 Open access peer-reviewed chapter
Open access peer-reviewed chapter
Abstract
Recently, the enthalpies of formation computed for different Lewis acid complexes of unsaturated molecules (e.g., aldehydes, imines, alkynes, and alkenes) offer a way to assess the suitability of a specific catalyst in a catalytic reaction. Main-group chemistry, in particular boron chemistry, which has resulted in a multitude of novel discoveries is one area of organometallic chemistry that has made significant strides during the past 20 years. In this review, the recent developments and applications of well-proven chemicals like tris(pentafluorophenyl)borane (B(C6F5)3) which has shown widespread applicability in a variety of chemistry have been discussed. Additionally, a number of brand-new Lewis acidic boranes and borocations have recently been reported, many of which are structurally suited for a particular use for main-group catalysis or borylation processes. These compounds undergo a variety of processes including borylation substitution and the addition of B-E across-bonds and they are used in the pharmaceutical and material sciences. Boron reagents also frequently make up the Lewis acid moiety of frustrated Lewis pairs (FLPs).
Keywords
- Lewis acid
- boron; catalysis
- tris(pentafluorophenyl)borane
- boron reagents
- elementoboration reactions
1. Introduction
Boron reagents exhibit powerful electrophilic nature and utilized in Lewis acids due to the presence of a p-orbital (vacant) that can readily accept donor molecules electrons. The variety of boron reagents is growing as a result of growth in the field of boron chemistry. A string of catalysts boron with increasing structural complexity and tunable acidity arose over the recent years comprising triaryl, trialkyl, and trihalo-boranes [1].
The boron-based catalysts have different properties, for instance, boron trihalides with a chemical formula of BX3 (X = F, Cl) are very sensitive to moisture and volatile in nature making them tough to handle. However, they are very effective as catalysts (Figure 1). According to studies conducted on perfluoroalkyl boranes are thermally unstable and are strong Lewis acid due to their highly electronegative fluorinated ligands [2]. Consequently, the emphasis was turned on halogenated triarylboranes, like tris(penta-fluorophenyl)-borane designated with a chemical formula, B(C6F5)3 and are commonly referred to as BCF (Figure 1). Tris(pentafluorophenyl)borane, also known as BCF, was first synthesized by Massey in the 1960s [3, 4].

Figure 1.
(a) Acidity of various Lewis acids (boron-based) (b) (BCF). Reproduced with permission from ref. [
The field has not received much attention by the community (scientific) in spite of its exciting features until the advent of its application in the polymerization of olefins with metallocenes as activators. It has been reported by [6] that B(C6F5)3 is among the well-defined and known activator in the area of polymerization (olefin) catalysis. Moreover, B(C6F5)3 (Lewis acid) is capable of abstracting alkyl groups from a metal (transition) center. In hydrocarbons, the main-group (e.g., Al and Ga) alkyls may also react quickly with this borane at ambient temperature, resulting in complete alkyl/aryl group exchange. This is a reliable and an easy approach to obtain compounds of galane and tris(pentafluorophenyl)alane [7].
According to the findings reported by [8], Frustrated Lewis Pairs (FLPs) (e.g., H2 and CO2
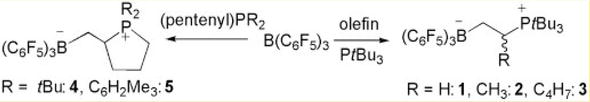
Figure 2.
Phosphine/borane pairs of olefins (inter- and intramolecular addition). Reproduced with permission from ref. [
2. Lewis acidic boron reagents synthesis
The preparation of halogenated triarylboranes (homoleptic) can be achieved by the application of either one of these two processes: the lithiation technique or the Grignard technique. Massey et al. [4] defined the BCF synthesis by employing a Grignard reagent with BCl3 for the first time. Moreover, the lithiation method was reported somewhere else [9]. The general procedure for both methods is presented in Figure 3.

Figure 3.
Grignard and lithiation methods for synthesis of homoleptic halogenated triarylboranes. Reproduced with permission from ref. [
Tris(perfluorotolyl)borane [B(4-(CF3)C6F4)3] has been synthesized by employing Grignard reagent to synthesis intermediate of arylcopper. This intermediate of copper then underwent reaction of transmetalation with BBr3 to produce the desire product (Figure 4) [10].

Figure 4.
Synthesis of [B(4-(CF3)C6F4)3] designated by No. 8 using the arylcopper intermediate designated by No. 7. Reproduced with permission from ref. [
Different purification strategies have been adopted for increasing the yield of the desired product. For instance, B(4-FC6H4)3, B(2-FC6H4)3, B(2,6-F2C6H3)3, B(3,4,5-F3C6H2)3, B(3,5-(CF3)2C6H3)3, and B(2,4,6-F3C6H2)3 have been purified using sublimation after synthesis with Grignard method [11, 12, 13, 14, 15]. Moreover, lithiation approach has been employed to prepare boranes with groups of trifluoromethyl on aryl ring (

Figure 5.
Synthesis of BCF designated by No. 11. Reproduced with permission from ref. [
As a result of the complexity of the procedures, few heteroleptic boranes have been synthesized. The heteroleptic borane (e.g., B(C6F5)2(2-(C6F5)C6F4)) that has been synthesized by substituting moiety of one perfluorophenyl by a motif of perfluorobiphenyl. The procedure for synthetic of 2-bromononofluoro-biphenyl portion comprised (C6F5)Li and C6F5Br reaction followed by BCl3 addition into Sn(Me)2(C6F5)2 and generated the B(C6F5)2Cl. Eventually, B(C6F5)2(2-(C6F5)C6F4) was prepared by the reaction between 2-lithiumnonofluoro-biphenyl and B(C6F5)2Cl (Figure 6) [19].

Figure 6.
Preparation of B(C6F5)2(2-(C6F5)C6F4) (simplest heteroleptic borane) designated by No. 16. Reproduced with permission from ref. [
The preparation of another heteroleptic boranes incorporating fluorinated and chlorinated aryl rings was reported somewhere else (Figure 7) [20]. The process included the dihalobenzene derivatives reacting with trimethyl borate and n-butyl lithium, resulting in boronic acids that were then transformed into salt of potassium trifluoroborate. Grignard reagents with various aryl frameworks reacted with the salt intermediates to yield the desired heteroleptic boranes. The resulting boranes exhibited an important property

Figure 7.
Preparation path for boranes (heteroleptic) bearing fluorinated and chlorinated aryl rings designated by No. 21. Reproduced with permission from ref. [
Halogenated triarylborane comprising three types of aryl rings was first synthetized using a five-step process starting from dimethylsulfide [21]. In the first step, Li(C6F5) (single equivalent) was formed at the temperature of -78°C and then H3B(C6F5) was obtained by reacting with borane dimethylsulfide. The hydride (

Figure 8.
The synthesizing process for B(C6F5)(C6Cl5)(3,5-(CF3)2C6H3) designated by No. 27. Reproduced with permission from ref. [
3. Application of novel boranes and borocations
Many new boranes and borocation are utilized in borylation reactions such as hydroboration, dehydroborylation, haloboration, and carboboration. Since each of these reactions has some conditions (most prominently the kind of group bonded to the boron reagent) along with a range of substrates, the potential to functionalize the structure and reduce the boron reagent’s reactivity is essential. The results of these reactions might be viewed as intermediates on the way to the more diverse and valuable molecules, as the addition of a boron species enables further functionalization through future cross-coupling reactions.
Direct borylation substitution reaction (e.g., dehydroborylation) is one field of chemistry of borylation in which C-B bond forms from C-H bond. And also, in this reaction B(C6F5)3 is inappropriate. It has been reported that trialkoxyboranes were appropriate reagents of borylation when joined with organolithiated species like alkynes to obtain alkynyl boronates (1) (Figure 9) [22, 23].

Figure 9.
Borylation of Lithiated alkynes. Reproduced with permission from refs. [
In a more contemporary method, the substrate is deprotonated using a base that can be integrated separately or, in the case of some borocations that can be added into the reagent structure and removing the need for lithiation. Certain species of borenium have been found to selectively dehydroborylate arenes and heteroarenes, according to recent research. Catechol-ligated borocations were used to catalyze arene borylation (intermolecular) (Figure 10) and generated a string of borylated arenes from benzene derivatives. [CatB][CbBr6](CbBr6 = [closo-1-H CB11H5Br6]) is a catalytic quantity of [CatB]+ [24].

Figure 10.
Dehydroborylation of Arenes. Reproduced with permission from ref. [
Several amine-ligated catechol boreniums were created using AlCl3 (halide abstraction catalyst) to obtain borocations [CatB (L)] [AlCl4] (

Figure 11.
Dehydroborylation of Arenes with Borocations. Reproduced with permission from ref. [
Furthermore, boreniums ([Cl2B(L)][AlCl4]) more reactive) were formed and used in arene dehydroborylation reactions (4) (Figure 12) [26, 27]. To boost the electrophilicity of the boreniums, the catechol ligand was replaced by two halides and allowed the reactions to continue rapidly at room temperature. The Lewis base alteration also provided access to many species of diborylated heteroaryl (5). These were isolated as esters of pinacolboronate, providing key synthetic molecules that can act as scaffolding for more complex molecules. For instance, Del Grosso et al. updated their research to include the haloarenes borylation [28] which was a crucial step in the creation of borylation chemistry (metal-free) as the deactivated arenes were not previously available with existing method [24].

Figure 12.
(Hetero) arenes Dehydroborylation. Reproduced with permission from Ref. method [
Furthermore, the borylation of monohaloarenes principally created the para-substituted isomer (6), single product, revealing its ability to control regioselectivity (Figure 13). It was reported that of the haloarene (10 equivalents) and AlCl3 (1 equivalent) were needed at the optimal conditions that were employed as the solvent [24].

Figure 13.
Borocation for arenes dehydroborylation. Reproduced with permission from ref. [
Recently, catalytic method (metal-free) to borylation of arene that employed the borane (1-TMP-2-BH2C6H4)2 as a C-H bond activator has been reported [29] (Figure 14). First, the borane was reacted with the N-methylpyrrole (arene) followed by H2 releasing after the C-H bond activation in the two positions. Then, the intermediate was reacted with H-BPin and regenerated the catalyst and produced the desire pinacolboronate ester (borylated arene) (8).

Figure 14.
Catalystic N-Methylpyrrole dehydroborylation. Reproduced with permission from ref. [
Indoles, furans, and electron-rich thiophene were added to the list of acceptable substrates besides N-substituted pyrroles (yield frequently exceeding 80%). In general, a single isomer of the products was produced with catalyst loading of 2.5 mol%. The employ of this system emphasizes the advantages over conventional pathways (
Moreover, 2-aminophenyl boranes (Figure 15) induced dehydroborylation of a variety of alkenes, arenes, and heteroarenes resulting in species of borylated [30]. This reaction is proposed to continue through an insertion of CH in an FLP-style, in which the amine and boron heterolytically split the CH bond. The close proximity of the Lewis base and acid promotes this reactivity, leading in a small kinetic barrier to reaction for thiophene with ΔG value of 21 Kcal/mol. The reaction of borylation was shown to be neutral (thermodynamically). Additionally, it was found that the equilibrium might be simply shifted by adding more substrate or might be eliminated the by-product (H-R2) in the reaction mixture.

Figure 15.
Alkenes dehydroborylation. Reproduced with permission from ref. [
3.1 Elementoboration reactions
Borocations and boranes can be involved in substitution reactions like dehydroborylations and addition reactions like hydro-, carbo-, and haloboration. In an elementoboration, boron with another group is added to a π-system excluding the necessity of a substitution reaction. There are studies on addition of (1, n) of boron reagents to alkynes (n = 1–4) reported somewhere else [31] which allows the integration of a unit of boron while creating a species of vinyl catalyst [32]. However, many historical examples have required the use of a metal catalyst [32]. With the help of cross-coupling reactions with the boron unit, these borylated alkenes are effective building blocks for more compounds [33].
The drawbacks of B(C6F5)3 become apparent when range of reactions is considered. For instance, despite the abundance of carboboration reaction the product utility was constrained by the need of transfer of C6F5 group. Moreover, further functionalizing challenge through boron group by employing cross-coupling reactions can be more difficult than those of conventional boronate esters and boronic acids [34].
In contrast, Lappert and Prokai’s research focused on haloboration reactions, which involve combining halogen and boron with a nucleophile like an alkyne to create a vinylboron species [35]. The halogen addition makes a new site for functionalization via cross-coupling reactions (Suzuki reaction), even though the final products resemble those of carboboration processes [36]. This is beneficial when the relevant R group can be added to the intermediate product after borylation instead of by direct carboboration.
Until recently, the reaction of haloboration was believed to be a specialized field of main-group chemistry involving terminal alkynes and haloboranes like BCl3 [37]. Recent findings have revealed that borocations with halides can resemble, if not surpass, the reactivity of trihalo-boranes. For instance, the structure and reactivity of a synthesized 2-dimethylaminopyridine (2-DMAP)-ligated borocation [Cl2B-(2-DMAP)][AlCl4] (10) were studied [38]. These classes were known as a boronium cation with acceptable moisture and air tolerance of 5% and decomposed after 24 hours. A low value of energy barrier to opening ring of the largely strained 4-membered ring molecule (

Figure 16.
Borocation energy barrier for opening ring. Reproduced with permission from ref. [
Boronium (10) was done via syn-1,2-haloboration (selective) through a variety of alkynes terminal resulting in vinylpinacolboranes (11) (63–88% yields with isolation of good to high) (Figure 17). Moreover, the analogue bromo [Br2B (2-DMAP)][BBr4] (12) has been presented to undertake haloboration through certain internal and terminal alkynes (

Figure 17.
Alkynes Haloboration through Borocations. Reproduced with permission from ref. [
Increasing the borocation of Lewis acidity resulted in a variety of internal alkynes that could be 1,2-haloborated. Several novel vinylboron molecules were identified as pinacolboronate esters by employing borenium [Cl2B(lut)] [AlCl4] (

Figure 18.
Internal alkynes haloboration. Reproduced with permission from ref. [
Cross-coupling reactions (sequential) of chloro and vinylboron substituents were used to obtain the alkene (highly functionalized) (15) (Figure 19) [39]. It was indicated that the reaction from alkyne to alkene (fully substituted) could be performed in a single pot. These alkenes (tetrasubstituted) are precursors and structural analogues to a number of drug molecules comprising Tamoxifen, Zuclomiphene, and a potent anticancer agent showing the significance of the reaction pathway [40].

Figure 19.
Haloboration products with cross-coupling (consecutive). Reproduced with permission from ref. [
Reactions of carboboration permit for quick creation of new bonds of C-B besides C-C across a p-system [41]. A range of trialkylboranes undergo carboboration with specific activated alkynes; nevertheless, B(C6F5)3 has been recently confirmed as a predilection of these reactions without the necessity of alkynes (activated) [42]. One of the basic examples is the reaction between phenylacetylene and B(C6F5)3 and 1,1-carboboration happens practically rapidly at room temperature but produces isomers of mixture of Z and E (Figure 20). Photoisomerization may be utilized to create a single isomer (16).

Figure 20.
Phenylacetylene carboboration by B(C6F5)3. Reproduced with permission from ref. [
Employing B(C6F5)3 for carboborations is inappropriate as the desired product lacks C6F5 group. B(C6F5)3 was modified with reagents of R-B(C6F5)2 that were prepared and used in reactions of carboboration by transfer of selective R-group (contrast to the transfer C6F5) [43]. However, this has left B(C6F5)2 group into the molecule and prevented additional functionalization due to the difficult behavior of the reaction (cross-coupling) by using boron species (requires higher temperature) and resulted in a small yield than boronate esters [32].
So far, research has presented a variety of heteroleptic borocations and boranes that may be prepared and used in different reactions of carboboration. For instance, the species of neutral borane (general formula: Cl2B-aryl) might be employed for 1,1-carboboration of trimethylsilyl (TMS) substituted alkynes as reported somewhere else [44]. In contrast, unlike other activated alkynes of carboborations, this process uses a group of BCl2 that may be changed into a boronate ester, which is more valuable for synthetic purposes, than trialkyl/arylboranes. Using a range of TMS-alkynes and cheaply available PhBCl2, single-products of vinylborane with outstanding regioselectivity and stereo were created. These products could then be isolated as pinacolboronates (17) (Figure 21). Along with arenes (triphenylamine and p-chlorobenzene) and heteroarenes (PhBCl2) numerous dichloroboranes were also produced.

Figure 21.
TMS-alkynes carboboration. Reproduced with permission from ref. [
In contrast to haloboration, only a few cases of metal-catalyzed carboboration of alkynes have been reported in the literature [45]. However, some recent examples using borocations and boranes have been reported. For instance, quinolatoboreniums were synthesized (19) and when it was exposed to 3-hexyne resulted in 1,2-carboboration and then transferred into either a moiety of thiophenyl or phenyl and the resulting products were esterified by pinacol to yield vinylboronates (isolated) (20) (Figure 22) [46].

Figure 22.
Borocations of 1,2-carboboration of 3-Hexyne. Reproduced with permission from ref. [
Furthermore, 1,2-carboboration was obtained by employing boreniums in which 3-hexyne readily reacted with a phosphorus-coordinated borenium (21) (Figure 23) [47]. As shown in Figure 23 a group mesityl was transferred and selectively generated the vinylboron species (22). The reagent comprises species of novel borenium that employs a scaffold of naphthalene to produce a strong interaction of intramolecular BP. Moreover, it has been shown to be stable, however, it showed an activation of H2, highly reactive and carboboration of 3-hexyne indicating the broad utility of borocations.

Figure 23.
3-Hexyne through 1,2-Carboboration with borocation. Reproduced with permission from refs. [
Recently, Melen et al. presented 1,2-carboboration of allenes by employing boranes [48]. The reactions of esters and allenyl ketones in the presence of B(C6F5)3 gave 1, 2-carboboration products (Figure 24). The reactions (stoichiometric) with allenyl ketones continued at room temperature (30 minutes) confirming the intramolecular structure of chelating cyclic dihydroxyborinine products as confirmed using X-ray crystallography (23).

Figure 24.
Allenyl ketones (1,2-Carboboration) employing B(C6F5)3. Reproduced with permission from ref. [
4. Main-group catalysis with boron reagents
Recently, there is a strong interest in using the main-group elements molecular catalysis than transition metals (TM) as there is a growing concern on the environmental and health impact of residues metals that remains in the products. The main group comprises Frustration Lewis Pair (FLP), single-site amphiphiles (e.g., singlet carbene (CR2) restricted phosphines, Di-coordinated borirene (LBR, L = Lewis donor), and di-coordination group with 14 cations) that begin to emerge as a powerful TM-free reagent for activation of inert substances and small molecules of interest as catalysts [49]. Boron reagents can function as Lewis acid due to their strong electrophilicity with vacancies of a p-orbital that can easily accept an electron from a donor molecule.
Numerous boranes (trialkyl-, triaryl-, and trihalo-boranes) have been produced and used over the past years. A series of boron-grounded catalysts with structural complexity and tunable acidity have been developed [1]. In 1950s, examinations were done on fluorinated ligands (largely electronegative) and perfluoroalkyl boranes and they showed a strong Lewis acidity to the boron center and less hydrolytically sensitivity of B-C bonds. Up to now, perfluoroalkyl boranes displayed thermal insecurity [5]. Catalysis is among the field of main-group chemistry that has flourished in recent times with borocations, boranes, and FLPs gaining features. This reagent has led to the development of carbon-carbon bond-forming responses, hydroboration, and hydrogenation which are conventionally essence-dominated paths [1].
Main-group catalysis comprises a rich soil in C-C bond-forming responses, hydroboration, hydrogenation, and numerous other metamorphoses which are conventionally attained by essence-grounded catalysis.
The low cost and less toxicity properties help in using the main-group elements for catalysis purposes. Recently, researchers are searching for the development of a new approach for greener catalysis. Moreover, less poisonous, earth-abundant, and first-row transition essence approaches are being developed to attain an environmentally-friendly catalysis [50]. Boranes can be used as Lewis acid catalysts as their central boron contains an empty p-orbital. The base of boranes intermediated Lewis acid catalysis is governed by the posterior release and attack of their empty p-orbital.
4.1 Boron catalysts for the formation of C-C bond
The development of a green method has fascinated tons attention all over the community (synthetic) as C-C bond construction in organic chemistry is among the vastest reactions. Among numerous C-C bonds protocols, compounds of organoboron are not simply confined to stoichiometric reagents. However, they owned an outstanding attainment as catalysts due to the easy change of the steric and electronic influences at the center of boron [51]. Tris(pentafluorophenyl)boran has attracted numerous attention among artificial chemists due to sterically hindered conformation at the center of boron (avoids the formation of Lewis adduct by Lewis base), stability (hydrolytic) (
Carboboration reaction allows the facile creation of new bonds of both C-B and C-C throughout a π machine. Historically, trialkylboranes spread have been indicated to undertake carboboration through certain alkynes (activated). Currently, B(C6F5)3 has been tested for a proclivity for this reaction without using of alkynes (activated). For instance, in B(C6F5)3 and phenyl acetylene, 1,1-carboboration has been attained (instantly at room temperature), however, it generated an aggregate of Z and E isomers (Figure 25). Photoisomerization may be employed to produce the unmarried isomer (16) [1].

Figure 25.
Phenylacetylene carboboration with B(C6F5)3. Reproduced with permission from ref. [
4.2 Boron-based catalyzed hydroborations
Hydroboration reactions have been extensively studied by employing metal catalysts. Currently, catalysis (metal-free) which provides a broader variety of substrates (borylated) access without eliminating impurities of trace metals has been reported. Many reactions of hydroboration are an atom-efficient with hydroboranes (e.g., pinacolborane (HBPin)) where selective addition is often detected. Now, numerous examples of boron-based catalysts have been reviewed somewhere else [52].
Hydroboration of alkenes by HBPin and catalyzed with tris[3,5-bis(trifluoromethyl)phenyl] borane (BArF3)6, B(C6F5)3 turned out to be unusable. A series of selective products (

Figure 26.
1,2-hydroboration of aliphatic alkenes and styrenes. Reproduced with permission from ref. [
5. Conclusions and outlook
This chapter presents the review and discussion of the use of boranes in catalysis and exploring new reactivities as well as catalysis of boron that permits to perform identified reactions in milder situations.
The conclusion of this review focuses on Lewis acidic boron reagents synthesis, application of novel boranes and borocations, main-group catalysis with boron reagents, and halogenated triarylboranes design with careful consideration of their Lewis acidity. this relatively new field of catalysis will contribute to synthetic chemistry.
References
- 1.
Lawson JR, Melen RL. Recent developments and applications of Lewis acidic boron reagents. 2017a; 41 :1-27. DOI: 10.1039/9781782626923-00001 - 2.
Piers WE, Chivers T. Pentafluorophenylboranes: From obscurity to applications. Chemical Society Reviews. 1997; 26 (5):345-354 - 3.
Massey AG, Park AJ. Perfluorophenyl derivatives of the elements: I. Tris (pentafluorophenyl) boron. Journal of Organometallic Chemistry. 1964; 2 (3):245-250 - 4.
Massey AG, Stone FGA, Park AJ. Tris (pentafluorophenyl) boron. In: Proceedings of the Chemical Society of London (Issue JUL, p. 212). Royal Soc Chemistry Thomas Graham House, Science Park, Milton Rd, Cambridge; 1963 - 5.
Nori V, Pesciaioli F, Sinibaldi A, Giorgianni G, Carlone A. Boron-based Lewis acid catalysis: Challenges and perspectives. Catalysts. 2022; 12 (1). DOI: 10.3390/catal12010005 - 6.
Klosin J, Fontaine PP, Figueroa R. Development of group IV molecular catalysts for high temperature ethylene-α-olefin copolymerization reactions. Accounts of Chemical Research. 2015; 48 (7):2004-2016 - 7.
Klosin J, Roof GR, Chen EY-X, Abboud KA. Ligand exchange and alkyl abstraction involving (perfluoroaryl) boranes and-alanes with aluminum and gallium alkyls. Organometallics. 2000; 19 (23):4684-4686 - 8.
McCahill JSJ, Welch GC, Stephan DW. Reactivity of “frustrated Lewis pairs”: Three-component reactions of phosphines, a borane, and olefins. Angewandte Chemie International Edition. 2007; 46 (26):4968-4971 - 9.
Ikeda Y, Yamane T, Kaji E, Ishimaru K. Method of Producing Tris (Pentafluorophenyl) Borane Using Pentafluorophenyl Alkali Metal Salt Prepared from Pentafluorobenzene. United States: Google Patents; 1996 - 10.
Körte LA, Schwabedissen J, Soffner M, Blomeyer S, Reuter CG, Vishnevskiy YV, et al. Tris (perfluorotolyl) borane—A boron Lewis superacid. Angewandte Chemie International Edition. 2017; 56 (29):8578-8582 - 11.
Carden JL, Gierlichs LJ, Wass DF, Browne DL, Melen RL. Unlocking the catalytic potential of tris (3, 4, 5-trifluorophenyl) borane with microwave irradiation. Chemical Communications. 2019; 55 (3):318-321 - 12.
Herrington TJ, Thom AJW, White AJP, Ashley AE. Novel H 2 activation by a tris [3, 5-bis (trifluoromethyl) phenyl] borane frustrated Lewis pair. Dalton Transactions. 2012; 41 (30):9019-9022 - 13.
Keess S, Simonneau A, Oestreich M. Direct and transfer hydrosilylation reactions catalyzed by fully or partially fluorinated triarylboranes: A systematic study. Organometallics. 2015; 34 (4):790-799 - 14.
Yin Q , Kemper S, Klare HFT, Oestreich M. Boron Lewis acid-catalyzed hydroboration of alkenes with Pinacolborane: BArF3 does what B (C6F5) 3 cannot do! Chemistry–a. European Journal. 2016b; 22 (39):13840-13844 - 15.
Yin Q , Soltani Y, Melen RL, Oestreich M. BArF3-catalyzed imine hydroboration with pinacolborane not requiring the assistance of an additional Lewis base. Organometallics. 2017; 36 (13):2381-2384 - 16.
Blagg RJ, Lawrence EJ, Resner K, Oganesyan VS, Herrington TJ, Ashley AE, et al. Exploring structural and electronic effects in three isomers of tris {bis (trifluoromethyl) phenyl} borane: Towards the combined electrochemical-frustrated Lewis pair activation of H 2. Dalton Transactions. 2016; 45 (14):6023-6031 - 17.
Cornet SM, Dillon KB, Entwistle CD, Fox MA, Goeta AE, Goodwin HP, et al. Synthesis and characterisation of some new boron compounds containing the 2, 4, 6-(CF 3) 3 C 6 H 2 (fluoromes= Ar), 2, 6-(CF 3) 2 C 6 H 3 (fluoroxyl= Ar′), or 2, 4-(CF 3) 2 C 6 H 3 (Ar ″) ligands. Dalton Transactions. 2003; 23 :4395-4405 - 18.
Toyota S, Asakura M, Oki M, Toda F. Structural and Stereochemical studies of Tris [2-(trifluoromethyl) phenyl] borane: Spontaneous resolution, Stereodynamics, and intramolecular CF··· B interactions. Bulletin of the Chemical Society of Japan. 2000; 73 (10):2357-2362 - 19.
Li L, Stern CL, Marks TJ. Bis (pentafluorophenyl)(2-perfluorobiphenylyl) borane. A new perfluoroarylborane cocatalyst for single-site olefin polymerization. Organometallics. 2000; 19 (17):3332-3337 - 20.
Gyömöre A, Bakos M, Földes T, Pápai I, Domján A, Soós T. Moisture-tolerant frustrated Lewis pair catalyst for hydrogenation of aldehydes and ketones. ACS Catalysis. 2015; 5 (9):5366-5372 - 21.
Blagg RJ, Wildgoose GG. H 2 activation using the first 1: 1: 1 hetero-tri (aryl) borane. RSC Advances. 2016; 6 (48):42421-42427 - 22.
Brown HC, Bhat NG, Srebnik M. A simple, general synthesis of 1-alkynyldiisopropoxyboranes. Tetrahedron Letters. 1988; 29 (22):2631-2634 - 23.
Brown HC, Cole TE. Organoboranes. 31. A simple preparation of boronic esters from organolithium reagents and selected trialkoxyboranes. Organometallics. 1983; 2 (10):1316-1319 - 24.
Del Grosso A, Pritchard RG, Muryn CA, Ingleson MJ. Chelate restrained boron cations for intermolecular electrophilic arene borylation. Organometallics. 2010; 29 (1):241-249 - 25.
Del Grosso A, Singleton PJ, Muryn CA, Ingleson MJ. Pinacol boronates by direct arene borylation with borenium cations. Angewandte Chemie International Edition. 2011b; 50 (9):2102-2106 - 26.
Del Grosso A, Helm MD, Solomon SA, Caras-Quintero D, Ingleson MJ. Simple inexpensive boron electrophiles for direct arene borylation. Chemical Communications. 2011a; 47 (46):12459-12461 - 27.
Bagutski V, Del Grosso A, Carrillo JA, Cade IA, Helm MD, Lawson JR, et al. Mechanistic studies into amine-mediated electrophilic arene borylation and its application in MIDA boronate synthesis. Journal of the American Chemical Society. 2013; 135 (1):474-487 - 28.
Del Grosso A, Carrillo JA, Ingleson MJ. Regioselective electrophilic borylation of haloarenes. Chemical Communications. 2015; 51 (14):2878-2881 - 29.
Légaré M-A, Courtemanche M-A, Rochette É, Fontaine F-G. Metal-free catalytic CH bond activation and borylation of heteroarenes. Science. 2015; 349 (6247):513-516 - 30.
Chernichenko K, Lindqvist M, Kotai B, Nieger M, Sorochkina K, Papai I, et al. Metal-free sp2-C–H borylation as a common reactivity pattern of frustrated 2-aminophenylboranes. Journal of the American Chemical Society. 2016; 138 (14):4860-4868 - 31.
Alfaro R, Parra A, Alemán J, Garcia Ruano JL, Tortosa M. Copper (I)-catalyzed formal carboboration of alkynes: Synthesis of tri-and tetrasubstituted vinylboronates. Journal of the American Chemical Society. 2012; 134 (37):15165-15168 - 32.
Nagao K, Ohmiya H, Sawamura M. Phosphine-catalyzed anti-carboboration of alkynoates with alkyl-, alkenyl-, and arylboranes. Journal of the American Chemical Society. 2014; 136 (30):10605-10608 - 33.
Lennox AJJ, Lloyd-Jones GC. Selection of boron reagents for Suzuki–Miyaura coupling. Chemical Society Reviews. 2014; 43 (1):412-443 - 34.
Chen C, Kehr G, Fröhlich R, Erker G. Carbon− carbon bond activation by 1, 1-carboboration of internal alkynes. Journal of the American Chemical Society. 2010; 132 (39):13594-13595 - 35.
Lappert MF, Prokai B. Chloroboration and allied reactions of unsaturated compounds II. Haloboration and phenylboration of acetylenes; and the preparation of some alkynylboranes. Journal of Organometallic Chemistry. 1964; 1 (5):384-400 - 36.
Miyaura N, Yamada K, Suzuki A. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Letters. 1979; 20 (36):3437-3440 - 37.
Joy F, Lappert MF, Prokai B. Chloroboration and allied reactions of unsaturated compounds: V. Haloboration and phenylboration of olefins; and the preparation of hexaphenyl-1, 4-diboracyclohexa-2, 5-diene. Journal of Organometallic Chemistry. 1966; 5 (6):506-519 - 38.
Lawson JR, Clark ER, Cade IA, Solomon SA, Ingleson MJ. Haloboration of internal alkynes with boronium and borenium cations as a route to tetrasubstituted alkenes. Angewandte Chemie International Edition. 2013; 52 (29):7518-7522 - 39.
De Vries TS, Prokofjevs A, Vedejs E. Cationic tricoordinate boron intermediates: Borenium chemistry from the organic perspective. Chemical Reviews. 2012; 112 (7):4246-4282 - 40.
Robertson DW, Katzenellenbogen JA, Hayes JR, Katzenellenbogen BS. Antiestrogen basicity-activity relationships: A comparison of the estrogen receptor binding and antiuterotrophic potencies of several analogs of (Z)-1, 2-diphenyl-1-[4-[2-(dimethylamino) ethoxy] phenyl]-1-butene (tamoxifen, Nolvadex) having altered basici. Journal of Medicinal Chemistry. 1982; 25 (2):167-171 - 41.
Suginome M. Catalytic carboborations. The Chemical Record. 2010; 10 (5):348-358 - 42.
Jiang C, Blacque O, Berke H. Activation of terminal alkynes by frustrated Lewis pairs. Organometallics. 2010; 29 (1):125-133 - 43.
Chen C, Voss T, Fröhlich R, Kehr G, Erker G. 1, 1-carboboration of 1-alkynes: A conceptual alternative to the hydroboration reaction. Organic Letters. 2011; 13 (1):62-65 - 44.
Lawson JR, Fasano V, Cid J, Vitorica-Yrezabal I, Ingleson MJ. The carboboration of me 3 Si-substituted alkynes and allenes with boranes and borocations. Dalton Transactions. 2016; 45 (14):6060-6070 - 45.
Okuno Y, Yamashita M, Nozaki K. Borylcyanocuprate in a one-pot Carboboration by a sequential reaction with an electron-deficient alkyne and an organic carbon electrophile. Angewandte Chemie. 2011; 123 (4):950-953 - 46.
Cade IA, Ingleson MJ. Syn-1, 2-Carboboration of alkynes with Borenium cations. Chemistry–a. European Journal. 2014; 20 (40):12874-12880 - 47.
Devillard M, Brousses R, Miqueu K, Bouhadir G, Bourissou D. A stable but highly reactive phosphine-coordinated borenium: Metal-free dihydrogen activation and alkyne 1, 2-carboboration. Angewandte Chemie International Edition. 2015; 54 (19):5722-5726 - 48.
Melen RL, Wilkins LC, Kariuki BM, Wadepohl H, Gade LH, Hashmi ASK, et al. Diverging pathways in the activation of allenes with Lewis acids and bases: Addition, 1, 2-carboboration, and cyclization. Organometallics. 2015; 34 (16):4127-4137 - 49.
Dewhurst RD, Légaré MA, Braunschweig H. Towards the catalytic activation of inert small molecules by main-group ambiphiles. Communications Chemistry. 2020; 3 (1):8-11. DOI: 10.1038/s42004-020-00371-4 - 50.
Kumar G, Roy S, Chatterjee I. Tris(pentafluorophenyl)borane catalyzed C-C and C-heteroatom bond formation. Organic and Biomolecular Chemistry. 2021; 19 (6):1230-1267. DOI: 10.1039/d0ob02478c - 51.
Rao B, Kinjo R. Boron-based catalysts for C−C bond-formation reactions. Chemistry - An Asian Journal. 2018; 13 (10):1279-1292. DOI: 10.1002/asia.201701796 - 52.
Lawson JR, Melen RL. Tris(pentafluorophenyl)borane and beyond: Modern advances in Borylation chemistry. Inorganic Chemistry. 2017b; 56 (15):8627-8643. DOI: 10.1021/acs.inorgchem.6b02911 - 53.
Yin Q , Kemper S, Klare HFT, Oestreich M. Boron Lewis acid-catalyzed hydroboration of alkenes with Pinacolborane: BArF3Does what B(C6F5)3Cannot do! Chemistry - A European Journal. 2016a; 22 (39):13840-13844. DOI: 10.1002/chem.201603466