Neurobiology. 2016; 53 :4094-4125. DOI: 10.1007/s12035-015-9337-5 - 12.
Wasik U, Milkiewicz M, Kempinska-Podhorodecka A, Milkiewicz P. Protection against oxidative stress mediated by the NRF2/Keap1 axis is impaired in primary biliary cholangitis. Scientific Reports. 2017; 7 :44769. DOI: 10.1038/srep44769 - 13.
Telkoparan-Akillilar P, Panieri E, Cevik D, Suzen S, Saso L. Therapeutic targeting of the NRF2 signaling pathway in cancer. Molecules. 2021; 26 :1417. DOI: 10.3390/molecules26051417 - 14.
Ma Q. Role of NRF2 in oxidative stress and toxicity. Annual Review of Pharmacology and Toxicology. 2013; 53 :401-426. DOI: 10.1146/annurev-pharmtox-011112-140320 - 15.
Stachurska A, Ciesla M, Kozakowska M, Wolffram S, Boesch-Saadatmandi C, Rimbach G, et al. Cross-talk between microRNAs, nuclear factor E2-related factor 2, and heme oxygenase-1 in ochratoxin A-induced toxic effects in renal proximal tubular epithelial cells. Molecular Nutrition & Food Research. 2013; 57 :504-515. DOI: 10.1002/mnfr.201200360 - 16.
Panieri E, Telkoparan-Akillilar P, Suzen S, Saso L. The NRF2/KEAP1 Axis in the regulation of tumor metabolism: Mechanisms and therapeutic perspectives. Biomolecules. 2020; 10 :791. DOI: 10.3390/biom10050791 - 17.
Esteras N, Dinkova-Kostova AT, Abramov AY. NRF2 activation in the treatment of neurodegenerative diseases: A focus on its role in mitochondrial bioenergetics and function. Biological Chemistry. 2016; 397 :383-400. DOI: 10.1515/hsz-2015-0270 - 18.
Alzheimer's Association. 2014 Alzheimer's disease facts and figures. Alzheimer's & Dementia. 2014; 10 (2):e47-e92. DOI: 10.1016/j.jalz.2014.02.001 - 19.
Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: The solution to an Alzheimer's disease conundrum? Trends in Neurosciences. 2001; 24 :219-224. DOI: 10.1016/s0166-2236(00)01749-5 - 20.
Sandbrink R, Hartmann T, Masters CL, Beyreuther K. Genes contributing to Alzheimer's disease. Molecular Psychiatry. 1996; 1 :27-40. DOI: 10.1038/sj.mp.4000236 - 21.
Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. The Journal of Neuroscience. 1997; 17 :2653-2657. DOI: 10.1523/JNEUROSCI.17-07-02653.1997 - 22.
Raina AK, Templeton DJ, Deak JC, Perry G, Smith MA. Quinone reductase (NQO1), a sensitive redox indicator, is increased in Alzheimer's disease. Redox Report. 1999; 4 :23-27. DOI: 10.1179/135100099101534890 - 23.
Wang Y, Santa-Cruz K, DeCarli C, Johnson JA. NAD(P)H:Quinone oxidoreductase activity is increased in hippocampal pyramidal neurons of patients with Alzheimer's disease. Neurobiology of Aging. 2000; 21 :525-531. DOI: 10.1016/s0197-4580(00)00126-0 - 24.
SantaCruz KS, Yazlovitskaya E, Collins J, Johnson J, DeCarli C. Regional NAD(P)H:Quinone oxidoreductase activity in Alzheimer's disease. Neurobiology of Aging. 2004; 25 :63-69. DOI: 10.1016/S0197-4580(03)00059-8 - 25.
Schipper HM, Cisse S, Stopa EG. Expression of heme oxygenase-1 in the senescent and Alzheimer-diseased brain. Annals of Neurology. 1995; 37 :758-768. DOI: 10.1002/ana.410370612 - 26.
Schipper HM, Chertkow H, Mehindate K, Frankel D, Melmed C, Bergman H. Evaluation of heme oxygenase-1 as a systemic biological marker of sporadic AD. Neurology. 2000; 54 :1297-1304. DOI: 10.1212/WNL.54.6.1297 - 27.
Aksenov MY, Markesbery WR. Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer's disease. Neuroscience Letters. 2001; 302 :141-145. DOI: 10.1016/s0304-3940(01)01537-4 - 28.
Tanji K, Maruyama A, Odagiri S, Mori F, Itoh K, Kakita A, et al. Keap1 is localized in neuronal and glial cytoplasmic inclusions in various neurodegenerative diseases. Journal of Neuropathology and Experimental Neurology. 2013; 72 :18-28. DOI: 10.1097/NEN.0b013e31827b1221 - 29.
Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, et al. Expression of NRF2 in neurodegenerative diseases. Journal of Neuropathology and Experimental Neurology. 2007; 66 :75-85. DOI: 10.1097/nen.0b013e31802f11bf - 30.
Lovell MA, Xie C, Markesbery WR. Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer's disease. Neurology. 1998; 51 :1562-1566. DOI: 10.1212/WNL.51.6.1562 - 31.
Ansari MA, Scheff SW. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. Journal of Neuropathology and Experimental Neurology. 2010; 69 :155-167. DOI: 10.1097/NEN.0b013e3181cb5af4 - 32.
Saharan S, Mandal PK. The emerging role of glutathione in Alzheimer's disease. Journal of Alzheimer's Disease. 2014; 40 :519-529. DOI: 10.3233/JAD-132483 - 33.
Kraft AD, Lee JM, Johnson DA, Kan YW, Johnson JA. Neuronal sensitivity to kainic acid is dependent on the NRF2-mediated actions of the antioxidant response element. Journal of Neurochemistry. 2006; 98 :1852-1865. DOI: 10.1111/j.1471-4159.2006.04001.x - 34.
Joshi G, Gan KA, Johnson DA, Johnson JA. Increased Alzheimer's disease-like pathology in the APP/PS1ΔE9 mouse model lacking NRF2 through modulation of autophagy. Neurobiology of Aging. 2015; 36 :664-679. DOI: 10.1016/j.neurobiolaging.2014.07.030 - 35.
Beal MF. Therapeutic approaches to mitochondrial dysfunction in Parkinson's disease. Parkinsonism & Related Disorders. 2009; 15 (Suppl 3):S189-S194. DOI: 10.1016/s1353-8020(09)70767-5 - 36.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ , Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997; 388 :839-840. DOI: 10.1038/42166 - 37.
Irizarry MC, Growdon W, Gomez-Isla T, Newell K, George JM, Clayton DF, et al. Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson's disease and cortical Lewy body disease contain alpha-synuclein immunoreactivity. Journal of Neuropathology and Experimental Neurology. 1998; 57 :334-337. DOI: 10.1097/00005072-199804000-00002 - 38.
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95 :6469-6473. DOI: 10.1073/pnas.95.11.6469 - 39.
Samii A, Nutt JG, Ransom BR. Parkinson's disease. Lancet. 2004; 363 :1783-1793. DOI: 10.1016/S0140-6736(04)16305-8 - 40.
Lesage S, Brice A. Parkinson's disease: From monogenic forms to genetic susceptibility factors. Human Molecular Genetics. 2009; 18 :R48-R59. DOI: 10.1093/hmg/ddp012 - 41.
van Muiswinkel FL, de Vos RA, Bol JG, Andringa G, Jansen Steur EN, Ross D, et al. Expression of NAD(P)H:Quinone oxidoreductase in the normal and Parkinsonian substantia nigra. Neurobiology of Aging. 2004; 25 :1253-1262. DOI: 10.1016/j.neurobiolaging.2003.12.014 - 42.
Castellani R, Smith MA, Richey PL, Perry G. Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Research. 1996; 737 :195-200. DOI: 10.1016/0006-8993(96)00890-9 - 43.
Dexter DT, Holley AE, Flitter WD, Slater TF, Wells FR, Daniel SE, et al. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: An HPLC and ESR study. Movement Disorders. 1994; 9 :92-97. DOI: 10.1002/mds.870090113 - 44.
Alam ZI, Daniel SE, Lees AJ, Marsden DC, Jenner P, Halliwell B. A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease. Journal of Neurochemistry. 1997; 69 :1326-1329. DOI: 10.1046/j.1471-4159.1997.69031326.x - 45.
Perry TL, Yong VW. Idiopathic Parkinson's disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neuroscience Letters. 1986; 67 :269-274. DOI: 10.1016/0304-3940(86)90355-7 - 46.
Zeevalk GD, Razmpour R, Bernard LP. Glutathione and Parkinson's disease: Is this the elephant in the room? Biomedicine & Pharmacotherapy. 2008; 62 :236-249. DOI: 10.1016/j.biopha.2007.06.005 - 47.
Imaizumi Y, Okada Y, Akamatsu W, Koike M, Kuzumaki N, Hayakawa H, et al. Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Molecular Brain. 2012; 5 :35. DOI: 10.1186/1756-6606-5-35 - 48.
Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. The New England Journal of Medicine. 2001; 344 :1688-1700. DOI: 10.1056/NEJM200105313442207 - 49.
Murayama S, Ookawa Y, Mori H, Nakano I, Ihara Y, Kuzuhara S, et al. Immunocytochemical and ultrastructural study of Lewy body-like hyaline inclusions in familial amyotrophic lateral sclerosis. Acta Neuropathologica. 1989; 78 :143-152. DOI: 10.1007/BF00691035 - 50.
Mizuno Y, Amari M, Takatama M, Aizawa H, Mihara B, Okamoto K. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. Journal of the Neurological Sciences. 2006; 249 :13-18. DOI: 10.1016/j.jns.2006.05.060 - 51.
Tan CF, Eguchi H, Tagawa A, Onodera O, Iwasaki T, Tsujino A, et al. TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathologica. 2007; 113 :535-542. DOI: 10.1007/s00401-007-0206-9 - 52.
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009; 323 :1208-1211. DOI: 10.1126/science.1165942 - 53.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993; 362 :59-62. DOI: 10.1038/362059a0 - 54.
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994; 264 :1772-1775. DOI: 10.1126/science.8209258 - 55.
Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proceedings of the National Academy of Sciences of the United States of America. 2002; 99 :1604-1609. DOI: 10.1073/pnas.032539299 - 56.
Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003; 302 :113-117. DOI: 10.1126/science.1086071 - 57.
Marangi G, Traynor BJ. Genetic causes of amyotrophic lateral sclerosis: New genetic analysis methodologies entailing new opportunities and challenges. Brain Research. 2015; 1607 :75-93. DOI: 10.1016/j.brainres.2015.02.003 - 58.
Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Archives of Neurology. 2011; 68 :1440-1446. DOI: 10.1001/archneurol.2011.250 - 59.
Sarlette A, Krampfl K, Grothe C, Neuhoff N, Dengler R, Petri S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. Journal of Neuropathology and Experimental Neurology. 2008; 67 :1055-1062. DOI: 10.1097/NEN.0b013e31818b4d40 - 60.
The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993; 72 :971-983. DOI: 10.1016/0092-8674(93)90585-E - 61.
Bates G. Huntingtin aggregation and toxicity in Huntington's disease. Lancet. 2003; 361 :1642-1644. DOI: 10.1016/S0140-6736(03)13348-4 - 62.
Browne SE, Beal MF. Oxidative damage in Huntington's disease pathogenesis. Antioxidants & Redox Signaling. 2006; 8 :2061-2073. DOI: 10.1089/ars.2006.8.2061 - 63.
Johri A, Chandra A, Beal MF. PGC-1alpha, mitochondrial dysfunction, and Huntington's disease. Free Radical Biology & Medicine. 2013; 62 :37-46. DOI: 10.1016/j.freeradbiomed.2012.09.034 - 64.
Reddy PH. Increased mitochondrial fission and neuronal dysfunction in Huntington's disease: Implications for molecular inhibitors of excessive mitochondrial fission. Drug Discovery Today. 2014; 19 :951-955. DOI: 10.1016/j.drudis.2014.01.021 - 65.
Browne SE, Ferrante RJ, Beal MF. Oxidative stress in Huntington's disease. Brain Pathology. 1999; 9 :147-163. DOI: 10.1111/j.1750-3639.1999.tb00215.x - 66.
National Multiple Sclerosis Society. Who Gets MS?. 2015. Available from: http://www.nationalmssociety.org/What-is-MS/Who-Gets-MS [Accessed: February 15, 2024] - 67.
Dhib-Jalbut S. Pathogenesis of myelin/oligodendrocyte damage in multiple sclerosis. Neurology. 2007; 68 :S13-S21. discussion S43-54. DOI: 10.1212/01.wnl.0000259402.47974.44 - 68.
Hofstetter H, Gold R, Hartung HP. Th17 cells in MS and experimental autoimmune encephalomyelitis. International MS Journal. 2009; 16 :12-18. DOI: 10.1177/1352458508101709 - 69.
Chiurchiu V. Novel targets in multiple sclerosis: To oxidative stress and beyond. Current Topics in Medicinal Chemistry. 2014; 14 :2590-2599. DOI: 10.2174/156802661 4666141029125739 - 70.
van Horssen J, Schreibelt G, Bo L, Montagne L, Drukarch B, van Muiswinkel FL, et al. NAD(P)H:quinone oxidoreductase 1 expression in multiple sclerosis lesions. Free Radical Biology & Medicine. 2006; 41 :311-317. DOI: 10.1016/j.freeradbiomed.2006.04.028 - 71.
van Horssen J, Schreibelt G, Drexhage J, Hazes T, Dijkstra CD, van der Valk P, et al. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radical Biology & Medicine. 2008; 45 :1729-1737. DOI: 10.1016/j.freeradbiomed.2008.09.036 - 72.
van Horssen J, Drexhage JA, Flor T, Gerritsen W, van der Valk P, de Vries HE. NRF2 and DJ1 are consistently upregulated in inflammatory multiple sclerosis lesions. Free Radical Biology & Medicine. 2010; 49 :1283-1289. DOI: 10.1016/j.freeradbiomed.2010.07.032 - 73.
Prasad KN. Simultaneous activation of Nrf2 and elevation of antioxidant compounds for reducing oxidative stress and chronic inflammation in human Alzheimer’s disease. Mechanisms of Ageing and Development. 2016; 153 :41-47. DOI: 10.1016/j.mad.2016.01.002 - 74.
Rojo AI, McBean G, Cindric M, Egea J, Lopez MG, Rada P, et al. Redox control of microglial function: Molecular mechanisms and functional significance. Antioxidants & Redox Signaling. 2014; 21 :1766-1801. DOI: 10.1089/ars.2013.5748 - 75.
Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990; 39 :151-170. DOI: 10.1016/0306-4522(90)90229-W - 76.
Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010; 7 :354-365. DOI: 10.1016/j.nurt.2010.05.014 - 77.
Innamorato NG, Rojo AI, García-Yagüe AJ, Yamamoto M, de Ceballos ML, Cuadrado A. The transcription factor NRF2 is a therapeutic target against brain inflammation. Journal of Immunology. 2008; 181 (1):680-689. DOI: 10.4049/jimmunol.181.1.680 - 78.
Wang X, de Rivero Vaccari JP, Wang H, et al. Activation of the nuclear factor E2-related factor 2/antioxidant response element pathway is neuroprotective after spinal cord injury. Journal of Neurotrauma. 2012; 29 (5):936-945. DOI: 10.1089/neu.2011.1922 - 79.
Rojo AI, Innamorato NG, Martín-Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A. NRF2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia. 2010; 58 (5):588-598. DOI: 10.1002/glia.20947 - 80.
Chen PC, Vargas MR, Pani AK, et al. NRF2-mediated neuroprotection in the MPTP mouse model of Parkinson's disease: Critical role for the astrocyte. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106 (8):2933-2938. DOI: 10.1073/pnas.0813361106 - 81.
Lastres-Becker I, Innamorato NG, Jaworski T, et al. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain. 2014; 137 (Pt 1):78-91. DOI: 10.1093/brain/awt323 - 82.
Li E, Noda M, Doi Y, et al. The neuroprotective effects of milk fat globule-EGF factor 8 against oligomeric amyloid β toxicity. Journal of Neuroinflammation. 2012; 9 :148. DOI: 10.1186/1742-2094-9-148 - 83.
Dinkova-Kostova AT, Rumen VK, Kazantsev AG. The role of Nrf2 signaling in counteracting neurodegenerative diseases. The FEBS Journal. 2018; 285 :3576-3590. DOI: 10.1111/febs.14379 - 84.
LoGerfo A, Chico L, Borgia L, et al. Lack of association between nuclear factor erythroid-derived 2-like 2 promoter gene polymorphisms and oxidative stress biomarkers in amyotrophic lateral sclerosis patients. Oxidative Medicine and Cellular Longevity. 2014; 2014 :432626. DOI: 10.1155/2014/432626 - 85.
von Otter M, Landgren S, Nilsson S, et al. Association of NRF2-encoding NFE2L2 haplotypes with Parkinson's disease. BMC Medical Genetics. 2010; 11 :36. DOI: 10.1186/1471-2350-11-36 - 86.
von Otter M, Bergstrom P, Quattrone A, De Marco EV, Annesi G, Soderkvist P, et al. Genetic associations of NRF2-encoding NFE2L2 variants with Parkinson's disease—A multicenter study. BMC Medical Genetics. 2014; 15 :131. DOI: 10.1186/s12881-014-0131-4 - 87.
Chen YC, Wu YR, Wu YC, Lee-Chen GJ, Chen CM. Genetic analysis of NFE2L2 promoter variation in Taiwanese Parkinson's disease. Parkinsonism & Related Disorders. 2013; 19 (2):247-250. DOI: 10.1016/j.parkreldis.2012.10.018 - 88.
Rojo AI, Pajares M, García-Yagüe AJ, et al. Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biology. 2018; 18 :173-180. DOI: 10.1016/j.redox.2018.07.006 - 89.
Bergström P, von Otter M, Nilsson S, et al. Association of NFE2L2 and KEAP1 haplotypes with amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. 2014; 15 (1-2):130-137. DOI: 10.3109/21678421.2013.839708 - 90.
Wang L, Lewis T, Zhang YL, et al. The identification and characterization of non-reactive inhibitor of Keap1-NRF2 interaction through HTS using a fluorescence polarization assay. In: Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): National Center for Biotechnology Information (US); 2010 - 91.
Hancock R, Bertrand HC, Tsujita T, et al. Peptide inhibitors of the Keap1-NRF2 protein-protein interaction. Free Radical Biology & Medicine. 2012; 52 (2):444-451. DOI: 10.1016/j.freeradbiomed.2011.10.486 - 92.
Zhuang C, Miao Z, Sheng C, Zhang W. Updated research and applications of small molecule inhibitors of Keap1-NRF2 protein-protein interaction: A review. Current Medicinal Chemistry. 2014; 21 (16):1861-1870. DOI: 10.2174/0929867321 666140217104648 - 93.
de Zeeuw D, Akizawa T, Audhya P, et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. The New England Journal of Medicine. 2013; 369 (26):2492-2503. DOI: 10.1056/NEJMoa1306033 - 94.
Reisman SA, Ward KW, Klaassen CD, Meyer CJ. CDDO-9,11-dihydro-trifluoroethyl amide (CDDO-dhTFEA) induces hepatic cytoprotective genes and increases bile flow in rats. Xenobiotica. 2013; 43 (7):571-578. DOI: 10.3109/00498254.2012.750022 - 95.
Li B, Abdalrahman A, Lai Y, Janicki JS, Ward KW, Meyer CJ, et al. Dihydro-CDDO-trifluoroethyl amide suppresses inflammatory responses in macrophages via activation of NRF2. Biochemical and Biophysical Research Communications. 2014; 444 :555-561. DOI: 10.1016/J.BBRC.2014.01.101 - 96.
Reisman SA, Lee CY, Meyer CJ, Proksch JW, Ward KW. Topical application of the synthetic triterpenoid RTA 408 activates NRF2 and induces cytoprotective genes in rat skin. Archives of Dermatological Research. 2014; 306 (5):447-454. DOI: 10.1007/s00403-013-1433-7 - 97.
Probst BL, Trevino I, McCauley L, Bumeister R, Dulubova I, Wigley WC, et al. A novel synthetic triterpenoid with broad anticancer and anti-inflammatory activity. PLoS ONE. 2015; 10 :e0122942. DOI: 10.1371/journal.pone.0122942