Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
To purchase hard copies of this book, please contact the representative in India:
CBS Publishers & Distributors Pvt. Ltd.
www.cbspd.com
|
customercare@cbspd.com
The most valuable application of next generation sequencing (NGS) technology is genome sequencing. Genomes of several aquatic models had been sequenced in the past few years due to their importance in genomics, development biology, toxicology, pathology, and cancer research. NGS technology is greatly advanced in sequencing length and accuracy, which facilitate the sequencing process, but sequence assembly, especially for the species with complicated genomes, is still the biggest challenge for bench-top scientists.
Xiphophorus Genetic Stock Center, Texas State University, San Marcos, TX, USA
Yingjia Shen
Xiamen University, Shenzhen, China
Wesley Warren
The Genome Institute at Washington University, St. Louis, MO, USA
Ronald Walter
Xiphophorus Genetic Stock Center, Texas State University, San Marcos, TX, USA
*Address all correspondence to: y_l54@txstate.edu
1. Introduction
Next generation sequencing (NGS) technology has been broadly used in biomedical research. The most valuable application of this technology is genome and transcriptome sequencing, which form a bridge to link fundamental discoveries in research using disease model systems to clinical application. Aquatic animal models are widely used in genomics, development biology, toxicology, pathology, and cancer research (for a recent review, see [1]). The genomes of several aquatic models had been sequenced using NGS technology over the past few years [2, 3]. NGS technology has been trending toward reduced cost with greater sequencing length and accuracy. While this has facilitated the sequencing process, sequence assembly remains a significant challenge for bench-top scientists, and especially for species with complicated genomes.
In this chapter, we will focus on the application of NGS in aquatic genome and transcriptome assemblies. Although our focus will be on the genome sequencing of aquatic models, the associated techniques, problems, concerns, and solutions can also be applied to genome sequencing of other model systems. Using Xiphophorus maculatus (X. maculatus), X. couchianus, and X. hellerii genome sequencing as examples, we will discuss the technical details of NGS, data processing, and genome assembly using guided approaches. We will also discuss the problems encountered in genome sequencing of several feral fish models (ice fish, blind cave fish, etc.) and alternative approaches to sequence and assemble these genomes. Some problems remain and these are causing a bottleneck to broadening the representation of aquatic models with genome assemblies. These problems are summarized and methods to address them in the next five years are proposed.
In recent years, aquatic animal models have been widely used in human disease research. These model systems have demonstrated the usefulness for improving our understanding of disease pathology at the molecular and cellular biology levels and have facilitated the development of new diagnostic and therapeutic methods. A few examples of diseases modeled by aquatic models are summarized in Table 1.
Table 1.
Aquatic models for human diseases
An example of the use of an aquatic model for human disease research is the Xiphophorus model. In the 1920s, it was found that F1 interspecies hybrids between X. maculatus (X. maculatus) and X. hellerii, when backcrossed to X. hellerii, result in melanoma development among 25% of the backcross progeny (Gordon-Kosswig cross [4–6]). The melanoma develops from naturally occurring macromelanophores that are found in Xiphophorus. In this cross, melanoma development is the result of interaction of a melanoma locus Tu and a tumor suppressor locus (R/Diff) that is capable of inhibiting Tu’s oncogenic effect in the parental X. maculatus fish. Since Tu and Diff are on different chromosomes, the segregation of Tu and Diff into backcross hybrids results in 25% of the animals with inherited Tu but do not inherit melanoma suppression by the R/Diff and thus exhibit melanomagenesis. The gene corresponding to Tu was discovered to be a mutant copy of the human epidermal growth factor receptor (EGFR) termed Xmrk, while a candidate gene for R/Diff is a Xiphophorus homologue of human cdkn2a/b (i.e., p15/16) [7–9]. It has been found that the mutational inactivation of human cdkn2a (p16) is associated with human melanoma (for a review, see [10]), and EGFR-driven downstream signaling by Ras-Raf-MAPK activation is a marker of human melanoma (for a review, see [11, 12]). This makes Xiphophorus a good model for genetic study of melanoma, a cancer that shows increasing worldwide incidence but has forwarded very few experimentally tractable animal models [13–15]. In addition to this spontaneous melanoma model, different Xiphophorus interspecies hybrids have been shown to be melanoma inducible after exposure to DNA damaging agents such as UVB light. Some of these inducible melanoma models involve hybridization of X. maculatus and X. couchianus with a following backcross of the F1 hybrid to the X. couchianus parent. Both the heavy pigmented backcross progeny and F1 hybrids can develop melanoma after UVB or MNU exposure in their early life stage [16–20].
Genomes of aquatic disease models serve as bridges to link phenotypic changes to genetic responses and allow physiological and pathophysiological discoveries from animal models to be applied to human disease research. The sequencing of model system genomes offers researchers great resources for biomedical research. Genome sequences allow researchers to (a) find sequence variation among genomes and transcriptomes between different species and populations; (b) compare genetic response between different phenotypes, development stages, disease conditions, drug treatment, etc.; and (c) discover gene/gene and gene/environment interactions and use these findings to direct medical applications.
For Xiphophorus, genome sequencing, assembly, and annotation for 3 Xiphophorus species (X. maculatus, X. couchianus, and X. hellerii) were accomplished in 2014 ([3, 21] and unpublished data). In the post-Xiphophorus genome era, these genomes resources have strengthened the Xiphophorus melanoma models by establishing high similarity in gene expression patterns for Xiphophorus and human melanoma tumors. The genome assemblies for both parents of an interspecific disease model are now allowing regulatory dissection of melanoma relevant gene expression in hybrids and after tumor-inducing treatments [22]. The gene expression features that characterize metastatic melanoma progression in humans closely mimic those found in Xiphophorus melanoma tumors (unpublished data). For the purpose of screening potential anti-melanoma compounds, a mutant Xmrk gene has been used to make a transgenic medaka (Oryzias latipes) fish model that develops melanoma very early after hatching [23, 24]. Whole transgenic melanoma medaka at 3–4 weeks post hatch are being utilized to characterize melanoma disease markers and for use in screening of small compounds for inhibitors of melanoma progression. In this way, several aquatic models systems represent a direct connection from “fish tank” discovery to “bedside” therapeutic application (for additional information on this topic, see https://dpcpsi.nih.gov/sites/default/files/orip/document/zebrafish_workshop_final_report_orip_website.pdf).
The NGS technique produces millions of short sequences (typical read length of 125 bp), which represent many unconnected small pieces of a genome or transcriptome, in each flow cell of the sequencing platform per sequence run. With these short sequences, one may de novo construct transcripts or genomes, characterize sequence variation (i.e., single nucleotide variation (SNV), insertion, and deletion), quantify sequence architecture (i.e., sequence repeats, copy numbers, and gene expression), and most importantly provide a sequence reference to expand discoveries from one species to another. Over the past decade, the sequence length of NGS (specifically Illumina technology) has significantly increased from 35 bp to current commonly produced 125 bp (Illumina HiSeq), and new long single sequence technology platforms are delivering sequence lengths of up to 40 kb in size (e.g., Pacific Bioscience RSII at 20 kb) that are changing the paradigm for whole genome de novo assembly.
It is beyond the scope of this chapter to examine all of the current and upcoming sequencing technologies, and thus we focus on the most common NGS platform that is currently being employed to establish genomic and transcriptomic resources in aquatic models systems.
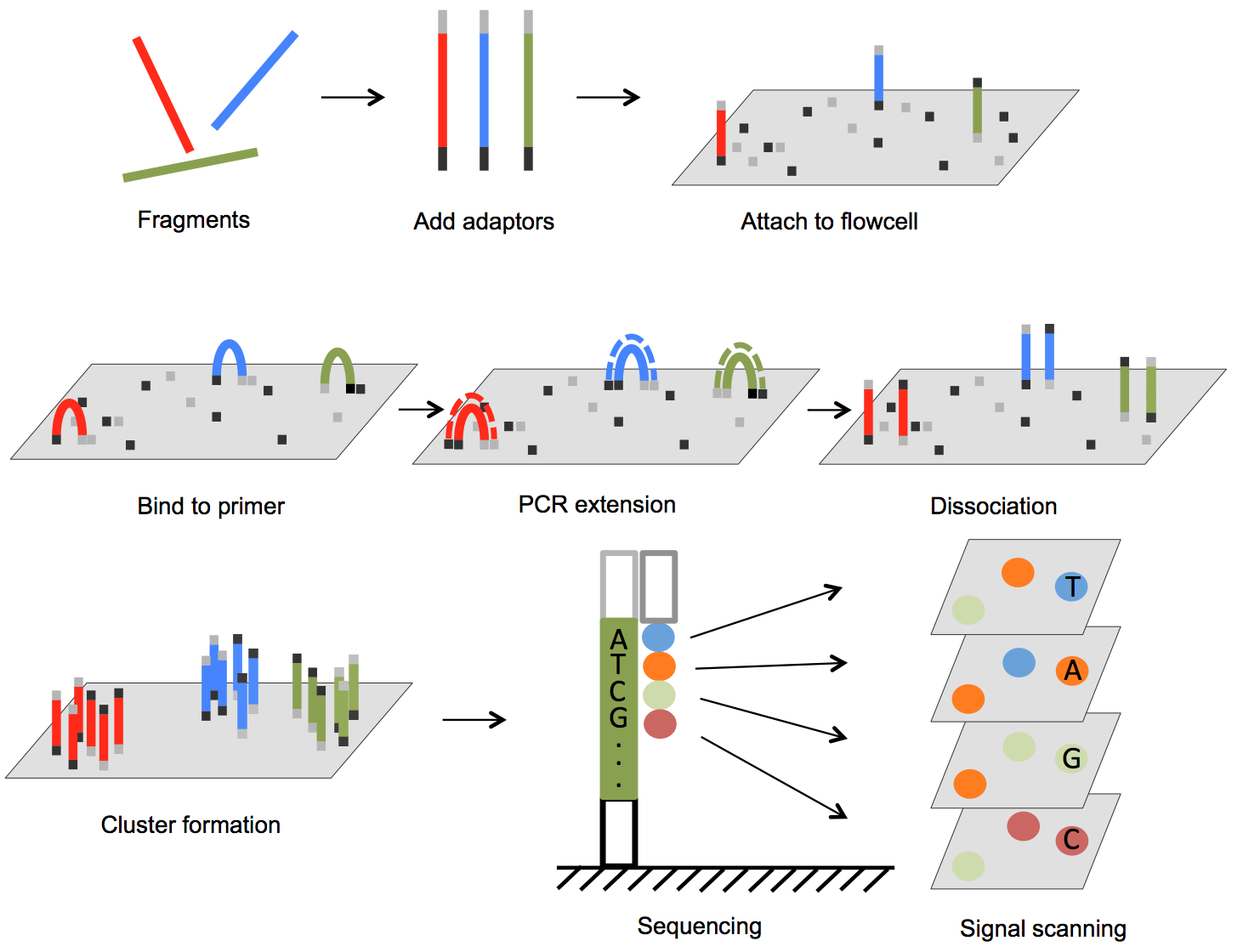
The Illumina genome analyzer platform is currently the most widely used NGS system accounting for over 70% of the NGS market [25]. In Figure 1, we illustrate the basic steps of Illumina sequencing technology. The sequencing process starts with preparation of a library. The DNA (for genomic sequencing) or cDNA (for RNA sequencing) sample is sheared, usually by physical, enzymatic, or chemical method, into short fragments predetermined to be a specific size, and then sequencing adaptors are ligated to both ends of each short fragment by annealing. The fragments are then loaded onto a flow cell. The flow cell has oligonucleotides bound to the surface of the flow cell, and their sequences are complementary to the adaptors such that the free end of the fragment is attached to the flow cell via base pairing. A PCR step converts the initial fragment to its complementary sequence, and now both the forward strand and the reverse strand of fragments are bound to the surface of the flow cell (Figure 1). To amplify the signal, PCR is repeated for several rounds resulting in a cluster of copies around the initial copy of a fragment. Cyclic sequencing of these fragment clusters is very similar to Sanger sequencing and utilizes a sequence-by-synthesis process. One of two unique primers is attached to the free end of the bound fragments, and then nucleotides that each carries a different fluorescent reporter tag and a reversible terminator are flowed onto the flow cell. Since each nucleotide contains an elongation terminator, only a single nucleotide can be incorporated into newly synthesized sequences per sequencing cycle. After the nucleotide incorporation, laser sources excite the fluorescent reporter, and an optical sensor scans the entire flow cell to capture colors that represent newly added bases in every cluster. This optical information is converted to a base call for each growing sequence. At the end of each cycle, the terminator is removed and the next cycle continues until the desired sequence length is attained. In paired-end sequencing, after the forward strand sequence is attained, another sequence primer initiates the sequencing of the reverse strand of each fragment.
Figure 1.
Outline of Illumina genome analyzer sequencing process. (1) Adaptors are annealed to the ends of sequence fragments. (2) Fragments bind to primer-loaded flow cell and bridge PCR reactions amplify each bound fragment to produce clusters of fragments. (3) During each sequencing cycle, one fluorophore attached nucleotide is added to the growing strands. Laser excites the fluorophores in all the fragments that are being sequenced and an optic scanner collects the signals from each fragment cluster. Then the sequencing terminator is removed and the next sequencing cycle starts.
This massively parallel sequencing platform allows high throughput sequencing. Each flow cell contains 8 lanes with each lane producing 250 million reads (i.e., up to 500 GB/flow cell) with length of each sequence read ranging from 35 bp to 250 (Illumina HiSeq-2500) or 300 bp (Illumina MiSeq). Each sequencing adaptor has incorporated into it a unique barcode in the format of oligonucleotides. Thus, multiple samples from different sources can be pooled together in one lane, and this greatly facilitates the sequencing throughput.
Before subsequent sequence assembly or reference sequence alignment, a quality control step is usually necessary to attain sequences that best represent the biology being studied. A short sequencing result file contains two types of “contaminants” that can hinder the sequence assembly and result in misrepresentation of actual nucleotide sequence: adaptor sequence and low quality base calls. For paired-end sequencing, the length of DNA fragment between the two adaptor sequences is defined as “insertion size.” When the desired sequencing length is longer than insertion size, the short sequencing can contain adaptor sequence in it. This artificial sequence must be trimmed off, so as not to produce significant sequence error in sequence assemblies. Another contaminant, the low quality base call, has many sources, from equipment to sequencing glitches. The quality of a base call is defined as Phred quality score (QPhred score). If we assign P as base calling error probabilities [26], then
QPhred=–10log10PBB1
To retain the most usable as high-quality sequencing reads, the adaptor sequences are first clipped off, subsequently trim off low-quality base calls at the end of sequencing reads, and finally filter out sequence reads that contain a certain percentage of base calls that are below a defined QPhred score. Several tool software packages are available that can be utilized to perform the read filtering steps (e.g., fastx_toolkit: http://hannonlab.cshl.edu/fastx_toolkit/).
3.2. Sequence assembler algorithms
There are two major types of sequence assembly methods, Overlap-Layout-Consensus assembly and De Bruijn graph assembly. Current efficient and successful sequence assembly programs, including the ones employed for Xiphophorus genome assemblies (i.e., ALLPATHS), utilize the De Bruijn graph as a central data processing structure (De Bruijn-based assemblers are summarized in Table 2).
Table 2.
De Bruijn-based sequence assembler
De Bruijn graph-based assembler begins the assembling process by breaking the sequencing reads into k-mers, which in a genome is defined as a sequence of k consecutive bases. To build a De Bruijn graph, each k-mer is split into two parts, the left (k– 1) base x and right (k– 1) base y. Then all the x and the possible y are joined together by directed edges (x → y). A De Bruijn graph is obtained by taking the x and the y as nodes and the adjacencies as edges. The edges represent (k– 1) overlap between the connected nodes. In DNA sequencing, each node can have 8 possible connections, 4 are from the upstream sequence and 4 are to the downstream sequence, respectively. Actual connections are recorded in the memory as they are observed in the sequencing data. As sequencing data runs through the graph-building algorithm, discrete seed graphs are joined as the reads connecting to them are identified. In Figure 2, we present a simplified assembly and a sequence feature that can lead to problems in the sequence assembling process.
Figure 2.
Outline of De Bruijn graph build during the sequence assembling process. A short model genome is sequenced. Four short reads were generated from template. The k-mer length of 5 was chose to be used in sequence assembly. For each k-mer, the left k– 1 and right k– 1 were represented as nodes in the De Bruijn graph, and all left parts are connected to possible right parts by directed edges. The red digit shows the number of occurrence of each node. The cyclic edge at the rightmost end of the graph causes the gap of contig assembly. Thus, the final assembly does not fully represent the “repeat” in the genome sequence.
In Figure 2, 4 short DNA fragments that were attained from a randomly sheared 21 nt genome are sequenced. The k-mer length of 5 was chosen for this assembly. In the De Bruijn graph, there are 11 balanced nodes, where the number of indegree equals that of outdegree, and two semibalanced nodes, where indegree differs from outdegree. This graph is directed, connected, and considered as Eulerian since it has and only has at most 2 semibalanced nodes. The node in this directed graph that has more outdegree than indegree is considered to be the staring site of the assembly, while the other semibalanced node is the end of the assembly. At the end of the graph, where a cyclic edge forms, a problem for short sequence assemblers when repetitive sequence regions are encountered is presented. De Bruijn algorithms cannot resolve this problem and will simply ignore it, resulting in gaps in the contigs assembled. Long repeats present in the genome constantly cause assembly issues in practice. A detailed solution to this will be discussed in the following part of this chapter.
Taking ALLPATHS for instance, the memory use is estimated to be roughly 1.7 bytes per read base, which equals to a 102-GB RAM of a 60× coverage 1-GB genome. This level of RAM requirement can be fully fulfilled nowadays. Alternatively, this RAM requirement can be solved by sharing memory from different computer nodes, or by distributing the workload to different nodes within a computer cluster, which is normally accessible in most universities and research institutions. In addition, the development of cloud computing allows one to gain access to high-speed computer clusters in a pay-as-you-go manner, and there are several recently developed cloud-based sequence assemblers (summarized in Table 3).
Table 3.
Cloud computing-based sequence assemblers
3.3. Xiphophorus genome sequencing and assembly
Sequencing of X. maculatus genome is of great value to the aquatic model community [3, 21]. A problem encountered by those using the Xiphophorus model was that a genome sequence of one single Xiphophorus parent used in an interspecies cross did not allow the regulation of allele-specific gene expression to be determined in interspecies hybrid. The interspecies crosses are important in disease model research for both spontaneous and induced melanoma and other life history traits that involve complex genetic interactions. Therefore, 2 additional Xiphophorus species genomes (X. couchianus and X. hellerii) have been sequenced and assembled. In this section, sequencing and assembling multiple Xiphophorus species genomes is used as real-world example of the process of genome sequencing.
3.3.1. Biological sample
X. couchianus were maintained by sibling inbreeding, and the fish that were sequenced were in their 77th generation of inbreeding. X. hellerii was maintained by reciprocal cross breeding between 2 distinct X. hellerii strains, differing by sword color. All the fish that were used for genome sequencing were female since the high degree of repetitive DNA generally found to make up Y-chromosomes can confound the downstream assembly.
3.3.2. Genome sequencing and assembly
The Illumina HiSeq-2000 platform was chosen for Xiphophorus genome sequencing. Sequencing libraries with different insert sizes (300 bp, 500 bp, 3 kb, and 8 kb) were prepared. The purpose of using different insert size libraries is to using the paired-end reads that span different lengths of genome to estimate the gap size in a higher level of assembly. Over 700 (X. couchianus) and 360 (X. hellerii) million 100 bp paired-end short sequence reads were obtained from sequencer.
Genomes of X. couchianus and X. hellerii were constructed at three stages: contig, scaffold, and chromosome. The contigs were assembled in a de novo manner to maximally capture any sequences that are not present in X. maculatus, while scaffolds and chromosomes were assembled using the X. maculatus genome as a reference to guide assembly.
The first stage contig assembly was carried out by ALLPATHS using only the Illumina sequencing reads. This step generated contig-level assembly with N50 of 60 kb and 30 kb for X. couchianus and X. hellerii, respectively.
These contigs were further grouped into scaffolds using the X. maculatus scaffolds assembly as reference. X. couchianus and X. hellerii de novo assembled contigs, as well as the sequencing reads, were aligned to X. maculatus genome scaffold assembly using a multi-phase aligner SRprism (ftp://ftp.ncbi.nlm.nih.gov/pub/agarwala/srprism/). The sequence gaps between consecutive contigs were filled with long-insertion paired-end Illumina reads that bridge the upstream ends and downstream ends of contigs that are right next to the gaps. Scaffolding of contigs and gap fillings increased the length of both assemblies to N50s of 1.8 Mb and 1.6 Mb, respectively.
The construction of chromosomal level genome was accomplished by aligning de novo assembled contigs to the X. maculatus chromosome assembly using Mummer 3 package Nucmer3.0 (http://mummer.sourceforge.net). For each species, sequences of contigs and the location of X. maculatus chromosome alignments were recorded. By using a customized Perl script, these sequences and alignment information were organized into chromosomes.
3.3.3. Genome annotation
To annotate the newly assembled X. couchianus and X. hellerii genome, two methods, rapid annotation of transfer tool (RATT) and de novo assembled transcriptome, were used and the result from each were compared to each other.
Transcript sequences and associated functional annotations can be transferred between closely related species. A modified gene annotation method, RATT, was applied using the X. maculatus genome and gene model as a reference to quickly transfer genome annotation [27]. Since the X. maculatus genome was already available, using RATT to transfer annotation can minimize computational and human resources that are required for genome annotation. Both X. couchianus and X. hellerii genomic scaffold sequences were used as query species to be aligned to the well annotated X. maculatus genome using Nucmer3.0 with parameters implemented by RATT for annotation transfer. To avoid frame shift between two species, the synteny between both species and reference was established and insertions/deletions were also identified, respectively. X. maculatus gene models were then transferred and corrected to both query species. Of the 20,482 gene models annotated in Xiphophorus genome, 20,300 and 20,325 of them were transferred to X. couchianus and X. hellerii, respectively (Table 4).
To compare to this RATT annotation transfer method, X. couchianus and X. hellerii genome annotations were also annotated with a different method using de novo assembled transcriptomes. This method is reference genome independent. Briefly, RNA samples from one month old whole fish of X. hellerii and X. couchianus and a collection of tissues of mature individuals of each species were sequenced using Illumina GAIIx platform as 60 bp paired-end reads as well as HiSeq-2000 platform as 100 bp paired-end reads. De novo transcript assemblies and reports of putative transcripts were performed using velvet v1.1.05 and Oases v0.1.22 [28, 29]. The transcriptome assembly resulted in 110,604 and 242,675 transcripts for X. couchianus and X. hellerii, respectively.
Table 4.
Comparisons between reference-based annotation and de novo-based annotation
Comparing these two methods of annotation to each other in perspective of transcriptome quality, de novo method produced very larger transcriptomes in number of transcripts and final assembly size (Table 4). Many transcripts produced this way are unverified isoforms of same genes and redundant splicing isoforms of the same gene. In contrast, the RATT gene model transfer produced transcriptomes are similar to the reference [27]. In addition, both methods produced comparable N50s; however, reference-based method had longer average length, suggesting this method is superior.
In conclusion, the de novo assembly of a species transcriptome and its use in biological inference studies is appropriate, when a reference genome is not available and assuming tissue diversity is adequately captured. Nonetheless, reference-based gene model transfer is a reliable, economical, and efficient means to annotate closely related species.
3.3.4. Transposable elements analysis
As found previously, X. maculatus transposable elements (TEs) make up ~5% of the transcriptome [3]. Although the percentage of TEs is only slightly higher than the compact genomes of puffer fishes and is close to that of chicken genome, there is a high diversity of TE families in X. maculatus genome [3, 30, 31].
To annotate the TEs in X. couchianus and X. hellerii genomes, a previously established library was further completed employing RepeatScount (http://bix.ucsd.edu/repeatscout/) and RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html) software. Redundant sequences were discarded, leaving 1019 sequences in the new library. RepeatMasker (http://www.repeatmasker.org/) was subsequently utilized to mask genome assemblies. Custom Perl script was then used to establish repeat coverage and copy numbers. After removing TE sequences that are smaller than 80 nt and share less than 80% identity with reference library, TEs were found to make up ~12% of each Xiphophorus genome (X. maculatus, 12.11%; X. couchianus, 12.61%; X. hellerii, 12.14%; unpublished data). A detailed classification of TEs in each Xiphophorus genome is shown in Table 5.
4. Problems and potential resolutions in genome assembly
4.1. Repetitive sequences in genome result in gaps of assembly
Several aquatic model genomes have been sequenced, assembled, and annotated for public use due to the activities of the aquatic model community. During the genome sequencing and assembling process for many of these model systems, several problems have been encountered. Specific sequence architecture (e.g., repetitive sequences) may confuse assembly algorithms and results in gaps in sequence contiguity that ultimately lead to a poorly-assembled genome or no assembly at all. For example, k-mer frequency estimation showed the toadfish genome consisted of ~48% repetitive sequences, which account for the rather high assembly fragmentation. Regions that have assembling difficulties typically include repeats (repetitive sequences of varied lengths, usually found in intergenic regions), telomere sequences (short sequence repeated thousands of times), centromere sequences (large array of repetitive DNA), segmental duplication of loci (segments of DNA with near-identical sequence), and closely organized gene families (portion of genome with genes of very similar sequences). The problems in assembling these regions are also present in genome sequencing projects of other model organisms. During the sequence assembly of aquatic models listed in Table 6, a conservative estimation of missing bases in each draft genome shows a range of 66 to 239 Mb within scaffolds, and 14 Mb to 26 Mb between scaffolds, respectively.
Table 6.
Reference assembly gap sequence estimates from NCBI or Ensembl
Although the length of sequencing reads continues to expand, repetitive sequences are still the main barrier encountered, toward a goal of uninterrupted consensus base counts. It is well known no graphical-based assembly method completely resolve repeat structure. Both graphical approaches, De Bruijn and Overlap-Layout-Consensus, will exclude repetitive sequence by truncating the assembly when certain repeat types are encountered or alternatively collapse unique repeats into a single representation (Figure 2). This leaves gaps in sequence assembly and collapses long repeat sequences. Some of the gaps can be closed by using proper oriented paired-end reads with long insertion sizes, such as bacteria artificial chromosome or P1-derived artificial chromosome clones. However, in most cases, such long insert resources are not available. During scaffold assembly of X. couchianus and X. hellerii genomes, consensus contigs were built by locating consecutive contigs bridged by mate pairs having 30-mers on each side of the gap, followed by de novo assembly in gaps using the bridged contigs and 30-mers from reads that were used in the first-level contig assembly. However, repetitive regions that expand hundreds of Mb can still not be resolved by this method.
4.2. Long sequencing reads are possible solution to assembly issues
Since repetitive sequences are the major causes of gaps in sequence assemblies, one way to maximize assembly contiguity is to employ long reads that are capable of covering the entire repetitive regions. The Pacific Bioscience (PacBio, www.pacificbiosciences.com) P6-C4 sequencing platform now offers the longest sequencing reads in the field, with longest sequence read length of 40 kbp and an average length of ~10 kbp (Figure 3).
Figure 3.
Outline of PacBio Single Molecule Real Time sequencing (SMRT) technology. Unlike Illumina sequencing platform, the sequencing adaptor form loops at the ends of double-stranded DNA fragments and ultimately form a circular sequencing template. After removing the adaptor sequences from raw reads, the genomic sequence information can be retained for de novo assembly. P6-C4 chemistry offers currently longest sequence reads. (The figure on the right is from Pacific Biosciences, http://www.pacificbiosciences.com/products/smrt-technology.)
Since PacBio long sequencing reads are capable of traveling through the repeat regions, therefore gaps are less likely to be present when assembling the genome. In several recent aquatic genome-sequencing projects, the incorporation of PacBio sequencing technology in concert with very deep Illumina 100 bp paired-end reads (60× coverage) significantly improved the quality of genome assembly. For example, using 8×–30× PacBio sequence coverage, 62% of gaps could be closed with a 2-fold increase in N50 contig length for the blind cavefish genome build (unpublished data). Similarly, gap filling using long sequencing reads almost tripled the N50 contig length (from 5 kb to 14 kb) for the ice fish genome, but this genome assembly remains plagued with difficult regions that have yet to be resolved (unpublished data).
The usage of long sequencing reads to improve the current genome builds is not limited to aquatic genome research as this application has also been utilized in the improvement of genome quality of other model organisms as well (e.g., avian models [32]). For example, the current chicken reference genome has 8106 gaps within scaffolds. After PacBio’s long sequence reads (10× coverage) were incorporated, 6888 of these gaps were closed, along with 6.3 Mb of new sequence added (unpublished data).
For small genomes (<200 Mb haploid size), long sequencing read technology has advanced to a stage where near complete genomes can be represented. For example, the Drosophila genome has 139.5 million base pairs located on 4 pairs of chromosomes that can be covered once by 10,000 averaged-length PacBio sequencing reads [33]. One concern of PacBio long sequencing technology is its high error rate (median error rare of ~11%) in base calls. However, this “error-prone” problem can be addressed. First, PacBio sequencing technology utilizes a circular template. It allows the polymerase to travel through the template multiple times, thus generating several copies of reads that represent the same genome fragment. Second, although the error rate of “single-pass” PacBio sequencing reads is high, the errors are distributed randomly and can be filtered out upon building consensus for all sequence copies of a given fragment. Quiver (www.pacbiodevnet.com/Quiver) was developed to deliver high-quality consensus sequences by averaging the sequence information for each base call vertically to each other. Based on the error rate, 9 out of 10 reads will contain a correctly sequenced base, making it straightforward to distinguish the correct base call. This error correction is capable of generating >99.9% accurate consensus sequence [34, 35].
In addition to improving current genome assembly quality, long sequencing reads are capable of sequencing full-length transcripts, thus facilitating gene expression analyses and transcriptome assembly. Current RNA-Seq tasks apply short reads (50 bp single-end to 125 bp paired-end depends on experiment design) to fragmented cDNA libraries. These short reads are then aligned to either reference genome or an array of reference transcripts for statistical analysis of gene expression. Uniquely aligned short reads provide solid evidence of the expression levels of the aligned genes. However, inappropriate treatment of ambiguously aligned reads can lead to biased or even mistaken expression profiles in complicated vertebrate genomes (e.g., zebrafish genome and human genome). This problem severely affects transcript variance discovery such as alternative splicing and relative expression of alternative splicing isoforms, which play significant roles in pathological processes (e.g., Bcl11b1). Alternative splicing isoform expression quantification heavily relies on distribution of short reads on each exon; thus, low-coverage splicing isoforms cannot be distinguished [36]. The utilization of PacBio long-read sequencing platform can eliminate this problem by providing long reads that are capable of covering all connected exons in one single read, thus avoiding mistakes in assigning reads to a certain exons [37].
The availability of aquatic genome models in the past few years significantly expends the resources for biological and biomedical discovery. However, as detailed, problems persist in the current aquatic model draft assemblies (i.e., gaps in and between scaffold and repetitive sequence). Over the next few years, there should be a concerted effort to (a) de novo assemble genomes by combining standard Illumina library builds with new PacBio long-read sequencing and (b) developing new assembly routines to resolve assembly errors and create chromosome builds for each species.
5.1. De novo genome assembly using long sequencing reads
In Table 6, we show estimated sequence gaps missing from within scaffolds. It is estimated that 2–5% of each genome is not sequenced or assembled outside of scaffold gaps (unpublished result). Previous tasks to close gaps in the assemblies of other species genomes have shown that structurally variant alleles, simple tandem repeats, and high GC content regions account for the majority of these gaps. The new PacBio sequencing technology, if used to produce high coverage (at least 60×) fragments, may be expected to overcome many of these assembly problems and should result in better-represented genome models. Assembling genomes using PacBio sequencing reads requires special treatment to the raw reads, as well as the sequence assembling processes. For example, the multiple-pass raw reads from circular sequencing template need to be clipped into subreads that represent the DNA fragment. The PacBio sequencing reads also need to be error-corrected using Quiver. The sequence assembling process with these very long reads requires different tools than what were discussed above. MinHash Alignment Process (MHAP) that is included in Celera Assembler PBcR pipeline is a reference implementation of a probabilistic sequence overlapping algorithm that is designed for detecting overlaps between long-read sequence data [33]. It is therefore a proper tool for sequence assembly that employs long sequencing reads.
During the process of de novo genome assembly using long sequencing read technology, higher-quality genome models are expected. This will provide animal disease model communities much better genome references (longer N50, less gaps and less missing bases) in newly developed draft de novo assemblies. In addition, re-sequencing to enhance the contiguity of current genome assemblies by incorporating PacBio reads promises to produce much improve reference genomes in the next few years.
5.2. Chromosome level aquatic genome assembly
Accurate chromosome assemblies require correctly ordered contigs in scaffolds for gene functional interpretation. During chromosome construction, the placement and order of scaffolds on chromosomes relies on a genetic map, which is based on meiotic recombination. Among the aquatic genome models created in the past few years, the Xiphophorus genome assembly has been aligned to chromosomes using a Rad-Tag approach to generate a meiotic gene map having over 16,000 markers ([21] and unpublished data). The RAD-tag markers and microsatellite makers from older studies were used to guide the placement of scaffolds into the Xiphophorus chromosomes (for RAD-tag method, see [38]). However, the RAD-tag map method is resource and labor intensive, for examples, 267 backcross Xiphophorus hybrids were used for genetic mapping and sequence alignment [21].
Recently, new optical mapping technology has been provided by BioNano (http://www.bionanogenomics.com). The optical mapping improves the process of constructing whole genome physical map. In this process, high molecular weight genomic DNA is immobilized onto the positively charged glass surface of a chip-like device having engraved nano-channels that are only wide enough to stretch a single DNA molecule. Buffer fluid that flows though the channel stretches a single DNA molecule to maintain its orientation and integrity. The DNA molecules are subsequently sheared by a restriction enzyme into fragments that are stained with florescent dye. An imaging system then measures the florescent light intensity that represents the length of each DNA fragment. Accompanied with the restriction enzyme site sequence, the length of each fragment is linked to form a single-molecule optical restriction map.
During chromosome assembly, the scaffold sequences can be converted to in silico restriction map. The location of the restriction enzyme digestion sequence and the distance between these sequences can then be used to assign scaffolds into chromosomes [39]. Using this approach, incorrect joining errors of contigs may be corrected to improve the current reference genome continuity concurrent with scaffolds alignment into chromosomes.
Figure 4.
Illustration of optic mapping technology. Genomic DNA is obtained from lysed cells and is loaded onto a chip-like channel-forming device. DNA molecules are stretched onto a positively changed glass surface by buffer fluid that flows through the channels. This step maintains the integrity and orientation of the DNA molecule for subsequent steps. The stretched and immobilized DNA molecules are digested with a restriction enzyme and subsequently stained with florescent dye. The florescent light intensity of each DNA fragment was imaged, and the images are analyzed to measure the size of DNA fragments. Using the restriction enzyme digestion site sequence and the distance between digestion sites, a single-molecule restriction map can be generated to guide scaffold assignment.
Aquatic models are proven to be as important and useful as other animal models to study the etiology and progression of human disease. Aquatic models have gained the attention of funding agencies, and the overall research community using aquatic models has grown rapidly. This growth has resulted in the availability of genome and reference transcriptome resources. The aquatic genome models that were constructed in the past few years are available through NCBI or Ensembl with new updates constantly being made. Although problems persist in genome assembly of complicated structures, newer sequencing platforms, mapping technologies, and sequence assembly algorithms are expected to rapidly address these problems and soon offer the community much improved resources.
References
1.Schartl, M., Beyond the zebrafish: diverse fish species for modeling human disease. Dis Model Mech, 2014. 7(2): p. 181–92.
2.McGaugh, S.E., et al., The cavefish genome reveals candidate genes for eye loss. Nat Commun, 2014. 5: p. 5307.
3.Schartl, M., et al., The genome of the platyfish, Xiphophorus maculatus, provides insights into evolutionary adaptation and several complex traits. Nat Genet, 2013. 45(5): p. 567–72.
4.Gordon, M., The genetics of a viviparous top-minnow platypoecilus; the inheritance of two kinds of melanophores. Genetics, 1927. 12(3): p. 253–83.
5.Häussler, G., Über Melanombildungen bei Bastarden von Xiphophorus Helleri und Platypoecilus Maculatus var. Rubra. J Mol Med, 1928. 7: p. 1561–1562.
6.K., K., Über Bastarde der Teleostier Platypoecilus und Xiphophorus. Z Indukt. Abstamm Vererbungsl, 1928. 44: p. 150–158.
7.Adam, D., W. Maueler, and M. Schartl, Transcriptional activation of the melanoma inducing Xmrk oncogene in Xiphophorus. Oncogene, 1991. 6(1): p. 73–80.
8.Kazianis, S., et al., Localization of a CDKN2 gene in linkage group V of Xiphophorus fishes defines it as a candidate for the DIFF tumor suppressor. Genes Chromosomes Cancer, 1998. 22(3): p. 210–20.
9.Kazianis, S., et al., Comparative structure and characterization of a CDKN2 gene in a Xiphophorus fish melanoma model. Oncogene, 1999. 18(36): p. 5088–99.
10.Mehnert, J.M. and H.M. Kluger, Driver mutations in melanoma: lessons learned from bench-to-bedside studies. Curr Oncol Rep, 2012. 14(5): p. 449–57.
11.Daud, A. and B.C. Bastian, Beyond BRAF in melanoma. Curr Top Microbiol Immunol, 2012. 355: p. 99–117.
12.Kraehn, G.M., M. Schartl, and R.U. Peter, Human malignant melanoma. A genetic disease? Cancer, 1995. 75(6): p. 1228–37.
13.Reed, D., et al., Controversies in the evaluation and management of atypical melanocytic proliferations in children, adolescents, and young adults. J Natl Compr Canc Netw, 2013. 11(6): p. 679–86.
14.Herlyn, M. and M. Fukunaga-Kalabis, What is a good model for melanoma? J Invest Dermatol, 2010. 130(4): p. 911–2.
15.Ha, L., et al., Animal models of melanoma. J Investig Dermatol Symp Proc, 2005. 10(2): p. 86–8.
16.Kazianis, S., et al., Genetic analysis of neoplasia induced by N-nitroso-N-methylurea in Xiphophorus hybrid fish. Mar Biotechnol (NY), 2001. 3(Supplement 1): p. S37–43.
17.Nairn, R.S., et al., A CDKN2-like polymorphism in Xiphophorus LG V is associated with UV-B-induced melanoma formation in platyfish-swordtail hybrids. Proc Natl Acad Sci U S A, 1996. 93(23): p. 13042–7.
18.Rahn, J.J., et al., Etiology of MNU-induced melanomas in Xiphophorus hybrids. Comp Biochem Physiol C Toxicol Pharmacol, 2009. 149(2): p. 129–33.
19.Setlow, R.B., et al., Wavelengths effective in induction of malignant melanoma. Proc Natl Acad Sci U S A, 1993. 90(14): p. 6666–70.
20.Setlow, R.B., A.D. Woodhead, and E. Grist, Animal model for ultraviolet radiation-induced melanoma: platyfish-swordtail hybrid. Proc Natl Acad Sci U S A, 1989. 86(22): p. 8922–6.
21.Amores, A., et al., A RAD-tag genetic map for the platyfish (Xiphophorus maculatus) reveals mechanisms of karyotype evolution among teleost fish. Genetics, 2014. 197(2): p. 625–41.
22.Lu, Y., et al., Molecular genetic response of Xiphophorus maculatus–X. couchianus interspecies hybrid skin to UVB exposure. Comp Biochem Physiol C Toxicol Pharmacol, 2015.
23.Schartl, M., et al., Conserved expression signatures between medaka and human pigment cell tumors. PLoS One, 2012. 7(5): p. e37880.
24.Schartl, M., et al., A mutated EGFR is sufficient to induce malignant melanoma with genetic background-dependent histopathologies. J Invest Dermatol, 2010. 130(1): p. 249–58.
25.Thayer, A.M., Next-Gen sequencing is a numbers game. Chem Eng News, 2014. 92(33): p. 11–15.
26.Ewing, B. and P. Green, Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res, 1998. 8(3): p. 186–94.
27.Otto, T.D., et al., RATT: Rapid Annotation Transfer Tool. Nucleic Acids Res, 2011. 39(9): p. e57.
28.Schulz, M.H., et al., Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics, 2012. 28(8): p. 1086–92.
29.Zerbino, D.R. and E. Birney, Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res, 2008. 18(5): p. 821–9.
30.Wicker, T., et al., The repetitive landscape of the chicken genome. Genome Res, 2005. 15(1): p. 126–36.
31.Aparicio, S., et al., Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science, 2002. 297(5585): p. 1301–10.
32.Ganapathy, G., et al., High-coverage sequencing and annotated assemblies of the budgerigar genome. Gigascience, 2014. 3: p. 11.
33.Berlin, K., et al., Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol, 2015. 33(6): p. 623–30.
34.Carneiro, M.O., et al., Pacific biosciences sequencing technology for genotyping and variation discovery in human data. BMC Genomics, 2012. 13: p. 375.
35.Koren, S., et al., Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol, 2012. 30(7): p. 693–700.
36.Hiller, D. and W.H. Wong, Simultaneous isoform discovery and quantification from RNA-seq. Stat Biosci, 2013. 5(1): p. 100–18.
37.Au, K.F., et al., Characterization of the human ESC transcriptome by hybrid sequencing. Proc Natl Acad Sci U S A, 2013. 110(50): p. E4821–30.
38.Lewis, Z.A., et al., High-density detection of restriction-site-associated DNA markers for rapid mapping of mutated loci in Neurospora. Genetics, 2007. 177(2): p. 1163–71.
39.Dong, Y., et al., Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus). Nat Biotechnol, 2013. 31(2): p. 135–41.
Written By
Yuan Lu, Yingjia Shen, Wesley Warren and Ronald Walter
Submitted: 14 April 2015Reviewed: 01 October 2015Published: 14 January 2016
 Open access peer-reviewed chapter
Open access peer-reviewed chapter