Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
Hepatitis C virus (HCV) is the main etiology of advanced liver fibrosis and cirrhosis with significant risk of progression to hepatocellular carcinoma (HCC). Several epidemiologic studies have documented a lot of risk factors related to the progression of HCC in chronic HCV patients. Factors that increase the risk of HCC development include obesity, diabetes mellitus, nonalcoholic fatty liver disease, aflatoxin exposure, alcohol consumption, occult hepatitis C infection, and genetic variations. HCV patients with genotypes 3 and 1 are also more liable to develop HCC. Also, male gender and higher age are considered as independent risk factors for HCC. Using the newly discovered direct-acting antivirals (DAAs), great improvement in sustained virological immune response (SVR) has occurred >90% in treated patients irrespective of their fibrosis level. Nevertheless, the progression to HCC in HCV patients who achieve SVR stays vulnerable to HCC development, especially patients with advanced fibrosis and/or cirrhosis.
Microbial Biotechnology Department, Biotechnology Institute, National Research Centre, Cairo, Egypt
Sally Farouk
Microbial Biotechnology Department, Biotechnology Institute, National Research Centre, Cairo, Egypt
Naiera Helmy
Microbial Biotechnology Department, Biotechnology Institute, National Research Centre, Cairo, Egypt
Noha Bader El Din
Microbial Biotechnology Department, Biotechnology Institute, National Research Centre, Cairo, Egypt
*Address all correspondence to: reem_elshanaweey@yahoo.com
1. Introduction
Infection with hepatitis C virus (HCV) is a worldwide major health problem, where its prevalence had an assessed 2.8% elevate through the last decade, to more than 185 million infections (3% of the world’s population) [1]. Chronic HCV infection is a principal cause of end-stage liver disease, HCC, and liver-related deaths. HCV has seven genotypes (gt 1–7) and about 100 subtypes. The infection rate and subtype predominance are country dependent. The infection rate variance between low and high-endemic countries is about 20% [2]. The lowest prevalence of HCV occurs in Australia, North America, North, and Western Europe. On the other hand, the highest prevalence of HCV infection occurs in African and Asian countries, where three-quarters of infected individuals are living in middle-income countries, including India, Nigeria, Pakistan, Egypt, China, and Russia together accounting for more than half of world HCV infection [3]. The highest virus prevalence was recorded in Egypt, due to community-wide mass anti-schistosomiasis treatment from the 1950s to the 1980s. At that time tartar emetic injections were the standard treatment therapy [4], and 22% of population were infected. Treatment especially targeted children and young adults, and more than two million shots were given yearly to almost 250,000 patients. Each patient was assumed to have a series of injections with the average number of injections per patient being nine in the 1960s, which then declined to six after 1975. HCV infection has a special situation in Egypt early in its history and will continue till elimination, hopefully, in the near future [1].
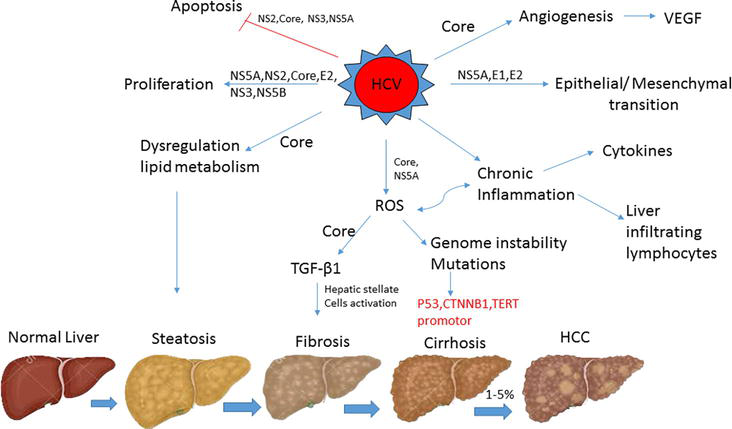
Chronic hepatitis and cirrhosis perform major risk factors for HCC development. Hepatic carcinogenesis is a multifactorial process that takes years, including chemical exposure or viral agents causing inflammatory reactions leading to mitochondrial oxidative damage, cytokine response, necrosis of hepatocytes, and finally malignant transformation and clonal expression. Hepatocytes become malignant through elevated liver cell turnover, due to chronic liver injury and renewal, during inflammation and oxidative stress states. Viral proteins may upregulate mitogenic pathways to prevent apoptosis and persuade the production of reactive oxygen species (ROS) [5]. Moreover, the virus prompts continuous inflammation with liver-infiltrating lymphocytes and cytokines, such as lymphotoxin (LTα and LTβ), that are closely related to development of HCC. Chronic inflammation aggravates ROS production which is a fundamental cause of genetic mutations. ROS are also correlated to inducing the transforming growth factor (TGF-β) pathway, activating hepatic stellate cell and fibrogenesis. Together, TGF-β and Toll-like receptor (TLR4) plays a vital role in the epithelial–mesenchymal transition. HCV dysregulates the patient’s lipid metabolism leading to the accumulation of liver fat that is highly correlated to HCC development. HCV also persuades angiogenic and metastatic pathways (Figure 1) [6].
3. Risk factors for HCC development in HCV patients
HCC etiology is generally supposed to be related to liver cirrhosis, viral hepatitis, alcoholic liver disease, metabolic-related fatty liver disease, and aflatoxin infection. Among them, viral hepatitis is the most important factor, of which chronic hepatitis B (HBV) and HCV infections are the most common [7].
3.1 Age and gender
Age and gender are independent risk factors for HCC patients with HCV infection. HCC represents the third most popular malignancy among men and 7th among women worldwide [8]. Men have a greater threat of HCC than women due to their exposure to other risk factors such as alcohol abuse, smoking plus persistent HCV or HBV infection [9]. Otherwise, sex hormones and pregnancy in women patients have a role in infection with HCV and its progression to HCC [10]. The age-standardized incidence rate (ASIR) in Eastern Mediterranean countries stated that the incidence of HCC in men was 8.1/100000 while in women was 4.7/100000. The occurrence of HCC is low before age 40 but then elevates exponentially. HCC investigation is recommended for Asian men greater than 40 years of age and Asian women greater than 50 years. The guidelines also suggest that surveillance should begin earlier at a younger age for African/North American blacks, without any precise specification of the age cut-off year [11]. Also, male patients who are older than 40 years and have elevated levels of alanine aminotransferase might benefit from HCC surveillance, irrespective of race [12].
3.2 Alcohol abuse
HCV infection rates have been shown to be significantly higher in alcoholic patients than in nonalcoholic ones [8]. Furthermore, alcohol consumption in chronic HCV patients has been associated with an accelerated rate of fibrosis and a higher risk of development of liver cirrhosis and eventually HCC [13]. Excessive alcohol consumption has been considered as the main reason for high HCC rates in various regions like Central and North Europe, while low alcohol consumption has been linked to low HCC mortality rates in countries like France [14]. Excessive alcohol intake leads to hepatocarcinogenesis due to the high production of acetaldehyde, which is mutagenic metabolite of ethanol. Additionally, it increases oxidative stress, DNA damage and causes a carcinogenic tissue microenvironment that might have a synergistic effect with viral hepatitis and metabolic syndrome [15].
HCC development might also be a direct outcome of increased HCV replication and weakening of the interferon’s antiviral activity resulting from alcohol consumption. Finally, weakened cellular immunity as a result of dendritic cell dysfunction, in addition to high oxidative stress and mitochondrial injury owing to alcohol consumption, all together confer to HCC progression [16].
3.3 Diabetes mellitus and non-alcoholic fatty liver disease
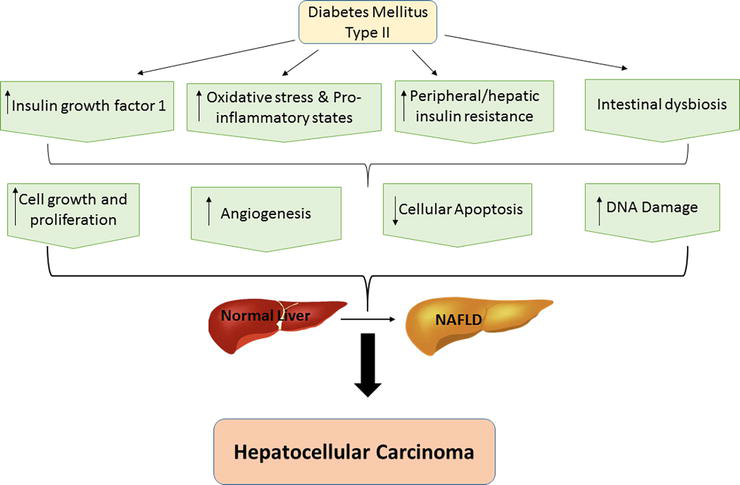
HCV-infected individuals who are obese, have diabetes mellitus (DM), and/or have nonalcoholic fatty liver disease (NAFLD) are more likely to develop HCC [8]. Around 25% of the global population are affected by NAFLD, among those 60% are affected by non-alcoholic steatohepatitis (NASH), evolving HCC at a rate of 5.29/1000 person per year [14]. Definitely, HCV patients in the US have been found to develop HCC more quickly than patients in China and fatty liver disease was reported to be a main provider of this difference [17]. According to epidemiological studies, DM has been associated with a two- or threefold rise in risk of HCC development in individuals with chronic HCV infection [18]. Type 2 diabetes mellitus (T2DM) can be linked to central obesity that induces carcinogenesis through the several mechanisms shown in Figure 2.
Figure 2.
Biological mechanisms linking type 2 diabetes mellitus and NAFLD to the development of HCC.
T2DM and NAFLD have been reported to be strongly associated with high hepatic/peripheral insulin resistance, and lipotoxicity, causing high secretion of various pro-inflammatory cytokines (e.g., C-reactive protein, interleukin-1, interleukin-6, tumor necrosis factor-alpha, tumor growth factor-beta), vasoactive factors and pro-oxidant molecules into bloodstream [19]. Many studies verified that all these factors with increased insulin-like growth factor-1 (IGF-1) production driver may participate in HCC development by induction of hepatocellular growth/proliferation and by inhibiting cellular apoptosis in liver [20]. Several studies reported that when hepatocytes become steatotic, they become capable of producing ROS causing cytotoxicity and DNA damage which in turn cause development of HCC [19].
Recently, accumulated data introduced that alterations in gut microbiota might have a role in obesity, T2DM pathogenesis, and NAFLD, which is implicated in hepatic carcinogenesis [21].
3.4 HCV genotypes
A main peculiarity of HCV is its high degree of heterogeneity. Recently, HCV has been classified into seven genotypes based on the sequence of the viral genome [22]. The rate of HCC development from chronic hepatitis varies and might be associated with multiple factors, such as old age, long duration of infection, gender or alcohol consumption >50 g/day, as well as viral factors like viral genotype/subtype or viral load [23].
Interestingly, patients infected with HCV genotype 3 are more likely to develop end-stage liver diseases and HCC, and eventually liver-related death than other genotypes [24]. However, even after eradication of the disease, the probability of developing HCC is still high. It has been proposed that this specific genotype undergoes a particular oncogenic mechanism that drives to HCC, even if patients are non-cirrhotic [25]. Likewise, HCV genotype 1b specifically may have a pivotal role in HCC progression, especially in patients with early-stage liver disease [26]. Therefore, combining genotypes 1 and 3 and the fast progression of liver damage might lead to low survival rates in HCV-related HCC patients [22]. Moreover, genotype 6 which is mainly prevalent in Southern China and Southeast Asian countries, including Vietnam, Malaysia, Cambodia, and Thailand, showed a higher risk of developing HCC among cirrhotic patients [27].
3.5 Strength of immune system
The immune system includes a wide variety of cells and mediators that interact in a sophisticated and dynamic network to confer protection against foreign pathogens while concurrently maintaining tolerance toward self-antigens [14]. One of the immune system components that elicited against foreign pathogens is “cytokines” which stimulate a host response aimed at controlling cellular stress and reducing cellular damage [28]. But, when host response failed to repair the injury, it stimulates excessive induction of immune cell infiltration leading to alteration and continual cytokine production which can impact several stages of cancer progression and development [29].
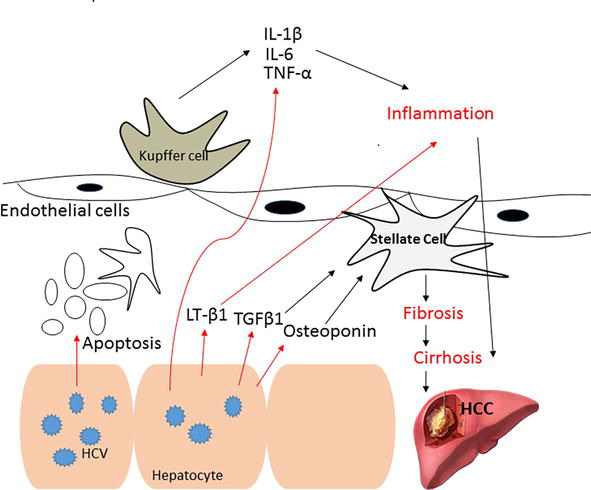
The HCV-mediated cellular immune response is generally weak, where the immune reactivity observed in HCV-infected patients’ livers is largely unspecific. Conversely, innate immune cells are thought to play a key role in HCV immunopathology [30]. Intra-hepatic production of cytokines and chemokines induced by HCV infection leads to the recruitment of non-specific lymphocytes. This pathway induces itself in the absence of viral clearance, causing necro-inflammatory and fibrotic liver disease. A significant lymphocyte infiltration was observed in the portal tracts in liver of HCV P21 core transgenic mice and was associated with elevated serum alanine transaminase (ALT) levels [31]. These changes in liver function are related to the HCC progression and development. Several examples of cytokine have been identified as indicator of elevated HCC risk as shown in Figure 3.
Figure 3.
Pro-inflammatory cytokines during HCV-related HCC.
Chronic hepatitis C is characterized by persistent hepatic inflammation. This is demonstrated by a higher production of pro-inflammatory cytokines and chemokines, which basically initiates from infected hepatocytes, circulating leukocytes, or Kupffer cells. These cells produce cytokines as response to cellular signaling cascades activation, virus-induced oxidative stress, apoptosis of infected cells, or direct activation of innate and adaptive immunity. Among these cytokines are interleukins 1β (IL-1β), 6 (IL-6), 8 (IL-8), and tumor necrosis factor α (TNF-α), in addition to lymphotoxin (LT).
3.6 Single nucleotide polymorphisms (SNPs)
Genetic alterations like SNPs, might change the disease risk and hence may be applied as predictive markers of the disease outcome. To that end, SNPs are useful indicators of the increased risk of HCC [8]. Accumulated data suggest a relationship between SNPs of certain genes and the vulnerability to HCC. For instance, the tolloid–like 1 gene (TLL1) on chromosome 4. A strong relation between TLL1 and HCC development in chronic HCV patients who achieved SVR after treatment was reported [28]. Furthermore, a transformation suppressor gene; the reversion-inducing-cysteine-rich protein with kazal motifs (RECK) gene polymorphism (rs11788747) has been shown to be involved in the pathogenesis of HCC and malignancies [32]. Nevertheless, a recent study performed on a group of Egyptian patients suggested that the RECK gene rs10814325 TT genotype might rather be linked to disease progression and metastasis than being a risk factor for HCC development in HCV patients [33].
Moreover, matrix metalloproteinases (MMPs) play an important role in tissue remodeling during the process of embryogenesis and tissue evolution as well as in wound healing in normal physiologic conditions [34]. Different variants of the MMP-11 gene could be related to clinical status and HCC progression. Subjects carrying the CT + TT allele of the MMP-11 SNP (rs738791) showed a higher risk of HCC development than wild-type (C/C) carriers. Thus, genetic variability in the MMP-11 gene represents a substantial predictor of early stages of HCC onset and a reliable biomarker for disease progression [35].
Additionally, IL-28B (rs12979860) TT genotype has been found to be more dominant in patients with progressive fibrosis, hepatic cirrhosis, HCC, and in the course of HCV recurrence after liver transplantation (LT) [36]. Hence, it seems to be associated with minor consequences in HCV chronic patients and increases the risk of HCC development. The T allele might be considered as a genetic risk factor for HCV-related carcinogenesis, antiviral therapy failure, and post-transplant fibrosis progression [37].
Likewise, transforming growth factor (TGF)-β is a multifunctional profibrotic cytokine that is found in three isoforms. There is a significant association between TGF-β1 869C/T and -509C/T polymorphisms and HCC risk [38]. It is important to note that TGF-β is considered a central mediator of fibrogenesis and has a vital role in the regulation of tumorigenesis [39]. Also, tumor necrosis factor-alpha (TNF-α), which is a potent antiviral cytokine possessing a broad spectrum of proinflammatory activities, plays a crucial role in host immunity against HCV infection, as cytotoxic T lymphocytes (CTLs) in the liver have been shown to secrete TNF-α [40]. Interestingly, HCV infection has been associated with high levels of circulating TNF-α and HCV infection stimulates the production of TNF-α in human hepatocytes. Thus, increased levels of TNF-α are correlated with the severity of tissue injury, hepatic inflammation, fibrosis, and eventually HCC [41]. Another SNP is major histocompatibility complex class I chain-related gene A (MICA) which plays a vital role in immune activation and surveillance against infection and tumorigenesis, and MICASNP rs2596542G > A is associated with HCC development among the Asian, Caucasian, and African ethnicity in certain genetic models [42].
3.7 Viral infection
3.7.1 Hepatitis B virus (HBV)
The leading cause of HCC in Southeast Asia and sub-Saharan Africa is chronic HBV infection, although the percentage of HBV-induced HCC is dropping [43]. Countries, in which HBV occurrence is higher than 2%, show raised mortality rates due to HCC. HBV chronically infected subjects are at high risk of developing HCC that might reach up to 30-fold. This is due to the caused liver inflammation and damage in addition to epigenetic defects leading to HCC progression [44]. The occurrence of HCC is higher in HBV-HCV co-infection than in HBV or HCV monoinfection. Factors, such as long disease duration, increased fibrosis levels, intolerance of carbohydrates as well as higher HCV RNA levels, increase the risk of HCC development [45].
In a recent study, the incidence of HCC in patients coinfected with HCV/HBV was 6.4 per 100 person/year, while monoinfection with either HBV or HCV was 2.0 and 3.7, respectively. Additionally, risk of progression to HCC after 10 years of infection was raised to 45% in case of HBV/HCV coinfection compared to 16 and 28% in case of HBV and HCV monoinfection, respectively. Multiple studies also suggested a synergistic effect of both viruses in case of HBV/HCV coinfection [44]. Due to the weak immune responses toward hepatitis viruses and their failure to completely inhibit or eradicate the virus, this causes persistent stimulation of antigen-specific immune response in chronic patients. The ongoing secretion of cytokines and recruitment of lymphocytes to the liver influences several cellular pathways leading to fibrosis, cirrhosis and eventually HCC [46].
3.7.2 Human immunodeficiency virus (HIV)
There is an increased rate of progression to cirrhosis and HCC in HIV/HCV co-infected patients [47]. One-third of HIV-infected patients have been reported to be coinfected with HCV, having an undesirable effect on HCV pathogenesis [48]. The percentage of liver fibrosis was reported as up to 3 times higher in cases of HIV/HCV coinfection than in HCV monoinfection. HIV also plays a role similar to HBV co-infection. Many studies depicted that 75% of HCV-related mortalities happen between the age of 45 and 65 years, especially in subjects co-infected with HIV or HBV and suffering from hepatic complications such as cirrhosis and HCC [49]. HCV has a necrotic effect on hepatocytes. HIV was found to speed up liver disease progression in HCV patients, as HCV replication augments HIV’s presence leading to high levels of HCV RNA. Furthermore, HIV has a destructive role on the immune system causing the exhaustion of CD4-T cells which cause a high diminution of CD4 cells, and the reduction of CD4 cell stimulates cirrhosis and HCC development [50].
3.7.3 Human cytomegalovirus (HCMV)
Human cytomegalovirus (HCMV) is a herpesvirus-specific species that infect a major part of the population in world and causes asymptomatic latent infection in healthy subjects [51]. However, it can cause severe disease in absence of a robust immune response which remains an important reason for morbidity in immune-compromised individuals where it may clear as symptomatic as hepatitis disease [52]. Most organs and tissues of the human body can be infected by HCMV, such as hepatocytes, fibroblasts, endothelial and neuronal cells, besides blood monocytes and macrophages [53]. HCC patients have a higher significant prevalence of HCMV than patients without HCC, HCMV is positively associated with serum IL-6 levels in cirrhotic patients, and is positively related to the presence of other hepatotropic viruses, such as HCV and HBV [54]. Higher incidence of HCMV among HCV genotype 4 infected patients with less response to IFN therapy has been reported in previous reports [55]. Most of previous reports on HCMV pathogenicity introduced some HCMV proteins as fibrogenesis inducers, such as human (CMV IE1 or IE2). Besides, several CMV proteins modulate the cellular apoptotic machinery like CMV UL97, CMV UL36, and CMV IE86 [54]. An important study on Egyptian patients with genotype 4 documented the dysregulation of the anti-fibrotic pathway (i.e., dysregulation of the JAK–STAT pathway). This dysregulation is likely to be a key molecular and immunological factor for increased susceptibility to HCC development in HCV/CMV co-infected patients with advanced-stage of liver fibrosis [55].
3.8 Effect of HCV treatment
Globally, HCC is the third important reason for cancer-related death, with more than half a million patients, being affected annually. Cirrhosis is the predominate risk factor for HCC, as the accumulative risk for HCC development ranges from 5 to 30% within 5 years among patients with cirrhosis [14], whereas the risk of HCC development in HCV-related cirrhotic patients is about 2–8% annually. Patients with established fibrosis are at higher risk of HCC development HCC than patients with a lower fibrosis level [56].
3.9 Treatment with interferon-based regimens
In the past decades, chronic hepatitis C patients were treated with IFN–based regimens for more than 20 years. Many studies have confirmed the long-term effects of this treatment protocol in achieving an SVR and curing about 50% of treated patients [57]. According to several studies, patients who achieved an SVR after IFN-α treatment, showed improved liver fibrosis, a lower risk of infection-related complications, and an overall reduced risk of de novo HCC development and mortality rates compared to those who failed to achieve SVR [58].
However, the remarkable benefits of this treatment protocol did not reduce HCC incidence, and the risk of developing HCC is not totally eliminated in patients with severe fibrosis or cirrhosis. Such conditions arise with an incidence of 0.3–4% [59].
In a Japanese study, two groups of patients were enrolled for treatment with IFN-α and ribavirin combination; a total of 863 non-cirrhotic patients and 150 cirrhotic patients developed HCC after a median 3.6-year follow-up period. In the non-cirrhotic group, the accumulative incidence rate of HCC development in the SVR group (1.7%) was significantly lower (P = 0.003) than in the non-responder group (7.6%). On the other hand, the accumulative incidence rates of HCC for the SVR group (18.9%) in the cirrhotic patients’ group were also significantly lower (P = 0.03) than in the non-responder group (39.4%) [60].
The key predictors of HCC have been found to include age (more than 60 years old); male sex; platelet count (below 150 × 109/L), levels of AFP (higher than 10 ng/mL); liver cirrhosis, and failure to achieve SVR [61]. In a study conducted by El-Seraget et al., the HCC risk after achieving SVR was reported to be 0.33% annually. Also, the annual HCC risk remained high among cirrhotic patients at the time of treatment (1.39%) and in cured patients older than age 64 (0.95%) with or without cirrhosis [62]. These outcomes recommend HCV treatment in early stage before the development of cirrhosis besides long-term monitoring of HCC in cirrhotic patients even after reaching the SVR [63].
3.9.1 Treatment with DAAs
3.9.1.1 Immunological mechanisms implicated in HCC development with DAA
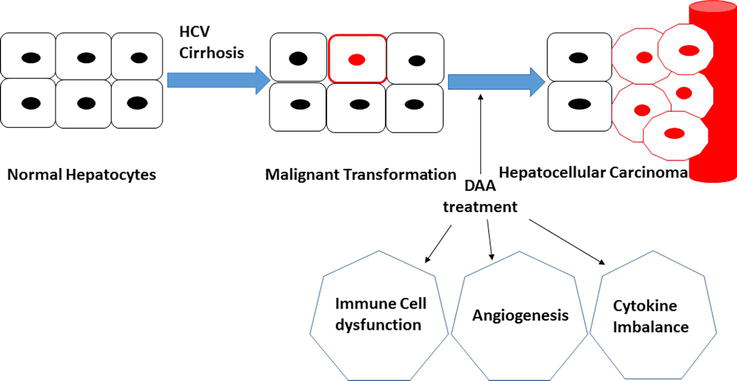
The exact reasons for high rates of tumor development or relapse after the DAAs treatment protocol remains unknown. However, there is one hypothesis that suggests that the anti-tumor host response might be dysregulated after the swift reduction of HCV viral load generated by DAAs, which may promote tumor recurrence [64]. In addition, the growth of existing precancerous lesions is due to immune distortion. HCV infection is activating the intra-hepatic cellular immune response, triggering the elevation of IFN-stimulated gene expression as well as stimulating the natural killer (NK) cells. Other studies suggested that DAA-mediated HCV clearance is associated with the intra-hepatic immune activation loss confirmed by reduced CXCL10 and CXCL11 chemokines levels, and normalization of NK cells phenotype and function. In the same context, Debes et al. has recognized a group of twelve immune mediators containing cytokines; growth factors and apoptosis markers elevated in the serum of patients that developed de novo or recurrent HCC before initiation of DAA treatment compared to treated patients who did not develop HCC [65]. These results suggest that patients with HCC history may previously express a different form of immune mediators before DAAs treatment (Figure 4) [66].
Figure 4.
Mechanisms involved in HCC recurrence after DAAs treatment for HCV chronic patients.
3.9.1.2 Risk of de novo HCC
DAAs treatment has proved to be successfully lowering the incidence of viral hepatitis worldwide, achieving SVR in more than 90% of patients enrolled for treatment regardless of the level of liver fibrosis [61]. There were several studies on the development of HCC following DAAs treatment (Table 1) [24, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76].
Studies assessing de novo HCC risk after DAA therapy.
DAA, Direct Acting Antivirals, NA, Not Available, SVR, Sustained Virological Response, HCC, Hepatocellular Carcinoma.
Undoubtedly, the HCV-treated population has significantly changed in the DAAs era, where it currently includes numerous patients with different HCC risk factors. This might explain the reason why the newer cohorts of patients treated with DAAs may encounter whole new HCC risks compared to those expected based on their medical history. HCV cirrhotic patients who reach SVR with DAAs treatment remain at high HCC risk, and for that, they should continue a long-run HCC screening.
3.9.1.3 Risk of HCC recurrence
A controversial issue is the impact of a DAAs-based SVR on the risks of HCC recurrence after early HCC treatment. In this area, several study cohorts have investigated the risk of HCC recurrence after DAAs treatment protocol as shown in Table 2 [59, 74, 76, 77, 78, 79].
29.8 with 12 months of DAA initiation (vs 31 in untreated patients)
Table 2.
Study cohorts investigating HCC recurrence risk after DAAs treatment protocol.
Well-designed studies with potent comparison arms are needed for solid assessment of DAAs impact on HCC recurrence. Presently, HCV-cirrhotic patients who underwent ablation or resection of HCC should not be dissuaded from receiving DAAs treatment aiming to restrain the liver disease progression. Otherwise, rigorous screening is a must to confirm tumor clearance in HCC patients prior to the initiation of DAAs treatment protocol. Also, monitoring HCC by liver imaging alongside AFP testing should be open-endedly continued at least twice a year after reaching SVR.
3.9.2 Occult HCV infection (OCI)
Occult HCV infection is defined as the persistence of viral RNA whether in liver cells or peripheral blood mononuclear cells (PBMCs) with no detectable HCV RNA levels in serum [80]. Recently, it was found that there is a markedly high prevalence of OCI-HCV following the DAAs treatment [61]. In spite of successful viral clearance, numerous patients continue to exhibit HCV-related disease progression [81]. Also, the risk of liver cancer development is not totally diminished, even in efficiently treated cirrhotic patients who reached the SVR. Although IFN-free treatment regimens are able to eradicate HCV infection more effectively, the replication of neoplastic clones continues in a setting of reduced inflammation because DAAs can eliminate HCV from serum rather than from cells. So, dual testing for HCV RNA in both serum and PBMCs is recommended at the end of HCV infection treatment with DDAs and during validation of the SVR after the initial response [82].
3.10 Aflatoxin B1 (AFB1)
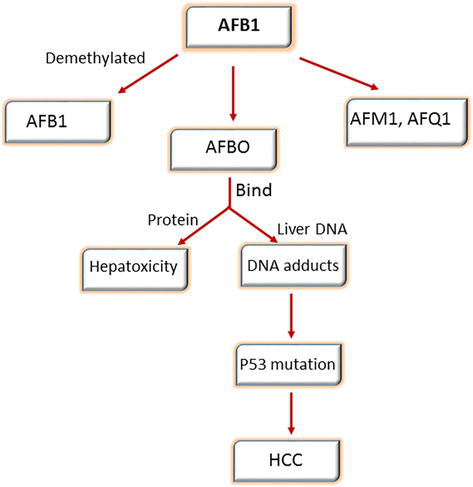
Aflatoxins are categorized into four types: B1, B2, G1, and G2; all known to be carcinogenic to both humans and animals. Among those, aflatoxin B1 is a known human carcinogen that has been shown to be a causal agent in the pathogenesis of HCC [83]. Aflatoxin is considered a food contaminant produced by the fungi Aspergillus flavus and Aspergillus parasiticus, which develop under special environmental conditions characterized by high temperature and moisture, which is widely common in areas of Southeast Asia and Sub-Saharan Africa under 40 southern and 40 northern equator [84]. Aflatoxin can affect a wide range of food commodities, such as corns, peanuts, spices, maize, rice, and legumes as well as meat, milk, and dried fruits. Additionally, the exposure to aflatoxin can accelerate HBV and HCV-associated carcinogenesis by introducing mutations [85]. Once AFB1 is ingested, it is metabolized by the cytochrome P-450 system into the intermediate reactive oxygen species (ROS): afatoxin-8, 9-expoxide, which can interfere with DNA forming. AFB1-guanine products can react with proteins forming AFB1-albumin which ultimately leads to acute toxicity (aflatoxicosis) and lesions, respectively [86]. Failing to repair these lesions triggers DNA mutations in key genes; particularly “G to T” transversions. In this domain, the mutation (AGG to AGT, R249S) has been recognized at codon 249 in the TP53 tumor suppressor gene, specific for exposure to aflatoxin other studies have confirmed that this hotspot in TP53 is a special site for AFB1 adducts formation. Consequently, the formation of AFB1-DNA adducts over time rises the HCC risk (Figure 5) [87].
All authors declare that there is no conflict of interest.
References
1.El-Ghitany EM, Hepatitis C. Virus infection in Egypt: Current situation and future perspective. Journal of High Institute of Public Health. 2019;49(1):1-9. DOI: 10.21608/jhiph.2019.29460
2.Morozov VA, Lagaye S. Hepatitis C virus: Morphogenesis, infection and therapy. World Journal of Hepatology. 2018;10(2):186-212. DOI: 10.4254/wjh.v10.i2.186
3.Shalimar PS, Gupta H, Bansal B, Elhence A, Krishna Kishore RV, et al. A systematic review of risk factors for hepatitis C virus infection among low-risk population in India. Journal of Clinical and Experimental Hepatology. 2022;12(6):1438-1444. DOI: 10.1016/j.jceh.2022.06.003
4.Elgharably A, Gomaa AI, Crossey MM, Norsworthy PJ, Waked I, Taylor-Robinson SD. Hepatitis C in Egypt - past, present, and future. International Journal of General Medicine. 2017;10:1-6. DOI: 10.2147/ijgm.s119301.s
5.Vescovo T, Refolo G, Vitagliano G, Fimia GM, Piacentini M. Molecular mechanisms of hepatitis C virus–induced hepatocellular carcinoma. Clinical Microbiology and Infection. 2016;22(10):853-861. DOI: 10.1016/j.cmi.2016.07.019
6.Miller FD, Elzalabany MS, Hassani S, Cuadros DF. Epidemiology of hepatitis C virus exposure in Egypt: Opportunities for prevention and evaluation. World Journal of Hepatology. 2015;7(28):2849-2858. DOI: 10.4254/wjh.v7.i28.2849
7.Zhang C-h, Cheng Y, Zhang S, Fan J, Gao Q. Changing epidemiology of hepatocellular carcinoma in Asia. Liver International. 2022;42(9):2029-2041. DOI: 10.1111/liv.15251
8.Abbas Z, Abbas M. Factors predicting hepatocellular carcinoma in hepatitis C infection. Hepatoma Research. 2018;4:43. DOI: 10.20517/2394-5079.2018.26
9.Liu P, Xie SH, Hu S, Cheng X, Gao T, Zhang C, et al. Age-specific sex difference in the incidence of hepatocellular carcinoma in the United States. Oncotarget. 2017;8(40):68131-68137. DOI: 10.18632/oncotarget.19245
10.Amr S, Iarocci E, Nasr G, Saleh D, Blancato J, Shetty K, et al. Multiple pregnancies, hepatitis C, and risk for hepatocellular carcinoma in Egyptian women. BMC Cancer. 2014;14:893. DOI: 10.1186/1471-2407-14-893
11.Mittal S, Kramer JR, Omino R, Chayanupatkul M, Richardson PA, El-Serag HB, et al. Role of age and race in the risk of hepatocellular carcinoma in veterans with hepatitis B virus infection. Clinical Gastroenterology and Hepatology. 2018;16(2):252-259. DOI: 10.1016/j.cgh.2017.08.042
12.Abdelmoez FA-b, Imam HM, Idriss NK, Wahid LA, Abbas WA, Abozaid MAA, et al. The role of hepatitis C virus and possible risk factors in development of hepatocellular carcinoma: 400 patients based study. The Egyptian Journal of Internal Medicine. 2019;31(1):64-72. DOI: 10.4103/ejim.ejim_50_18
13.Crisan D, Grigorescu MD, Radu C, Suciu A, Grigorescu M. Interferon-γ-inducible protein-10 in chronic hepatitis C: Correlations with insulin resistance, histological features & sustained virological response. The Indian Journal of Medical Research. 2017;145(4):543-550. DOI: 10.4103/ijmr.IJMR_1410_14
14.Fujiwara N, Friedman SL, Goossens N, Hoshida Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. Journal of Hepatology. 2018;68(3):526-549. DOI: 10.1016/j.jhep.2017.09.016
15.Heckley GA, Jarl J, Asamoah BO, G-Gerdtham U. How the risk of liver cancer changes after alcohol cessation: A review and meta-analysis of the current literature. BMC Cancer. 2011;11(1):446. DOI: 10.1186/1471-2407-11-446
16.McCartney EM, Semendric L, Helbig KJ, Hinze S, Jones B, Weinman SA, et al. Alcohol metabolism increases the replication of hepatitis C virus and attenuates the antiviral action of interferon. The Journal of Infectious Diseases. 2008;198(12):1766-1775. DOI: 10.1086/593216
17.Schlesinger S, Aleksandrova K, Pischon T, Jenab M, Fedirko V, Trepo E, et al. Diabetes mellitus, insulin treatment, diabetes duration, and risk of biliary tract cancer and hepatocellular carcinoma in a European cohort. Annals of Oncology. 2013;24(9):2449-2455. DOI: 10.1093/annonc/mdt204
18.Huang C-F, Yeh M-L, Huang C-I, Lin Y-J, Tsai P-C, Lin Z-Y, et al. Risk of hepatitis C virus related hepatocellular carcinoma between subjects with spontaneous and treatment-induced viral clearance. Oncotarget. 2016;8(27):43925-43933
19.Mantovani A, Targher G. Type 2 diabetes mellitus and risk of hepatocellular carcinoma: Spotlight on nonalcoholic fatty liver disease. Annals of Translational Medicine. 2017;5(13):270
20.Li C, Deng M, Hu J, Li X, Chen L, Ju Y, et al. Chronic inflammation contributes to the development of hepatocellular carcinoma by decreasing miR-122 levels. Oncotarget. 2016;7(13):17021-17034. DOI: 10.18632/oncotarget.7740
21.Cholankeril G, Patel R, Khurana S, Satapathy SK. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Current knowledge and implications for management. World Journal of Hepatology. 2017;9(11):533-543. DOI: 10.4254/wjh.v9.i11.533
22.Petruzziello A, Marigliano S, Loquercio G, Coppola N, Piccirillo M, Leongito M, et al. Hepatitis C Virus (HCV) genotypes distribution among hepatocellular carcinoma patients in Southern Italy: A three year retrospective study. Infectious Agents and Cancer. 2017;12(1):52. DOI: 10.1186/s13027-017-0162-5
23.Bruno S, Savojardo D, Almasio PL, Mondelli MU. Critical reappraisal of risk factors for occurrence of hepatocellular carcinoma in patients with hepatitis C virus. Hepatic Medicine: Evidence And Research. 2011;3:21-28. DOI: 10.2147/hmer.s16991
24.Kanwal F, Kramer J, Asch SM, Chayanupatkul M, Cao Y, El-Serag HB. Risk of hepatocellular cancer in HCV patients treated with direct-acting antiviral agents. Gastroenterology. 2017;153(4):996-1005.e1. DOI: 10.1053/j.gastro.2017.06.012
25.Baumert TF, Jühling F, Ono A, Hoshida Y. Hepatitis C-related hepatocellular carcinoma in the era of new generation antivirals. BMC Medicine. 2017;15(1):52. DOI: 10.1186/s12916-017-0815-7
26.Park HK, Lee SS, Im CB, Im C, Cha RR, Kim WS, et al. Hepatitis C virus genotype affects survival in patients with hepatocellular carcinoma. BMC Cancer. 2019;19(1):822. DOI: 10.1186/s12885-019-6040-3
27.Lee MH, Hsiao TI, Subramaniam SR, Le AK, Vu VD, Trinh HN, et al. HCV genotype 6 increased the risk for hepatocellular carcinoma among asian patients with liver cirrhosis. The American Journal of Gastroenterology. 2017;112(7):1111-1119. DOI: 10.1038/ajg.2017.123
28.Walker AJ, Peacock CJ, Pedergnana V, Consortium S-H, Irving WL. Host genetic factors associated with hepatocellular carcinoma in patients with hepatitis C virus infection: A systematic review. Journal of Viral Hepatitis. 2018;25(5):442-456. DOI: 10.1111/jvh.12871
29.Zhong JH, You XM, Gong WF, Ma L, Zhang Y, Mo QG, et al. Epidermal growth factor gene polymorphism and risk of hepatocellular carcinoma: A meta-analysis. PLoS One. 2012;7(3):e32159. DOI: 10.1371/journal.pone.0032159
30.Lee HL, Jang JW, Lee SW, Yoo SH, Kwon JH, Nam SW, et al. Inflammatory cytokines and change of Th1/Th2 balance as prognostic indicators for hepatocellular carcinoma in patients treated with transarterial chemoembolization. Scientific Reports. 2019;9(1):3260. DOI: 10.1038/s41598-019-40078-8
31.Wakita T, Katsume A, Kato J, Taya C, Yonekawa H, Kanegae Y, et al. Possible role of cytotoxic T cells in acute liver injury in hepatitis C virus cDNA transgenic mice mediated by Cre/loxP system. Journal of Medical Virology. 2000;62(3):308-317. DOI: 10.1002/1096-9071(200011)62:3<308::aid-jmv2>3.0.co;2-6
32.Chung TT, Yeh CB, Li YC, Su SC, Chien MH, Yang SF, et al. Effect of RECK gene polymorphisms on hepatocellular carcinoma susceptibility and clinicopathologic features. PLoS One. 2012;7(3):e33517. DOI: 10.1371/journal.pone.0033517
33.Yu Y, Hu Y, Li K, Chen Z, Zhang H, Zhang L. RECK gene polymorphism is associated with susceptibility and prognosis of wilms' tumor in Chinese children. Medical Science Monitor. 2015;21:1928-1933. DOI: 10.12659/msm.893606
34.Wang B, Hsu CJ, Lee HL, Chou CH, Su CM, Yang SF, et al. Impact of matrix metalloproteinase-11 gene polymorphisms upon the development and progression of hepatocellular carcinoma. International Journal of Medical Sciences. 2018;15(6):653-658. DOI: 10.7150/ijms.23733
35.Koleck TA, Bender CM, Clark BZ, Ryan CM, Ghotkar P, Brufsky A, et al. An exploratory study of host polymorphisms in genes that clinically characterize breast cancer tumors and pretreatment cognitive performance in breast cancer survivors. Breast Cancer. 2017;9:95-110. DOI: 10.2147/bctt.s123785
36.El-Awady MK, Mostafa L, Tabll AA, Abdelhafez TH, Bader El Din NG, Zayed N, et al. Association of IL28B SNP With progression of Egyptian HCV genotype 4 patients to end stage liver disease. Hepatitis Monthly. 2012;12(4):271-277. DOI: 10.5812/hepatmon.835
37.Attallah AM, Omran D, Marie MS, Abdelrazek M, Salama A, El Essawey R, et al. IL-28B rs12979860 polymorphism affect the course of chronic hepatitis and the development of HCC in Egyptian patients with hepatitis C type 4. British Journal of Biomedical Science. 2018;75(4):157-162. DOI: 10.1080/09674845.2018.1489599
38.Bader El Din NG, Farouk S, El-Shenawy R, Elhady MM, Ibrahim MK, Dawood RM, et al. The synergistic effect of TNFalpha −308 G/A and TGFbeta1-509 C/T polymorphisms on hepatic fibrosis progression in hepatitis C virus genotype 4 patients. Viral Immunology. 2017;30(2):127-135. DOI: 10.1089/vim.2016.0083
39.Bader El Din NG, Anany MA, Dawood RM, Ibrahim MK, El-Shenawy R, El Abd YS, et al. Impact of OAS1 Exon 7 rs10774671 genetic variation on liver fibrosis progression in Egyptian HCV genotype 4 patients. Viral Immunology. 2015;28(9):509-516. DOI: 10.1089/vim.2015.0041
40.Dong LM, Potter JD, White E, Ulrich CM, Cardon LR, Peters U. Genetic susceptibility to cancer: Thse role of polymorphisms in candidate genes. Journal of the American Medical Association. 2008;299(20):2423-2436
41.Nahon P, Zucman-Rossi J. Single nucleotide polymorphisms and risk of hepatocellular carcinoma in cirrhosis. Journal of Hepatology. 2012;57(3):663-674. DOI: 10.1016/j.jhep.2012.02.035
42.Wang H, Cao H, Xu Z, Wang D, Zeng Y. SNP rs2596542G>A in MICA is associated with risk of hepatocellular carcinoma: A meta-analysis. Bioscience Reports. 2019;39(5):1-13. DOI: 10.1042/bsr20181400
43.Yapali S, Tozun N. Epidemiology and viral risk factors for hepatocellular carcinoma in the Eastern Mediterranean countries. Hepatoma Research. 2018;4:24. DOI: 10.20517/2394-5079.2018.57
44.Yang JD, Kim WR, Coelho R, Mettler TA, Benson JT, Sanderson SO, et al. Cirrhosis is present in most patients with hepatitis B and hepatocellular carcinoma. Clinical Gastroenterology and Hepatology. 2011;9(1):64-70. DOI: 10.1016/j.cgh.2010.08.019
45.Marcon PDS, Tovo CV, Kliemann DA, Fisch P, de Mattos AA. Incidence of hepatocellular carcinoma in patients with chronic liver disease due to hepatitis B or C and coinfected with the human immunodeficiency virus: A retrospective cohort study. World Journal of Gastroenterology. 2018;24(5):613-622. DOI: 10.3748/wjg.v24.i5.613
46.Zampino R, Pisaturo MA, Cirillo G, Marrone A, Macera M, Rinaldi L, et al. Hepatocellular carcinoma in chronic HBV-HCV co-infection is correlated to fibrosis and disease duration. Annals of Hepatology. 2015;14(1):75-82
47.Neesgaard B, Ruhwald M, Weis N. Inducible protein-10 as a predictive marker of antiviral hepatitis C treatment: A systematic review. World Journal of Hepatology. 2017;9(14):677-688. DOI: 10.4254/wjh.v9.i14.677
48.Shenge JA, Odaibo GN, Olaleye DO. Phylogenetic analysis of hepatitis C virus among HIV/HCV co-infected patients in Nigeria. PLoS One. 2019;14(2):e0210724. DOI: 10.1371/journal.pone.0210724
49.Thomson EC, Nastouli E, Main J, Karayiannis P, Eliahoo J, Muir D, et al. Delayed anti-HCV antibody response in HIV-positive men acutely infected with HCV. AIDS. 2009;23(1):89-93. DOI: 10.1097/QAD.0b013e32831940a3
50.Kumar S, Stecher G, Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution. 2016;33(7):1870-1874. DOI: 10.1093/molbev/msw054
51.Herbein G. The human cytomegalovirus, from oncomodulation to oncogenesis. Viruses. 2018;10(8):1-13. DOI: 10.3390/v10080408
52.Shimamura M, Murphy-Ullrich JE, Britt WJ. Human cytomegalovirus induces TGF-β1 activation in renal tubular epithelial cells after epithelial-to-mesenchymal transition. PLoS Pathogens. 2010;6(11):e1001170. DOI: 10.1371/journal.ppat.1001170
53.Johnsen JI, Baryawno N, Söderberg-Nauclér C. Is human cytomegalovirus a target in cancer therapy? Oncotarget. 2011;2(12):1329
54.Lepiller Q , Abbas W, Kumar A, Tripathy MK, Herbein G. HCMV activates the IL-6-JAK-STAT3 axis in HepG2 cells and primary human hepatocytes. PLoS One. 2013;8(3):e59591. DOI: 10.1371/journal.pone.0059591
55.Ibrahim MK, Khedr A, Bader El Din NG, Khairy A, El Awady MK. Increased incidence of cytomegalovirus coinfection in HCV-infected patients with late liver fibrosis is associated with dysregulation of JAK-STAT pathway. Scientific Reports. 2017;7(1):10364. DOI: 10.1038/s41598-017-10604-7
56.Blach S, Zeuzem S, Manns M, Altraif I, Duberg A-S, Muljono DH, et al. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: A modelling study. The lancet Gastroenterology & hepatology. 2017;2(3):161-176
57.Villani R, Vendemiale G, Serviddio G. Molecular mechanisms involved in HCC recurrence after direct-acting antiviral therapy. International Journal of Molecular Sciences. 2019;20(1):49
58.Innes HA, McDonald SA, Dillon JF, Allen S, Hayes PC, Goldberg D, et al. Toward a more complete understanding of the association between a hepatitis C sustained viral response and cause-specific outcomes. Hepatology. 2015;62(2):355-364. DOI: 10.1002/hep.27766
59.Reig M, Mariño Z, Perelló C, Iñarrairaegui M, Ribeiro A, Lens S, et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. Journal of Hepatology. 2016;65(4):719-726. DOI: 10.1016/j.jhep.2016.04.008
60.Ogawa E, Furusyo N, Kajiwara E, Takahashi K, Nomura H, Maruyama T, et al. Efficacy of pegylated interferon alpha-2b and ribavirin treatment on the risk of hepatocellular carcinoma in patients with chronic hepatitis C: A prospective, multicenter study. Journal of Hepatology. 2013;58(3):495-501. DOI: 10.1016/j.jhep.2012. DOI: 10.017
61.Roche B, Coilly A, Duclos-Vallee JC, Samuel D. The impact of treatment of hepatitis C with DAAs on the occurrence of HCC. Liver International: Official Journal of the International Association for the Study of the Liver. 2018;38(Suppl. 1):139-145. DOI: 10.1111/liv.13659
62.El-Serag HB, Kanwal F, Richardson P, Kramer J. Risk of hepatocellular carcinoma after sustained virological response in veterans with hepatitis C virus infection. Hepatology. 2016;64(1):130-137. DOI: 10.1002/hep.28535
63.Nahon P, Bourcier V, Layese R, Audureau E, Cagnot C, Marcellin P, et al. Eradication of hepatitis C virus infection in patients with cirrhosis reduces risk of liver and non-liver complications. Gastroenterology. 2017;152(1):142-56.e2. DOI: 10.1053/j.gastro.2016.09.009
64.Nault JC, Colombo M. Hepatocellular carcinoma and direct acting antiviral treatments: Controversy after the revolution. Journal of Hepatology. 2016;65(4):663-665. DOI: 10.1016/j.jhep.2016.07.004
65.Debes JD, van Tilborg M, Groothuismink ZMA, Hansen BE, Schulze Zur Wiesch J, von Felden J, et al. Levels of cytokines in serum associate with development of hepatocellular carcinoma in patients with HCV infection treated with direct-acting antivirals. Gastroenterology. 2018;154(3):515-7.e3. DOI: 10.1053/j.gastro.2017.10.035
66.Mereness JA, Bhattacharya S, Ren Y, Wang Q , Anderson CS, Donlon K, et al. Collagen VI deficiency results in structural abnormalities in the mouse lung. The American Journal of Pathology. 2020;190(2):426-441. DOI: 10.1016/j.ajpath.2019.10.014
67.Kobayashi M, Suzuki F, Fujiyama S, Kawamura Y, Sezaki H, Hosaka T, et al. Sustained virologic response by direct antiviral agents reduces the incidence of hepatocellular carcinoma in patients with HCV infection. Journal of Medical Virology. 2017;89(3):476-483. DOI: 10.1002/jmv.24663
68.Kozbial K, Moser S, Schwarzer R, Laferl H, Al-Zoairy R, Stauber R, et al. Unexpected high incidence of hepatocellular carcinoma in cirrhotic patients with sustained virologic response following interferon-free direct-acting antiviral treatment. Journal of Hepatology. 2016;65(4):856-858. DOI: 10.1016/j.jhep.2016.06.009
69.Ravi S, Axley P, Jones D, Kodali S, Simpson H, McGuire BM, et al. Unusually high rates of hepatocellular carcinoma after treatment with direct-acting antiviral therapy for hepatitis C related cirrhosis. Gastroenterology. 2017;152(4):911-912. DOI: 10.1053/j.gastro.2016.12.021
70.Cheung MCM, Walker AJ, Hudson BE, Verma S, McLauchlan J, Mutimer DJ, et al. Outcomes after successful direct-acting antiviral therapy for patients with chronic hepatitis C and decompensated cirrhosis. Journal of Hepatology. 2016;65(4):741-747. DOI: 10.1016/j.jhep.2016.06.019
71.Foster GR, Irving WL, Cheung MC, Walker AJ, Hudson BE, Verma S, et al. Impact of direct acting antiviral therapy in patients with chronic hepatitis C and decompensated cirrhosis. Journal of Hepatology. 2016;64(6):1224-1231. DOI: 10.1016/j.jhep.2016.01.029
72.Ioannou GN, Green PK, Berry K. HCV eradication induced by direct-acting antiviral agents reduces the risk of hepatocellular carcinoma. Journal of Hepatology. 2017;68(1):25-32. DOI: 10.1016/j.jhep.2017.08.030
73.Cardoso H, Vale AM, Rodrigues S, Gonçalves R, Albuquerque A, Pereira P, et al. High incidence of hepatocellular carcinoma following successful interferon-free antiviral therapy for hepatitis C associated cirrhosis. Journal of Hepatology. 2016;65(5):1070-1071. DOI: 10.1016/j.jhep.2016.07.027
74.Conti F, Buonfiglioli F, Scuteri A, Crespi C, Bolondi L, Caraceni P, et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. Journal of Hepatology. 2016;65(4):727-733. DOI: 10.1016/j.jhep.2016.06.015
75.Toyoda H, Tada T, Takaguchi K, Senoh T, Shimada N, Hiraoka A, et al. Differences in background characteristics of patients with chronic hepatitis C who achieved sustained virologic response with interferon-free versus interferon-based therapy and the risk of developing hepatocellular carcinoma after eradication of hepatitis C virus in Japan. Journal of Viral Hepatitis. 2017;24(6):472-476. DOI: 10.1111/jvh.12665
76.Calleja JL, Crespo J, Rincón D, Ruiz-Antorán B, Fernandez I, Perelló C, et al. Effectiveness, safety and clinical outcomes of direct-acting antiviral therapy in HCV genotype 1 infection: Results from a Spanish real-world cohort. Journal of Hepatology. 2017;66(6):1138-1148. DOI: 10.1016/j.jhep.2017.01.028
77.Lack of evidence of an effect of direct-acting antivirals on the recurrence of hepatocellular carcinoma: Data from three ANRS cohorts. Journal of Hepatology. 2016;65(4):734-740. DOI: 10.1016/j.jhep.2016.05.045
78.Cabibbo G, Petta S, Calvaruso V, Cacciola I, Cannavò MR, Madonia S, et al. Is early recurrence of hepatocellular carcinoma in HCV cirrhotic patients affected by treatment with direct-acting antivirals? A prospective multicentre study. Alimentary Pharmacology & Therapeutics. 2017;46(7):688-695. DOI: 10.1111/apt.14256
79.Minami T, Tateishi R, Nakagomi R, Fujiwara N, Sato M, Enooku K, et al. The impact of direct-acting antivirals on early tumor recurrence after radiofrequency ablation in hepatitis C-related hepatocellular carcinoma. Journal of Hepatology. 2016;65(6):1272-1273. DOI: 10.1016/j.jhep.2016.07.043
80.Austria A, Wu GY. Occult hepatitis C virus infection: A review. Journal of Clinical and Translational Hepatology. 2018;6(2):155-160. DOI: 10.14218/jcth.2017.00053
81.El Kassas M, Funk AL, Salaheldin M, Shimakawa Y, Eltabbakh M, Jean K, et al. Increased recurrence rates of hepatocellular carcinoma after DAA therapy in a hepatitis C-infected Egyptian cohort: A comparative analysis. Journal of Viral Hepatitis. 2018;25(6):623-630. DOI: 10.1111/jvh.12854
82.Schietroma I, Scheri GC, Pinacchio C, Statzu M, Petruzziello A, Vullo V. Hepatitis C virus and hepatocellular carcinoma: Pathogenetic mechanisms and impact of direct-acting antivirals. The Open Virology Journal. 2018;12:16-25. DOI: 10.2174/1874357901812010016
83.Shi J, He J, Lin J, Sun X, Sun F, Ou C, et al. Distinct response of the hepatic transcriptome to Aflatoxin B1 induced hepatocellular carcinogenesis and resistance in rats. Scientific Reports. 2016;6:31898. DOI: 10.1038/srep31898
84.Hamid AS, Tesfamariam IG, Zhang Y, Zhang ZG. Aflatoxin B1-induced hepatocellular carcinoma in developing countries: Geographical distribution, mechanism of action and prevention. Oncology Letters. 2013;5(4):1087-1092. DOI: 10.3892/ol.2013.1169
85.Wu HC, Santella R. The role of aflatoxins in hepatocellular carcinoma. Hepatitis Monthly. 2012;12(10 hcc):e7238. DOI: 10.5812/hepatmon.7238
86.Lopez-Valdes S, Medinilla-Cruz M. The relationship of aflatoxin b1 and hepatocellular carcinoma: A mini review. Journal of Liver Research, Disorders & Therapy. 2017;3(6):00073
87.Lv J, Yu YQ , Li SQ , Luo L, Wang Q. Aflatoxin B1 promotes cell growth and invasion in hepatocellular carcinoma HepG2 cells through H19 and E2F1. Asian Pacific Journal of Cancer Prevention. 2014;15(6):2565-2570. DOI: 10.7314/apjcp.2014.15.6.2565
Written By
Reem El-Shenawy, Sally Farouk, Naiera Helmy and Noha Bader El Din
Submitted: 27 December 2022Reviewed: 28 December 2022Published: 06 February 2023
 Open access peer-reviewed chapter
Open access peer-reviewed chapter