
Abstract
Many commonly known antioxidants, from probucol to vitamin E, are fat-soluble and have been shown to be most effective when administered with meals. Following meal consumption, these compounds are incorporated into intestinal lipoproteins, known as chylomicrons, and secreted into the circulation. These lipid-carrying particles are responsible for the transport of newly absorbed dietary fat for delivery to peripheral tissues. In the bloodstream, chylomicrons interact with heparin-releasable lipases common known as lipoprotein lipase and hepatic triglyceride lipase. Bothe lipases are anchored along the endothelial wall via heparan sulfate proteoglycans and have triglycerides as their preferred substrate. During this process, as dietary triglycerides are hydrolyzed and transported across the endothelium, we hypothesize that antioxidants carried in chylomicrons would be delivered directly to the arterial wall where they would be most effective in quenching reactive oxygen species generated by activated macrophages. Thus, the metabolism of postprandial lipoproteins is a key process in the defense against oxidative stress and may provide the path for effective antioxidant management. In this chapter, we will review the evidence in support of the hypothesis that postprandial lipoproteins may contribute to the delivery of fat-soluble antioxidants that are administered orally.
Keywords
- lipoproteins
- chylomicrons
- postprandial
- lipoprotein lipase
- reactive oxygen species
- vitamin E
1. Introduction
Atherosclerosis is a chronic condition associated with the accumulation of cholesterol along the wall of blood vessels leading to the occlusion of normal blood flow [1]. In his 1978 Lyman Duff lecture, Zilversmit reviewed his work on the development of atherosclerosis in the rabbit model with emphasis on the role of postprandial lipoproteins [2]. The data suggested a close interaction between the endothelial lipoprotein lipase and triglyceride-rich (TG-rich) lipoproteins [3]. While subsequent research implicated the role of oxidative stress in the development and progression of atherosclerotic disease [1, 4, 5, 6], the role of TG-rich lipoproteins in this process is not understood.
Oxidative stress, a state of imbalance between oxidants and antioxidants in favor of oxidants, is associated with disruption of redox signaling and metabolic functions [7] resulting in many chronic conditions, from type 2 diabetes mellitus [8], atherosclerosis [4] to neurodegenerative diseases [9]. Multiple lines of research have suggested a role for pharmacologic as well as nutritional antioxidants in the management of these conditions. While several observational studies with various agents have reported promising results, several case-control studies as well as large prospective studies have yielded conflicting results. Several explanations have been suggested, including differences in the mode and time of delivery, as well as differences in plasma levels versus concentrations at the site(s) of action.
In this section we will review the evidence that suggest a role of TG-rich lipoproteins, in general, and specifically postprandial lipoproteins as a double-edged sword in the propagation of oxidant radicals from a local site of inflammation to other tissues as well as in the delivery of fat-soluble antioxidants.
2. Metabolism of postprandial lipoproteins
For the routine medical examination, most diagnostic blood tests are based on measurement of the levels of metabolites in fasting plasma. Considering the fact that a typical individual would be consuming three main meals each day, 4 to 6 hours apart, with possible snacks in-between, one is constantly in a non-fasted state. Throughout the waking hours, there are postprandial lipoproteins carrying newly absorbed nutrients for delivery throughout the body. Dietary fats and fat-soluble vitamins are packaged into large spherical particles, ranging from 80 to 1200 nm in diameter, and are known as intestinal lipoproteins or chylomicrons. Figure 1 illustrates the different organs that are involved in the postprandial response.
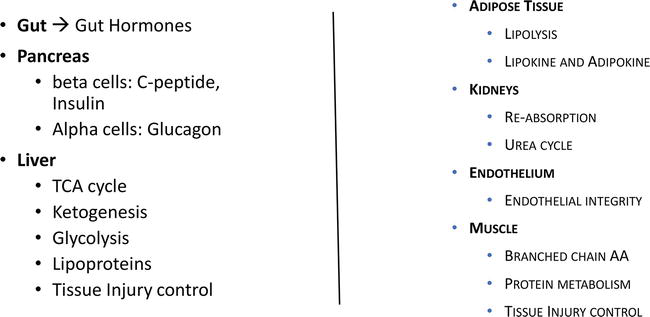
Figure 1.
Organs involved in the postprandial response.
Plasma chylomicrons are constituted of triglycerides (TG, 85–92%), phospholipids (PL, 6–12%), cholesterol (CHOL, 1–3%) and protein (1–2%). The protein component serves as solubilizing coat to allow these lipid-rich particles to circulate in the aqueous environment and be delivered to the appropriate peripheral tissues [10]. These proteins are referred to as apolipoproteins and consist of apoAs, apoB-48, apoCs, and apoE. ApoAs serve primarily as part of the solubilizing coat and do not play a role in the metabolism of these intestinal lipoproteins. On the other hand, apoB-48 is the major structural protein and is required for the formation and secretion of chylomicrons. Genetic deficiency in apoB-48, known as abetalipoproteinemia, is associated with fat malabsorption [11]. ApoCs are responsible for the modulation of lipoprotein lipase activity in the hydrolysis of triglycerides. ApoC-II that is present on the surface of chylomicrons is a required activator for lipoprotein lipase (LPL) [12]. Genetic deficiency in apoC-II is associated with significant elevations in plasma TG and the clinical condition known as type I hyperlipoproteinemia or hyperchylomicronemia. On the other hand, apoC-III, also present on the surface of chylomicrons, acts as an inhibitor of LPL activity [13]. ApoE serves as the ligand for the remnant receptor that is responsible for the uptake of the TG-depleted chylomicrons [14, 15].
Chylomicrons are assembled in enterocytes from dietary lipids and enter the systemic circulation via the lacteals. In the circulation they interact with lipoprotein lipase (LPL) that is attached to the endothelium throughout the body. LPL is responsible for the cleavage of TG into monoglycerides and fatty acids that can freely move into the cells for storage and energy utilization. Zilversmit and co-workers have previously demonstrated a link between the localization of lipoprotein lipase and accumulation of cholesterol transported in TG-rich chylomicrons [3]. Figure 2A schematizes the interactions between TG-rich lipoproteins and endothelial lipoprotein lipase. As the result of the opposite actions of apoC-II and apoC-III, partially delipidated chylomicrons are released back into the circulation and are available to interact with other lipoprotein lipase sites downstream.
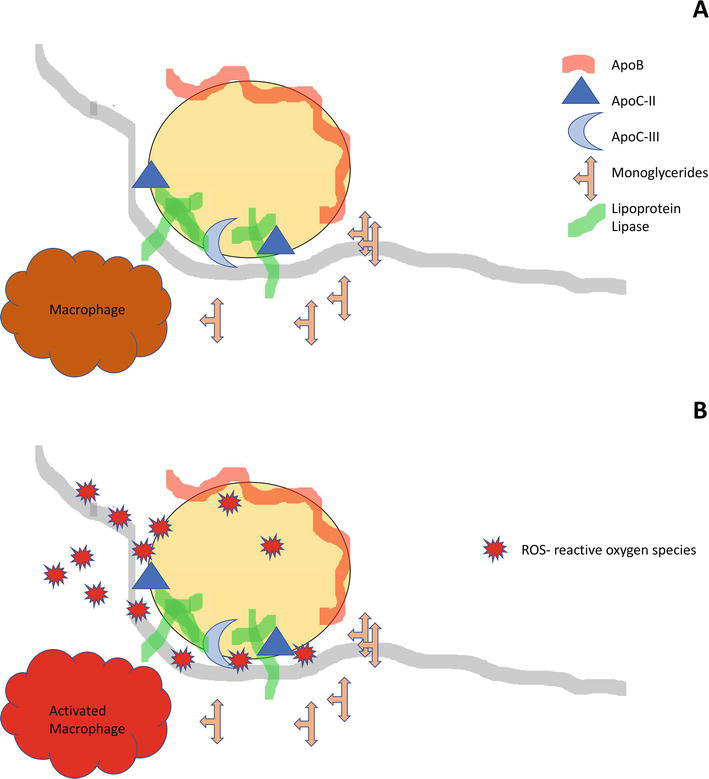
Figure 2.
Interactions of TG-rich lipoproteins with endothelial lipoprotein lipase.
The tight junction formed between TG-rich lipoproteins and the endothelium during TG hydrolysis are suitable for the flow of newly released monoglycerides and free fatty acids into the sub-endothelium. This flow of metabolites, however, could be bi-directional. We postulate that certain metabolites could also diffuse from the sub-endothelium onto the lipoproteins that are still in the circulation. In the presence of an inflamed arterial wall, excess reactive oxygen species (ROS) that are generated by resident activated macrophages could potentially seed fatty acids, preferentially highly oxidizable polyunsaturated fatty acids present on plasma TG-rich lipoproteins (Figure 2B). These particles could subsequently deliver oxidatively modified fatty acids to other tissues and thus propagating the oxidative process.
To examine this hypothesis, we carried out a number of studies looking at the meal-induced changes in oxidative markers. In a group of patients with documented coronary artery disease (CAD) we reported an acute and transient reduction in the levels of circulating autoantibodies (AAb) against malondialdehyde (MDA)-modified low-density lipoproteins (LDL), MDA-LDL following meal consumption [16]. This meal-induced reduction in AAb levels was not observed in a group a young healthy individuals with no history of heart disease [16]. In a group of patients with hypercholesterolemia, we subsequently reported that this transient response could not be demonstrated following oral challenges enriched either saturated or monounsaturated fatty acids but was specific for test meal containing polyunsaturated fatty acids that are highly susceptible to oxidative modification [17].
More recent unpublished data from our laboratory further support the significance of oxidative modification during the interactions between plasma lipoproteins and the arterial wall. In a group of 57 men and women we were able to analyze baseline plasma and plasma obtained at 15 minutes following an intravenous bolus injection of heparin (60 IU/kg body weight). Participants were enrolled in a clinical trial conducted at the University of Miami (courtesy of Dr. Ronald Goldberg and his team) and received the heparin to assess the activities of heparin-releasable lipolytic enzymes. Participants were classified as diabetes mellitus (DM), impaired glucose tolerance (IGT), and normal glucose tolerance (NGT) according to the response to the standard oral glucose tolerance test. Pre- and post-heparin plasma were immediately frozen at −80°C and shipped in dry ice to our laboratory at the end of the study. Plasma levels of autoantibodies against MDA-LDL (AAb) and LDL-IgG immune complexes (IC) were measured by ELISA as previously described [16]. Plasma levels of oxLDL were determined by enzyme-linked immunoassay (ELISA) using a commercial kit (www.mercodia.com). Figure 3 illustrates the percent increased (%D) in plasma levels of oxLDL, AAb, and IC following heparin injection. There was a 25% increase in plasma oxLDL for NGT as compared to 22% for DM + IGT (not significant). In fact, while the increase from pre-heparin levels were statistically significant (p < 0.01), diabetic status did not affect the changes in any measured parameter following heparin. This observation suggests that there are significant markers of oxidatively modified lipoproteins attached to the arterial wall that could be released by heparin.
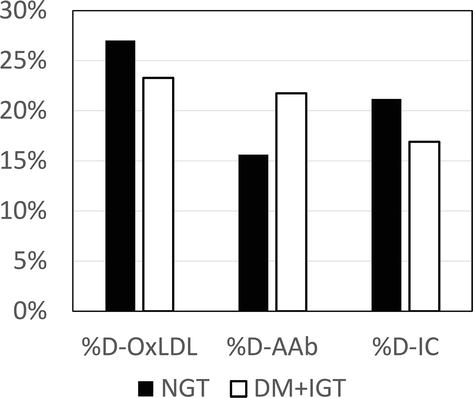
Figure 3.
Increases in markers of lipoprotein oxidation following intravenous heparin.
Using the same ELISA assay for oxLDL (www.mercodia.com), we measured plasma levels of oxLDL and apoB in frozen samples previously collected from our postprandial studies [16, 17]. Plasma apoB was determined by immunoturbidometric method on the AU480 chemistry autoanalyzer (Beckman Diagnostics, LaBrea, CA) using reagents and calibrators from Sekisui Diagnostics (www.sekisui-dx.com). Figure 4 illustrates the fold-increases in plasma apoB and the ratio of oxLDL/apoB following meal consumption. While there was minimal change in plasma apoB during the postprandial period for these two representative individuals, there was statistically significant increase in oxLDL/apoB at 4 hours following the meal. In fact, in individuals with documented CAD, it would appear that the oxLDL levels were elevated by 2 hours after the meal and remained elevated at 6 hours in some individuals. Additional studies are needed to confirm these increases in direct markers of lipoprotein oxidation following heparin and meal consumption.
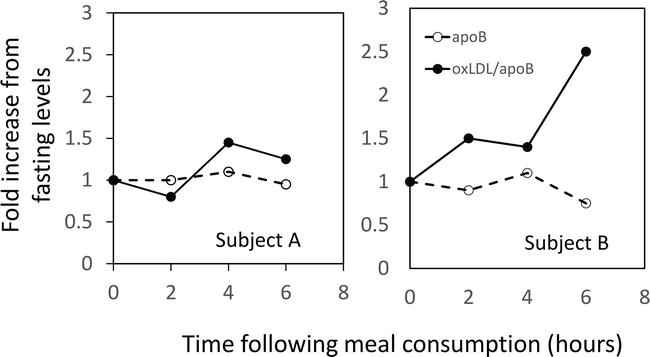
Figure 4.
Increases in oxLDL following meal consumption.
As summarized in this section, the interactions of plasma lipoproteins, in particular TG-rich lipoproteins with endothelial lipase could potentially play a central role in atherogenesis.
3. Probucol as a fat-soluble antioxidant
While hypolipidemic drugs, in particular bile acid resins [18] and 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors [19], have been the focus of pharmaceutical approach to the management of atherosclerosis [20], recent studies would suggest that certain antioxidants may also play a role in the prevention of atherosclerosis.
Probucol is a diphenolic compound that has a modest cholesterol-lowering effect before the introduction of the statins [21]. In vitro experiments indicated that probucol can inhibit oxidative modification of LDL [22], inhibit lipid accumulation in THP-1 cells, and enhance the release of cholesterol from macrophages [23]. In the landmark study using the Watanabe heritable hyperlipidemic (WHHL) rabbit model, Carew et al. [24] demonstrated that in spite of negligeable reduction in plasma cholesterol, animals treated with probucol had significantly less arterial disease as compared to animals treated with the HMG CoA reductase inhibitor lovastatin. In view of the observation that probucol is a lipid-soluble compound and is found associated with plasma LDL, subsequent studies demonstrated that LDL isolated from patients treated with probucol was resistant to ex vivo oxidation [25]. The ability of probucol to protect LDL from oxidative modification appears to be concentration dependent [26]. Furthermore, lipoprotein oxidation does not proceed until available probucol has been degraded [26].
While the mechanism responsible for the in vivo oxidative modification of LDL is still poorly understood, several in vitro processes have been demonstrated, including exposure to lipid oxidation products [27], monocyte-macrophages [28], endothelial cells [29] and smooth muscle cells [30]. Damaged endothelium can generate reactive intermediates that result in the oxidation of LDL [4] which trigger the release of growth factors responsible for tissue proliferation. Several trials with probucol in the prevention of restenosis after coronary angioplasty highlighted the importance of the timing of antioxidant therapy in the management of atherosclerosis. In an earlier trial when probucol + lovastatin was started 48 hours prior to angioplasty, minimal protection from restenosis was noted [31]. When probucol was administered 30 days prior to angioplasty, significant improvement in all indices of restenosis was observed [32]. While plasma levels of both probucol and alpha-tocopherol were at plateau level at the time of angioplasty, treatment with multivitamins (beta carotene + vitamin E + vitamin C) did not have any effect on the rate of restenosis.
The key take-away points from these studies are (1) the antioxidant protection depends on the concentration of probucol on the LDL particles and (2) the efficacy of the antioxidant therapy depends on prior exposure of the arterial wall to lipoproteins that are protected from oxidative modification. It should also point out that the modest cholesterol-lowering effect (10–20%) achieved with probucol was accounted by reduction in both low-density lipoproteins (LDL) and high-density lipoproteins (HDL). The reduction in HDL-cholesterol was reported to be the result of reduced production of the primary proteins of HDL [21] and is considered undesirable as HDL is regarded as a negative risk factor for coronary heart disease [33].
4. AGI-1067 as a fat-soluble antioxidant
Succinobucol also known as AGI-1067 is a synthetic analogue of probucol with strong anti-oxidant properties [34, 35]. In the Canadian Antioxidant Restenosis Trial (CART-1), treatment with either AGI-1067 or probucol before and after percutaneous coronary intervention (PCI) resulted in increases in luminal area at the PCI site [36].
In a series of animal studies including cholesterol-fed cynomolgus monkeys, cholesterol-fed rabbits, LDL-receptor (LDLr) −/− and apoE −/− mice, lipoproteins fractionated from frozen plasma by fast-phase liquid chromatography [37, 38] were shown to contain AGI-1067 [34]. LDL fractions from animals treated with AGI-1067 had reduced reactivity against a monoclonal antibody specific to oxidatively modified LDL and were protected from ex vivo oxidation in the presence of Cu2SO4 [34].
Consistent with these observations, an investigator-initiated, nested case-control study with frozen plasma from participants in the CART-1 trial also noted that both very low density lipoproteins (VLDL) and LDL from individuals receiving AGI-1067 were protected from ex vivo oxidative modification. Frozen plasma obtained from 40 participants at baseline and at 6-month of treatment with either AGI-1067 or placebo were adjusted to a density of 1.21 gm/mL to isolate the lipoprotein-rich fraction by ultracentrifugation. The lipoprotein-rich supernatants were subsequently fractionated by fast-phase liquid chromatography [37, 38] and VLDL and LDL fractions were pooled based on the cholesterol profile as previously described. ApoB contents in the pooled fractions corresponding to VLDL and LDL was quantitated by immunoassay and used to determine the amount of Cu2SO4 required for the ex vivo oxidation assay [39]. The detailed methodology has been published previously [40]. Figure 5 illustrates the decrease in oxidative susceptibility of LDL isolated from a participant receiving AGI-1067 (Figure 5A) as compared to the lack of change for LDL isolated from an individual receiving placebo (Figure 5B). Figure 6 illustrates the decrease in oxidative susceptibility of VLDL after 6-month therapy with AGI-1067 (Figure 6A) as compared to VLDL from an individual treated with placebo (Figure 6B). It should be noted that the ratio of Cu2SO4 to apoB was the same for all incubations and differences in maximum OD (270 nm) reflected total lipid contents in the incubation mixture. The mean increase in lag times with AGI-1067 was 260% (±50%) for isolated LDL and 110% (±35%) for isolated VLDL (unpublished data).
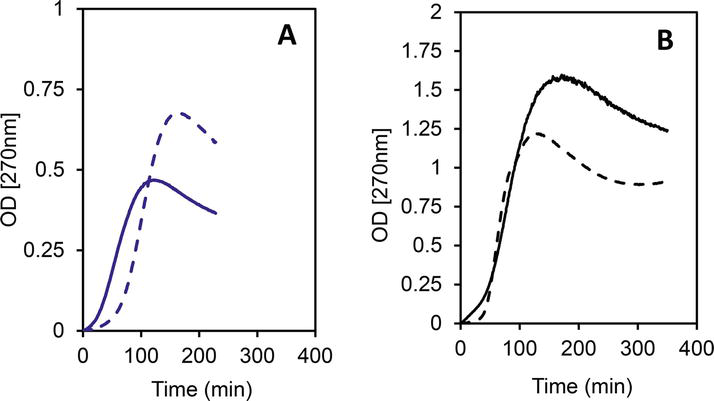
Figure 5.
Ex vivo oxidative susceptibility of LDL with AGI-1067 (Panel A) v. placebo (Panel B). Baseline (solid line) post-treatment (dashed line).
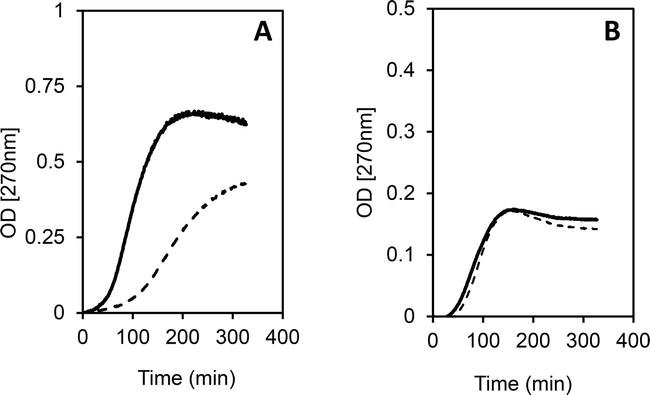
Figure 6.
Ex vivo oxidative susceptibility of isolated VLDL with AGI-1067 (Panel A) v. placebo (Panel B). Baseline (solid line) post-treatment (dashed line).
5. Vitamin E as a fat-soluble antioxidant
With this backdrop of beneficial effect of antioxidant agents on the progression of coronary artery atherosclerosis, there was considerable interest in trials with nutritional antioxidants [41, 42]. In the Basel Study [43] of healthy males monitored over a 7-year period, while there was no association between coronary death and vitamin E levels at baseline, risk of coronary death was higher in the lowest quartile of vitamin C and beta-carotene. Several studies have suggested that antioxidant vitamins, specifically vitamin E, in humans may decrease the susceptibility of LDL to oxidation ex vivo [44, 45, 46]. In a non-randomized trial with serial coronary angiography, Hodis et al. [47] reported that men with previously coronary artery bypass graft surgery demonstrated less lesion progression when colestipol-niacin therapy was supplemented with vitamin E and C. They also found no benefit of vitamin A and C supplementation in patients receiving placebo [47].
In a double-blind, placebo controlled trial with α-tocopherol or placebo, administered after successful percutaneous transluminal coronary angioplasty, the 31% reduction in incidence of restenosis with tocopherol did not reach statistical significance [48] in spite of a high dose of vitamin E supplementation (1200 IU/day). Possible explanations for the lack of significance ranged from small sample size to short-term follow-up, timing of the supplementation, and mode of administration. A substudy of the Heart Outcomes Prevention Evaluation (HOPE) trial in patients with known cardiovascular disease, supplementation with vitamin E at 400 IU/day had no effect on atherosclerosis progression as assessed by ultrasound measurements [49]. In the secondary prevention trial (CHAOS, Cambridge Heart Antioxidant Study) of patients with angiographically proven CAD, there was a 47% reduction in the primary end-point (cardiovascular death and nonfatal myocardial infarction) with vitamin E supplementation as compared to placebo [50]. In fact, this result was attributable to a reduction in the risk for nonfatal myocardial infarction with an 18% increase in cardiovascular death in the vitamin E group. Subsequent analysis suggested that only a small number of the deaths was actually compliant with the vitamin E regimen [51]. A number of studies also failed to demonstrate conclusive benefit of vitamin E supplementation, including the Gruppo Italiano per lo Studio Della Sopravvivvenza nell’Infarto Miocardico (GISSI)-Prevenzione Trial [52], the Heart Protection Study [53], and the Alpha-Tocopherol/Beta-Carotene Cancer (ATBC) prevention study [54].
A review of vitamin E metabolism may shed some light into these somewhat conflicting observations [55]. There are several natural forms of vitamin E typically classified as either tocopherols or tocotrienols, with α, β, γ, δ isoforms. The activity of these various forma depends on its absorption, transport, and delivery to the tissues. Dietary vitamin E are absorbed, secreted into the circulation as chylomicrons carrying newly absorbed TG. As chylomicrons are metabolized in the circulation, vitamin E are transferred to peripheral tissues. This association between chylomicron metabolism and vitamin E might explain the benefit of dietary but not supplemental vitamin E in heart disease outcome [56]. The importance of fat absorption and vitamin E bioavailability was further confirmed by a series of study by Traber and co-workers [57, 58, 59].
In the liver, α-tocopherol transfer protein (α-TTP) preferentially enriched hepatic very low density lipoproteins (VLDL) with α-tocopherol. This process explains why α-tocopherol is the most abundant form of vitamin E in plasma, but γ-tocopherol is the most abundant form in western diet. As chylomicrons are responsible for the delivery of dietary TG to peripheral tissues, VLDL are responsible for the delivery of newly synthesized TG to peripheral tissues. As VLDL are metabolized, α-tocopherol are transferred to other plasma lipoproteins, including low density lipoproteins (LDL) and high density lipoproteins (HDL). Impaired metabolism of VLDL would be expecting to affect the transfer of α-tocopherol to LDL and thus affect its resistance to oxidative modification.
In a recent review, Vardi et al. emphasized the importance of proper patient selection in the assessment of the role of vitamin E in the prevention of cardiovascular disease [60]. To ensure the optimal bioavailability of fat-soluble antioxidants, it is critical to have an adequate amount of dietary fats to enhance the absorption and incorporation of antioxidants in the TG-rich chylomicrons for secretion into the bloodstream.
6. Role of postprandial lipoproteins in the delivery of antioxidants
As in the case of both probucol and AGI-1067 which are fat soluble and are most effective when administered with meals, the bioavailability of vitamin E depends on fat absorption and incorporation of vitamin E in chylomicrons for delivery to the peripheral tissues. Fat-soluble antioxidants are secreted by the intestine as chylomicrons and are transferred to peripheral tissues as the TG-rich lipoproteins are hydrolyzed by lipoprotein lipase along the arterial wall. Thus, by delivering antioxidant agents directly to the peripheral tissues, it may be possible to affect the oxidative status of the arterial wall. This process may explain the increased benefit observed when treatment with probucol was initiated several weeks prior to angioplasty [31, 32] and allowed to continue for extended period after the procedure.
In view of the data from Traber and co-workers [58] on the bioavailability of vitamin E with different administration regimens, new clinical trials with specific instructions for participants to take the vitamin E supplement with meals would be required to demonstrate the benefits of vitamin E in cardiovascular disease prevention.
7. Conclusion
The bioactivity of dietary supplements and oral agents depends on several factors including absorption, transport and delivery. We have reviewed studies that support the hypothesis that meal consumption is associated with in vivo generation of lipoprotein-associated biomarkers of oxidative stress [16] as well as changes in ex vivo oxidative modification of lipoproteins [40]. We also presented preliminary data suggesting that subpopulation of lipoproteins that are retained along the arterial wall consists primarily of oxidatively modified particles. The in vivo generation of lipoprotein-associated lipoproteins reported in patients with inflamed endothelium suggests that the interactions of TG-rich lipoproteins during postprandial lipemia could allow the transfer of ROS from the sub-endothelium to plasma lipoproteins [16]. The observation that this postprandial response is transient and specific for meals enriched in polyunsaturated fatty acids [17] further suggest that the type of fatty acids may affect antioxidant property. Without the double bonds, monounsaturated fatty acids are not susceptible to oxidative modification from ROS transferred to plasma lipoproteins during and would not promote the propagation of ROS to other tissues. In conclusion, for optimal benefit from interventions with fat-soluble antioxidants, it is important that the antioxidant be administered with meal and the consideration for the type and quantity of fatty acids be taken into account to ensure the optimal synthesis and secretion of intestinal chylomicrons. Impaired metabolism of these intestinal lipoproteins would affect the delivery of antioxidants to the peripheral tissues and thus the beneficial effect of the treatment.
References
- 1.
Ross R. The pathogenesis of atherosclerosis: A perspective for the 1990's. Nature. 1993; 362 :801-809 - 2.
Zilversmit DB. Atherogenesis: A postprandial phenomenon. Circulation. 1979; 60 :473-485 - 3.
Zilversmit DB. A proposal linking atherogenesis to the interaction of endothelial lipase with TG-rich lipoproteins. Circulation Research. 1973; 33 :633-638 - 4.
Steinberg D, Parthasarathy S, Carew TE, Khoo JD, Witztum JL. Beyond cholesterol: Modifications of LDL that increase its atherogenicity. The New England Journal of Medicine. 1989; 320 :915-924 - 5.
Ross R. Atherosclerosis: An inflammatory disease. The New England Journal of Medicine. 2002; 340 :115-126 - 6.
Steinberg D, Witztum JL. Oxidized low-density lipoprotein and atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010; 30 (12):2311-2316 - 7.
Sies H. Oxidative stress: A concept in redox biology and medicine. Redox Biology. 2015; 4 :180-183 - 8.
Tousoulis D, Papageorgiou N, Andourlakis E, Siasos G, Latsios G, Tentolouris K, et al. Diabetes mellitus-associated vascular impairment. Journal of the American College of Cardiology. 2013; 62 :667-678 - 9.
Moreira P, Sayre L, Zhu X, Nunomura A, Smith MA, Perry G. Detection and localization of markers of oxidative stress by in situ methods: Application in the study of Alzheimer disease. Methods in Molecular Biology. 2010; 610 :419-434 - 10.
Dominiczak M. In: Rifai N, Warnick G, Domniczak M, editors. Handbook of Lipoprotein Testing. Washington, DC: Amer Assoc Clin Chem; 2000. pp. 1-29 - 11.
Schaefer E, McNamara J. In: Rifai N, Warnick G, Dominiczak M, editors. Handbook of Lipoprotein Testing. Washington, DC: AACC Press; 2000. pp. 77-101 - 12.
Brown WV, Levy RI, Fredrickson DS. Further characterization of apoproteins from the human plasma very low density lipoproteins. The Journal of Biological Chemistry. 1970; 245 :6588-6594 - 13.
Brown WV, Baginsky ML. Inhibition of lipoprotein lipase by an apoprotein of very-low density lipoproteins. Biochemical and Biophysical Research Communications. 1972; 46 :375-382 - 14.
Rensen PCN, Herijgers N, Netscher MH, et al. Particle size determines the specificity of apoE-containing TG-rich emulsions for the LDL receptor versus hepatic remnant receptor in vivo. Journal of Lipid Research. 1997; 38 :1070-1084 - 15.
Havel RJ. Receptor and non-receptor mediated uptake of chylomicron remnants by the liver. Atherosclerosis. 1998; 141 :S1-S7 - 16.
Le N-A, Li X, Kyung S, Brown WV. Evidence for the in vivo generation of oxidatively modified epitopes in patients with documented CAD. Metabolism. 2000; 49 :1271-1277 - 17.
Gradek Q , Harris M, Yahia N, Davis W, Le N-A, Brown W. Polyunsaturated fatty acids acutely suppress antibodies to malondialdehyde-modified LDL in patients with vascular disease. The American Journal of Cardiology. 2004; 93 :881-885 - 18.
Group L-CW. The lipid research clinics coronary primary prevention trial results: I. Reduction in incidence of coronary heart disease. Journal of the American Medical Association. 1984; 251 :351-364 - 19.
Brown G, Albers JJ, Fisher LD, Schaefer SM, Lin JT, Kaplan C, et al. Regression of CAD as a result of intensive lipid-lowering therapy in men with high levels of apoB. The New England Journal of Medicine. 1990; 323 :1289-1298 - 20.
Dujovne C, Harris W. The pharmacological treatment of dyslipidemia. Annual Review of Pharmacology and Toxicology. 1989; 29 :265-288 - 21.
Atmeh R, Stewart J, Boag D, Packard C, Lorimer A, Shepherd J. The hypolipidemic action of probucol: A study of its effects on high and low density lipoproteins. Journal of Lipid Research. 1983; 24 :588-595 - 22.
Parthasarathy S, Young SG, Witztum JL. Probucol inhibits oxidative modification of LDL. The Journal of Clinical Investigation. 1986; 73 :641-644 - 23.
Yamamoto A, Hara H, Takaichi S, Wakasugi J, Tommikawa M. Effect of probucol on macrophages, leading to regression of xanthomas and atheromatous vascular lesions. The American Journal of Cardiology. 1988; 62 :31B-36B - 24.
Carew T, Schwenke D, Steinberg D. Antiatherogenic effect of probucol unrelated to its hypocholesterolemic effects. Proceedings of the National Academy of Sciences of the United States of America. 1987; 84 :7725-7729 - 25.
Regnstrom J, Walldius G, Carlson L, Nilsson J. Effect of probucol treatment on the susceptibility of LDL isolated from hypercholesterolemic patients to become oxidatively modified in vitro. Atherosclerosis. 1990; 82 :43-51 - 26.
Barnhart RL, Busch SJ, Jackson RL. Concentration-dependent antioxidant activity of probucol in LDL in vitro: Probucol degradation precedes lipoprotein oxidation. Journal of Lipid Research. 1989; 30 :1703-1710 - 27.
Fogelman AM, Schechter I, Seager J. Malondialdehyde alteration of LDL leads to cholesteryl ester accumulation in human monocyte macrophages. Proceedings of the National Academy of Sciences of the United States of America. 1980; 77 :2214-2218 - 28.
Cathcart M, Morel D, Chilsom G. Monocytes and neutrophils oxidize LDL making it cytotoxic. Journal of Leukocyte Biology. 1985; 38 :341-350 - 29.
Steinbrecher U, Parsarathy S, Leake D, Witztum JL, Steinberg D. Modification of LDL by endothelial cells involves lipid peroxidation and degradation of LDL phospholipids. Proceedings of the National Academy of Sciences of the United States of America. 1984; 81 :3883-3887 - 30.
Heinecke J, Baker L, Rosen H, Chait A. Superoxide-mediated modification of LDL by arterial smooth muscle cells. The Journal of Clinical Investigation. 1986; 77 :757-761 - 31.
O'Keefe JJ, Stone G, McCallister BJ, Maddex C, Ligon R, Kacich R, et al. Lovastatin plus probucol for prevention of restenosis after percutaneous transluminal coronary angioplasty. The American Journal of Cardiology. 1996; 77 :649-652 - 32.
Tardif J, Cote G, Lesperance J, Bourassa M, Lambert J, Doucet S, et al. Probucol and multivitamins in the prevention of restenosis after coronary angioplasty. New England Journal of Medicine. 1997; 337 :365-372 - 33.
Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. HDL as a protective factor against CHD. The American Journal of Medicine. 1977; 62 :707-714 - 34.
Sundell C, Somers P, Meng C, Hoong L, Suen K, Hill R, et al. AGI-1067: A multifunctional phenolic antioxidant, lipid modulator, anti-inflammatory and antiatherosclerotic agent. The Journal of Pharmacology and Experimental Therapeutics. 2003; 305 :1116-1123 - 35.
Meng C, Somers P, Hoong L, Zheng X, Ye Z, Worsencroft K, et al. Discovery of novel phenolic antioxidants as inhibitors of vascular adhesion molecule-1 expression for use in chronic inflammatory diseases. Journal of Medicinal Chemistry. 2004; 47 :6420-6432 - 36.
Tardif J, Gregoire J, Schwatrz L, Title L, Laramee L, Reeves F, et al. Effects of AGI-1067 and probucol after percutaneous coronary interventions. Circulation. 2003; 107 :552-558 - 37.
Innis-Whitehouse W, Li X, Brown WV, Le NA. An efficient chromatographic system for lipoprotein fractionation using whole plasma. Journal of Lipid Research. 1998; 40 :679-690 - 38.
Le NA, Innis-Whitehouse W, Li X, Bakker-Arkema R, Black D, Brown WV. Lipid and apolipoprotein levels and distribution in patients with hypertriglyceridemia: Effect of triglyceride reductions with atorvastatin. Metabolism. 2000; 49 (2):167-177 - 39.
Esterbauer H, Striegl G, Puhl H, Rotheneder M. Continuous monitoring of in vitro oxidation of human LDL. Free Radical Biology & Medicine. 1989; 6 :67-75 - 40.
Le N-A, Farkas-Epperson M, Sweeney ME, Wilson PWF, Brown WV. Effect of ABT-335 (fenofibric acid) on meal-induced oxidative stress in patients with metabolic syndrome. Atherosclerosis. 2013; 231 :268-273 - 41.
Chilsolm G. Antioxidants and atherosclerosis: A current assessment. Clinical Cardiology. 1991; 14 :25-30 - 42.
Harris W. The prevention of atherosclerosis with antioxidants. Clinical Cardiology. 1992; 15 :636-640 - 43.
Gey K, Stagelin H, Eichholzer M. Poor plasma status of carotene and vitamin C is associated with higher mortality from ischemic heart disease and stroke. Basel Prospective Study. The Clinical Investigator. 1993; 71 :3-6 - 44.
Dieber-Rotheneder M, Puhl H, Waeg G, Striegl G, Esterbauer H. Effect of oral supplementation with d-a-tocopherol on the vitamin E content of human LDL and its oxidation resistance. Journal of Lipid Research. 1991; 32 :1325-1332 - 45.
Princen H, van Poppel G, Vogelezang C, Buytenhek R, HJ K. Supplementation with vitamin E but not beta-carotene in vivo protects LDL from lipid peroxidation in vitro: Effect of cigarette smoking. Arteriosclerosis and Thrombosis. 1992; 12 :554-562 - 46.
Jialal I, Grundy S. Effect of dietary supplementation with alpha-tocopherol on the oxidative modification of LDL. Journal of Lipid Research. 1992; 87 :597-601 - 47.
Hodis H, Mack W, LaBree L, Cashin-Hemphill L, Sevanian A, Johnson R, et al. Serial coronary angiographic evidence that antioxidant vitamin intake reduces progression of coronary atherosclerosis. Journal of the American Medical Association. 1995; 273 :1849-1854 - 48.
DeMaio S, King S, Lembo N, Roubin G, Hearn J, Bhagavan H, et al. Vitamin E supplementation, plasma lipids and incidence of restenosis after percutaneous transluminal coronary angioplasty. Journal of the American College of Nutrition. 1992; 11 :68-73 - 49.
Lonn E, Yusuf S, Dzavik V, Doris I, Yi Q , Smith S, et al. Effects of ramipril and vitamin E on atherosclerosis. Circulation. 2001; 103 :919-925 - 50.
Stephens N, Parsons A, Brown M, Schofield P, Kelly F, Cheeseman K, et al. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet. 1996; 347 :781-786 - 51.
Mitchinson M, Stephens N, Parsons A, Bligh E, Schofield P, Brown M. Mortality in the CHAOS trial. Lancet. 1999; 353 :381-382 - 52.
Anonymous. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after MI: Results of the GISSI-Prevenzione trial. Lancet. 1999; 354 :447-455 - 53.
MRC/BHF. MRC/BHF Heart Protection Study of cholesterol-lowering therapy and of antioxidant vitamin supplementation in a wide range of patients at increased risk of coronary heart disease: Early safety and efficacy experience. European Heart Journal. 1999; 20 :725-741 - 54.
Virtamo J, Rapola J, Ripatti S, Heinonen O, Taylor P, Albanes D, et al. Effect of vitamin E and beta carotene on the incidence of primary nonfatal myocardial infarction and fatal coronary heart disease. Archives of Internal Medicine. 1998; 158 :668-675 - 55.
Schmolz L, Birringer M, Lorkowski S, Wallert M. Complexity of vitamin E metabolism. World Journal of Biological Chemistry. 2016; 7 :14-43 - 56.
Kushi L, Folsom A, Prineas R, Mink P, Wu Y, Bostick R. Dietary antioxidant vitamins and death from coronary heart disease in postmenopausal women. The New England Journal of Medicine. 1996; 334 :1156-1162 - 57.
Traber M, Goldberg I, Davidson E, Lagmay N, Kayden HJ. Vitamin E uptake by human intestinal cells during lipolysis in vitro. Gastroenterology. 1990; 98 :96-103 - 58.
Leonard S, Good C, Gugger E, Traber M. Vitamin E bioavailability from fortified breakfast cereal is greater than that from encapsulated supplements. The American Journal of Clinical Nutrition. 2004; 79 :86-92 - 59.
Traber M, Leonard S, Ebenuwa I, Violet P, Niyyati M, Padayatty S, et al. Vitamin E catabolism in women as modulated by food and by fat studied using 2 deuterium-labeled alpha-tocopherols in a 3-phase nonrandomized crossover study. The American Journal of Clinical Nutrition. 2021; 113 :92-103 - 60.
Vardi M, Levy N, Levy A. Vitamin E in the prevention of cardiovascular disease: The importance of proper patient selection. Journal of Lipid Research. 2013; 54 :2307-2314